

Abstract
Major histocompatibility complex (MHC) class I variants H-2Kb and H-2Kbm8 differ primarily in the B pocket of the peptide-binding groove, which serves to sequester the P2 secondary anchor residue. This polymorphism determines resistance to lethal herpes simplex virus (HSV-1) infection by modulating T cell responses to the immunodominant glycoprotein B498-505 epitope, HSV8. We studied the molecular basis of these effects and confirmed that T cell receptors raised against Kb–HSV8 cannot recognize H-2Kbm8–HSV8. However, substitution of SerP2 to GluP2 (peptide H2E) reversed T cell receptor (TCR) recognition; H-2Kbm8–H2E was recognized whereas H-2Kb–H2E was not. Insight into the structural basis of this discrimination was obtained by determining the crystal structures of all four MHC class I molecules in complex with bound peptide (pMHCs). Surprisingly, we find no concerted pMHC surface differences that can explain the differential TCR recognition. However, a correlation is apparent between the recognition data and the underlying peptide-binding groove chemistry of the B pocket, revealing that secondary anchor residues can profoundly affect TCR engagement through mechanisms distinct from the alteration of the resting state conformation of the pMHC surface.
Keywords: major histocompatibility complex, crystallography, antigen presentation, herpes simplex virus 1, T cells
Introduction
Cytotoxic CD8+ T cells are the main effector arm of the adaptive immune system in charge of combating intracellular pathogens. These cells recognize pathogen-derived peptides presented by surface-expressed MHC class I molecules and lyse the infected cell that bears them. Analysis of the three-dimensional crystal structures of MHC class I molecules in complex with bound peptide (pMHCs) has revealed in great molecular detail the mechanisms these proteins use to bind a vast number of chemically distinct peptides (1, 2). The peptide-binding groove formed by the α1/α2 domains is structured to bind peptides 8–10 amino acids long in an extended conformation. Eight β-strands establish the platform floor of the groove, with two antiparallel α-helices serving as its walls. The peptide binds between these helices and uses its main chain and terminal atoms to form extensive hydrogen bonds to MHC side chains, enabling peptide binding in a predominantly sequence-independent manner. Peptide selectivity is achieved via discrete peptide-binding pockets present in the groove that are tailored to preferentially accept a subset of peptide side chains dubbed binding anchors. The high degree of polymorphism observed in class I alleles is clustered to residues that form these binding pockets, providing the basis for allele-specific peptide-binding motifs (3). The motifs for most MHC class I alleles can be described as a combination of preferred residues located at primary and secondary anchor positions (4). Primary anchor residues are typically completely buried within the peptide-binding groove pockets in a tightly coordinated fashion; only a highly restricted set of amino acids can typically serve as the primary anchor of a given pocket, allowing for highly stable peptide binding. In contrast, secondary anchor residues tend to be less restrictive in allowable sequence due to flexibility of the engaging pocket, and their substitution to less favorable residues often does not markedly affect peptide-binding affinity or kinetics.
TCR recognition of pMHC has also been studied extensively (5). The recent crystal structures of TCR–pMHC complexes have demonstrated that the TCR binds in a roughly diagonal orientation over the groove-flanking α-helices, positioning its CDR loops over the peptide (6–8). Numerous studies examining the role of solvent-exposed, TCR-contacting peptide side chains in TCR recognition have shown that even minor chemical differences in them can be discerned (9–12). In contrast, the peptide anchor side chains are buried and are thought unable to directly contact the TCR. These, and other similar observations, suggest that modulation of peptide-binding affinity is the dominant way anchor side chains affect TCR recognition. However, our group and others have recently reexamined this issue and have shown that peptide anchor side chains, especially secondary anchors, can have a more direct effect, independent of peptide-binding affinity, on TCR recognition (13–15).
One of the best-studied models in which discrete variation affects one of the minor anchor pockets of an MHC class I molecule is provided by allelic variant molecules H-2Kb (Kb) and its spontaneous mutant, H-2Kbm8 (Kbm8). Kbm8 arose from Kb by a gene conversion event (16) that changed four amino acids [Tyr22 to Phe22; Met23 to Ile23; Glu24 to Ser24; and Asp30 to Asn30] all located on the platform floor of the peptide-binding groove. Of these four residues, two (MHC 23 and 30) are thought to be largely irrelevant to antigen processing and presentation, as their side chains point away from the peptide-binding groove, are not solvent accessible, and are not analogous to residues shown previously to contact TCRs (17). The two remaining residues, MHC 22 and 24, point into the groove and serve to create the B pocket, where they contact the side chain of the second amino acid of the bound peptide (P2). Indeed, site-directed mutagenesis has shown that most, if not all, of the effects of the Kbm8 mutation upon presentation of the antigenic peptide OVA-8 can be recapitulated by isolated mutations at MHC 22 and 24 (18). Notably, Kb and Kbm8 have identical sequences at their membrane distal TCR-contacting residues. Therefore, the bulk of biological differences between them are likely a consequence of indirect effects of the changes in the B pocket, which could be manifest as differences in peptide-binding preferences or alternate conformations of identically presented peptides.
The hallmark of peptides naturally presented by Kb is the presence of two primary anchors: a phenylalanine or tyrosine as the fifth (or sixth) residue and a leucine as the last of eight (or nine) residues (4, 19). These two primary anchors find themselves deeply buried in the C and F pockets of the Kb peptide-binding groove, respectively, and their substitution to other amino acids typically results in a dramatic reduction in affinity and pMHC stability at the cell surface (20). We have previously characterized an important secondary anchor located at the peptide P2 position that lies sequestered in the B pocket of the Kb groove that, in this allele, is directly adjacent to the C pocket (21). Interestingly, the preferred residue at P2 depends on which primary anchor occupies the C pocket; Ala and Gly are preferred if the C pocket anchor is Tyr, whereas medium-sized residues predominate for primary Phe anchors (15, 21). Peptide elution experiments demonstrated that many peptides ignore the Kbm8 mutation and are able to bind to both molecules with high stability, although a subset does bind differentially (20, 22). Despite the discrete nature of the change in Kbm8, the two molecules exhibit differences in several interrelated biological properties, including peptide-dependent antibody recognition (23), cross-reactivity (24, 25), peptide binding and presentation (11, 14, 25, 26), and intrathymic repertoire selection (27, 28). Most importantly, the coisogeneic mouse strains C57BL/6 (B6, H-2b) and B6.C-H-2bm8 (bm8, H-2bm8), which differ from each other solely by expression of Kb or Kbm8, also differ in susceptibility to HSV-1, due to the fact that the dominant epitope of this virus, the glycoprotein B epitope 498-505 (called here HSV8), elicits a markedly more efficient CTL response in bm8 mice (29).
Previously, we reported that B6 mice immunized with HSV8 produce CTLs, which readily recognize Kb–HSV8, but are unable to recognize Kbm8–HSV8 (14). This difference was not due to differential affinity or stability of peptide binding, nor was it due to gross conformational changes, as assayed by a panel of conformation-sensitive antibodies. As the alteration in Kbm8 potentially changes the hydrogen bond network between the peptide P2 side chain and the MHC B pocket, we hypothesized that this change might somehow be manifest at the pMHC surface, where it could be directly detected by TCR. To test this hypothesis, we performed second-site reversion of the Kbm8 mutation by introducing compensatory P2 peptide mutations that could restore the lost hydrogen bond network. Indeed, changing the peptide SerP2 to a GluP2 (H2E peptide) compensated for the reciprocal Glu24 to Ser24 mutation present in Kbm8, so that the same Kb–HSV8-reactive TCR that could not recognize Kbm8–HSV8 was able to recognize Kbm8–H2E. However, when this H2E peptide was bound to Kb, there was no detectable TCR recognition. Although our paper highlighted an important role for secondary anchor residues in TCR recognition, it remained unclear how exactly these B pocket interactions were being discerned.
To resolve this issue at the atomic level, we have now undertaken the study of these pMHCs by protein crystallography, solving the crystal structures of four distinct complexes: Kb–HSV8, Kbm8–HSV8, Kb–H2E, and Kbm8–H2E. Examining the surface properties of each pMHC revealed, to our surprise, only nonconcerted differences that do not appear to correlate with T cell activation profiles. However, our structures did reveal localized features of the pMHC B pockets that do correspond with our TCR recognition data. When compared with Kb–HSV8, Kbm8–HSV8 and Kb–H2E contained alternate hydrogen bonding and chemical environments in their B pockets, resulting in loss of B6-derived TCR recognition. Alternatively, Kbm8 in complex with the H2E peptide containing the secondary anchor substitution resulted in a B pocket environment closely approximating the one present in Kb–HSV8. Thus, our results indicate that pMHC binding pocket modifications can profoundly affect TCR activation without significantly altering the resting state (TCR unliganded) conformation of the pMHC surface, demonstrating that they can be significant determinants in an immune response.
Materials and Methods
Protein Expression and Purification.
The Kb ectodomain (1–274) and mβ2m were expressed and purified from cell supernatants of Drosophila melanogaster S2 cells using established protocols (17, 19). In brief, purification consisted of Ni+2 chelation chromatography followed by incubation with molar excess of either HSV8 or H2E peptides. Each peptide was synthesized using standard protocols and tested for identity by electrospray MS. The resulting complexes were subjected to ion-exchange chromatography (MonoQ) followed by size exclusion chromatography. Native gel electrophoresis and isoelectric focusing were used to ensure the homogeneous nature of these pMHC samples. Additionally, previously published protocols (30) for bacterial expression and subsequent oxidative refolding was used to produce Kb (1–274) and Kbm8 (1–280) ectodomains in complex with mβ2m.
Circular Dichroism (CD) and Thermal Denaturation.
Experiments were performed in a manner similar to what has been published previously for other MHC class I peptide complexes (31, 32). All four bacterially expressed, oxidatively refolded pMHCs were buffer exchanged to 0.2 mg/ml (5 μm) in 10 mM KH2PO4/K2HPO4 pH 7.5, 150 mM NaCl, and 0.01% sodium azide using Centricon filtration devices (Amicon, Inc.). Three independent thermal denaturation experiments per pMHC were undertaken at the CD shared equipment facility at the University of Medicine and Dentistry of New Jersey. The midpoint of thermal denaturation (Tm) for each protein was calculated by taking the first derivative of the ellipticity data at 218 nm and identifying the inflexion point.
Crystallization and Structure Determinations.
Purified Kbm8 and Kb peptide complexes were concentrated to 5–6 mg/ml in 20 mM ammonium acetate, pH 6.9, and 0.01% sodium azide. Crystals of insect cell–expressed Kb peptide complexes were produced in hanging drops by vapor diffusion at 20°C against wells filled with 3–5% MPD, 2.0 M Na/K2PO4, pH 6.5, and molar excess HSV8 or H2E peptide. Bacterially expressed, oxidatively refolded Kbm8 peptide complex crystals were grown similarly in 12–15% PEG 8000, 100 mM sodium cacodylate, pH 6.6, and 100–150 mM calcium acetate. Diffraction quality crystals appeared within 48 h and were cryoprotected just before flash cooling through the 1:1 addition of well solution plus 20% ethylene glycol (Kbm8) or 25% glycerol (Kb). Diffraction data for Kbm8–H2E crystals were collected using a Rigaku X-ray source and an R-axis IV image plate detector, while data for the other three pMHCs were obtained at the Advanced Photon Source (APS), beamline ID-19 (SBC-CAT). Data were indexed and processed using DENZO and SCALEPACK (see Table I and reference 33). Kb peptide complexes crystallized in the P21212 space group with one pMHC in the asymmetric unit (ASU), whereas Kbm8 peptide complexes crystallized in the P21 space group and had two pMHCs in the ASU. A nearly isomorphous Kb structure, PDB ID code 1KJ3, without its peptide and with its mutant residues truncated to alanine, was rigid body refined into Kbm8–HSV8 data. The HSV8 peptide was traced and the model was built to high confidence after several iterative rounds of model building in O (34) coupled to atomic refinement and map generation with the CNS program (35). This high confidence Kbm8–HSV8 model was used as a starting point for the Kbm8–H2E model. A similar procedure was performed for the Kb peptide complex structures, with PDB ID code 2VAA used as the starting point. Each Kb–peptide complex model contained mβ2m (residues 1–99), Kb (residues 1–274), and the respective full-length peptide. The Kb–HSV8 complex model contains one N-linked carbohydrate at Asn86, whereas the Kb–H2E complex model contains two N-linked carbohydrates at Asn86 and Asn176. The mβ2m present in the Kbm8–peptide complexes contained an additional NH2-terminal Met residue numbered residue zero. Kbm8 models contained residues 1–278; poor electron density for Ser279 and Thr280 prevented them from being modeled.
Table I.
Summary of Data Collection, Phasing, and Refinement
| Data collection for H-2Kb and H2-Kbm8 peptide complexes | ||||
|---|---|---|---|---|
| Data set | Kbm8–H2E | Kbm8–HSV8 | Kb–H2E | Kb–HSV8 |
| Space group | P21 | P21212 | ||
| Unit cell (abc, Å; αβγ, °) | a = 66.57; b = 90.18; c = 88.98; β = 111.32; α,γ = 90 |
a = 66.56; b = 90.13; c = 89.01; β = 111.39; α,γ = 90 |
a = 135.24; b = 89.54; c = 45.25; α,β,γ = 90 |
a = 134.89; b = 90.22; c = 45.45; α,β,γ = 90 |
| Wavelength (Å) | 1.5418 | 1.0332 | 0.97945 | |
| X-ray source | Rigaku | APS-19-ID | APS-19-ID | APS-19-ID |
| Resolution, outer shell (Å) | 20-2.6 (2.7-2.6) | 20-1.9 (1.97-1.9) | 20-2.1 (2.17-2.1) | 20-2.6 (2.69-2.6) |
| Observations/unique | 70,233/29,043 | 411,580/72,537 | 229,268/32,839 | 79,892/17,348 |
| Completeness (%) | 95.6 (93.5) | 94.0 (77.7) | 99.9 (100) | 99.1 (99.9) |
| Rsym (%) | 10.2 (42.1) | 7.6 (36.0) | 7.0 (35.4) | 9.1 (36.9) |
| I/σ | 8.8 (1.8) | 19.9 (3.1) | 27.3 (5.2) | 15.0 (3.9) |
| Refinement statistics | ||||
| Molecules in ASU | 2 | 2 | 1 | 1 |
| Rcrystal, outer shell (%) | 23.0 (38.9) | 22.9 (33.5) | 20.7 (24.2) | 19.8 (26.5) |
| Rfree, outer shell (%) | 28.7 (43.1) | 25.7 (35.4) | 23.1 (27.1) | 23.9 (35.1) |
| Avg. temperature factor (Å2) | 36.0 | 35.0 | 32.8 | 35.0 |
| Rms deviations | ||||
| Bonds (Å), angles (°) | 0.007/1.33 | 0.005/1.29 | 0.005/1.30 | 0.006/1.30 |
| Coordinate error estimates | ||||
| Luzzati, SigmaA | 0.37/0.49 | 0.27/0.31 | 0.25/0.16 | 0.30/0.23 |
| Ramachandran plot | ||||
| Favored, allow., gener. (%) | 85.4/13.7/1.0 | 91.7/7.7/0.6 | 89.1/10.3/0.6 | 90.9/8.5/0.6 |
| PDB ID code | 1RJZ | 1RJY | 1RK1 | 1RK0 |
| Structural analysis | ||||
| α1/α2/peptide RMSD (Å) | –/0.72 | 0.75/0.92 | 1.23 | 1.20 |
| Peptide SC | 0.71/0.68 | 0.71/0.72 | 0.72 | 0.70 |
Computational Analysis.
Graphical structure representations were created using Ribbons (36). The molecular surfaces of the B pockets were generated using sph-ms, a component program of Ribbons, using a 1.4 Å probe. Specifically, the P2 peptide Cα and side chain atoms were removed from the structure, and sph-ms was used to generate the dot surface of the B pocket with the rest of the peptide bound. The geometric surface complementarity of the peptide–MHC interfaces was calculated with the program SC (37). Electrostatic potential maps were generated with Delphi, and Grasp was used to read in these maps and graphically display each pMHC's electrostatic molecular surface potential (38). The 2C TCR from the Kb–2C complex (PDB idcode 2CKB) was docked via the MHC α1/α2 regions onto our pMHCs. PDBDIST was used to determine TCR–pMHC contacts within 4.2 Å and these delineated the TCR contact box present in Fig. 2 (39). HBplus was used to determine the hydrogen bonding in each pMHC's B pocket (40). RMSD calculations between structures were calculated with the CNS program using pMHCs superimposed via their α1/α2 domains (35). RMSDs of mutant residues were calculated using positionally equivalent atoms (i.e., side chain Oγ of SerP2 considered equivalent to GluP2 side chain Cγ). The reference structure for these calculations was one of the Kbm8–H2E structures (chains D and Q) in the ASU. Simulated annealing electron density omit maps of B pocket residues for each complex were generated using the CNS program and displayed in Ribbons (35, 36).
Figure 2.

pMHC electrostatic surface properties. The membrane-distal, peptide-binding platforms of all four pMHCs are depicted. Electrostatic properties were mapped to the molecular surfaces using the program GRASP. Surface colors are contoured from red (−8kT) to blue (+8kT). Elements of the surface corresponding to the peptides are approximately enclosed with dashed lines. (solid lines) The portion of the pMHC surface containing modeled 2C TCR contacts is boxed.
Results
pMHC Crystal Structure Determinations.
We reported previously that B6-derived T cells exhibit a restricted, mono-specific pattern of recognition of the dominant peptide epitope of the HSV-1, HSV8; this peptide was recognized only when presented by Kb, but not when bound to its variant, Kbm8 (14). At that time, we hypothesized that this was due to the loss of hydrogen bonds between SerP2 of HSV8 and the altered B pocket of Kbm8 (particularly due to the key mutations Tyr22 to Phe22 and Glu24 to Ser24). We supported this hypothesis by performing second-site reversion of TCR recognition: introduction of a glutamic acid at position P2 of HSV8 (peptide H2E) led to reconstitution of T cell recognition of the new pMHC, Kbm8–H2E. Since then, we have confirmed these observations using several CTL lines and clones (41).
In this paper, we set out to solve the crystal structures of all four pMHCs to ascertain whether any differences exist that could explain our differential TCR recognition. Kb–HSV8 and Kb–H2E were expressed in D. melanogaster cells and recovered from cell supernatants, whereas Kbm8–HSV8 and Kbm8–H2E were oxidatively refolded from Escherichia coli inclusion bodies. The Kb–peptide complexes crystallized in the primitive orthorhombic space group P21212 with one pMHC in the ASU. The Kbm8–peptide complexes, which lack N-linked carbohydrate additions at residues Asn86 and Asn176, crystallized in the primitive monoclinic space group P21 and have two pMHCs per ASU. Each structure was solved by molecular replacement and after several rounds of model building and refinement, quality atomic models were obtained (Fig. 1). Kb–HSV8 was solved to 2.6 Å resolution, whereas the Kb–H2E complex was solved to 2.1 Å resolution. The estimated coordinate error (Luzzati) for each complex is 0.30 and 0.25 Å, respectively. Similarly, Kbm8–HSV8 was solved to 1.9 Å resolution and Kbm8–H2E was solved to 2.6 Å resolution with estimated coordinate errors of 0.27 and 0.37 Å, respectively. A summary of the data collection and refinement results are presented in Table I.
Figure 1.
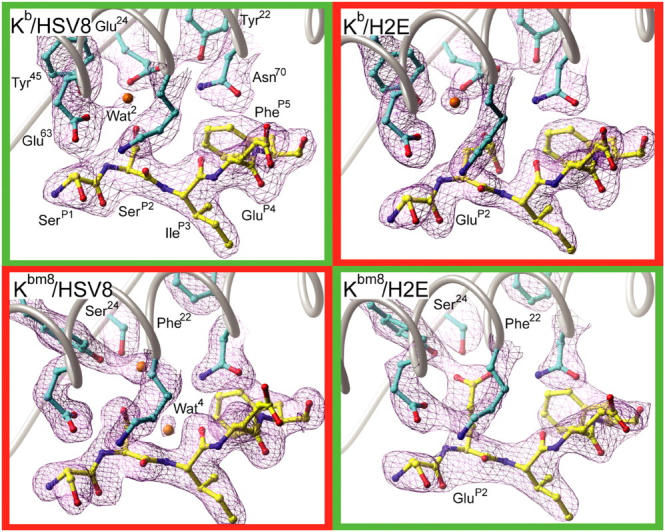
Crystallographic electron density maps of the pMHC B pockets. Sigma-weighted simulated annealing Fo-Fc omit electron density maps contoured at 3.5σ around the B pocket regions of all four pMHCs. (cyan) Kb and Kbm8 carbon; (yellow) HSV8 and H2E peptide carbon; (red) oxygen; (blue) nitrogen; (orange) water. Recognized pMHCs are in green boxes and unrecognized pMHCs are in red boxes.
The structures of Kb and Kbm8 that we have resolved are very similar to those that have been described previously (17, 19). The HSV8 and H2E peptides bind in a canonical fashion to both MHC molecules. The side chains of residues PheP5 and LeuP8 are deeply buried into the MHC binding groove, serving as primary anchors and sequestering into the C and F pockets, respectively. Similarly, the Ser/GluP2 and IleP3 side chains are also buried, acting as secondary anchors at the B and D pockets, respectively. Pointed away from the peptide-binding groove are the side chains of SerP1, GluP4, AlaP6, and ArgP7. Although the SerP1 side chain is solvent inaccessible due to its small size, the other three side chains have significant solvent exposure. These observations are consistent with studies that demonstrate P4, P6, and P7 residues of HSV8 are major TCR determinants, with larger substitutions at P1 also perturbing TCR recognition (11, 14, 23).
Comparison of pMHC Surface Properties.
It has been shown previously that small but significant differences in pMHC surface properties can occur between peptides presented by Kb and its natural occurring variants (17). SEV9 and VSV8 peptides in complex with Kbm1 have a notably different electrostatic surface compared with analogous Kb complexes, whereas the Kbm8 complexes were very similar to those of Kb. We initiated a structural analysis of our pMHCs to determine if any significant differences could be identified. The electrostatic surface properties of each pMHC were calculated using Delphi and visualized using GRASP. This analysis reveals the potential TCR interacting surface of each pMHC to be virtually indistinguishable from one another, with the exception of the surface surrounding the ArgP7 peptide residue (Fig. 2). The significance of this difference is likely minimal as it is due to alternate crystal contacts that, in Kb, are positioned over the COOH terminus of the peptide. This results in a different conformation of the ArgP7 side chain as well as a small local rearrangement that is centered around Ser73. It is unlikely that this rearrangement plays a significant role in TCR recognition, as the conformation of ArgP7 is nearly isostructural between respective recognized and unrecognized pMHC structures (i.e., Kbm8–H2E vs. Kbm8–HSV8).
Next, we undertook a comparative analysis of the pMHC structures to ascertain whether there is any correlative structure variation. Analysis of α1/α2 domain similarity revealed an RMSD deviation of ∼0.8 Å between the four Kbm8 α1/α2 domains, whereas RMSD deviation between the two Kb α1/α2 domains was 0.45 Å. However, the RMSD deviation between the Kb structures and Kbm8–H2E complex was ∼1.2 Å (Table I). These differences in global α1/α2 structure can be largely attributed to differences in loop conformations and, in the case of Kb to Kbm8 comparisons, differences in crystal packing and solvent conditions. Specifically, loops between β strands 1/2, 3/4, and 5/6 were highly divergent, even between both molecules in the ASU of the Kbm8 structures. Differential crystal packing at the β-strand 4/helix 1 region (MHC 52–57) in the Kbm8 structures results in another area of significant conformational difference. These regions are not known to have any influence on conventional TCR recognition. Further inspection of MHC residues did not reveal other residues with significant, correlated deviations that could potentially make direct contact with the TCR.
Comparison of the four peptides from these pMHCs demonstrates that small, but significant, positional differences occur at the amino terminal ends of the peptides. When the peptides are superimposed, via the respective MHC α1/α2 domains, these differences become clear (Fig. 3). Remarkably, the largest of these structural differences occurs between the two complexes, Kb–HSV8 and Kbm8–H2E (Fig. 3, green), which are recognized equivalently. Seen from a top-down perspective, the most noticeable deviations occur at the P1 and P2 positions, highlighting a lateral displacement of these residues (Fig. 3 A). In contrast, the two unrecognized complexes, Kb–H2E and Kbm8–HSV8 (Fig. 3, red), are fairly similar in conformation to the Kbm8–H2E peptide structure. A side view perspective reveals an additional downward shift of the SerP2 in Kb–HSV8 from the GluP2 of Kbm8–H2E (Fig. 3 B). This displacement is propagated through the peptide main chain, causing a diminishing shift through P3 and a prominent movement of P1. The P3–P8 residues, common to all peptides, are nearly isostructural between Kb and Kbm8 structures, with the exception of the P7 side chain conformer difference, which is due to differential crystal packing between Kb and Kbm8.
Figure 3.
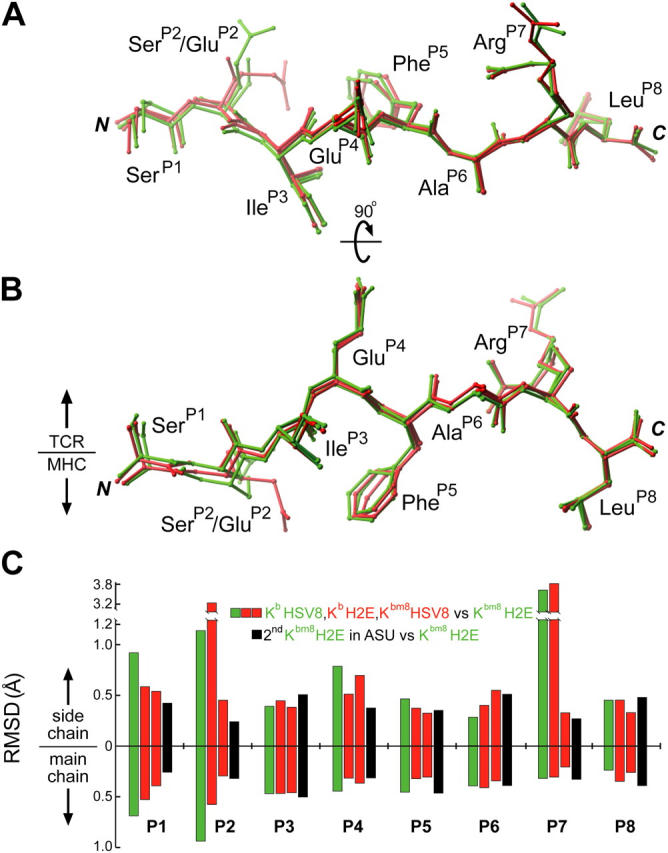
Positional deviation analysis of MHC presented peptides. Peptides from all four pMHCs have been superimposed via their α1/α2 domains. They are shown from a top-down perspective (A) and a side view (B). (green) Peptides from recognized pMHCs. (red) Peptides from unrecognized pMHCs. The large difference observed for ArgP7 is due to alternate crystal packing arising from different space groups. (C) The positional differences of the peptides were quantitated and graphed to denote per residue main and side chain differences in reference to Kbm8–H2E. Recognized peptides (green) and unrecognized peptides (red) have been compared with one of the peptides from the Kbm8–H2E crystal structure (chain Q of 1RJZ), with the differences between the two peptides in the Kbm8–H2E ASU (black) representing a measure of experimental error.
When these positional deviations were quantitated, we found that only the peptide amino terminal variations of Kb–HSV8 and Kbm8–H2E (Fig. 3 C, green bars) were significant, albeit small. Their displacements were two to three times larger than those observed between Kbm8–H2E and its replicate in the ASU (Fig. 3 C, black bars), a measure of experimental error. However, these results are in stark contrast with the TCR recognition data, as the magnitude of the positional deviation does not correlate to recognition. Together, it is clear that TCR recognition in this system does not correlate to evident surface properties of the pMHCs, indicating that the TCR may well be discriminating between other biophysical properties of the complexes.
Comparison of Kb and Kbm8 B Pocket Environments.
Kb and Kbm8 differ in two residues, MHC 22 and MHC 24, which serve to create B pockets that are unique to each molecule. A comparison of the B pockets from each of our pMHCs demonstrates that the bm8 mutations increase the size and alter the chemical environment, allowing for different peptide-binding modes (Fig. 4). The B pockets in the Kb–peptide complexes are elongated, but shallow, due mainly to residue Glu24 that sits directly underneath the P2 anchor (Fig. 5). The Kb B pocket accommodates HSV8 very well, with the SerP2 directly interacting with Glu24. In contrast, the GluP2 mutation in the H2E peptide is forced into an unfavorable chemical environment. This GluP2 side chain is too large to occupy the space where Wat2 is located in the Kb–HSV8 structure; its accommodation would require rearrangement of several Kb B pocket side chains. Additionally, that conformation would put the negatively charged GluP2 in very close proximity to the negatively charged Glu24 side chain, creating an unfavorable electrostatic interaction given that peptide loading occurs at near neutral pH. Instead, the GluP2 side chain turns away from Glu24 and tucks itself underneath the main chain of the peptide. Surprisingly, the H2E peptide provides a similar “fit” for the Kb binding groove as the HSV8 peptide, as assessed by comparable shape complementarity (SC) coefficients (Table I and reference 37).
Figure 4.

Surface representation of pMHC B pockets. The Tyr22 to Phe22, and Glu24 to Ser24 mutations present in Kbm8 relative to Kb result in a larger, more accommodating, and neutral B pocket. Residues are represented as CPK models. Peptide atoms are in grayscale. (Yellow) P2 side chain carbon; (cyan) MHC carbon; (red) MHC oxygen; (orange) waters. The molecular surfaces of the B pockets are represented with purple dots.
Figure 5.

pMHC B pocket hydrogen bonding schemes. The B pocket hydrogen bonding networks of all four pMHCs are depicted. The specific interactions of peptide P2 residues with MHC are highlighted. Recognized pMHCs are in green boxes and the unrecognized pMHCs are in red boxes. (cyan) MHC carbon atoms; (yellow) peptide carbon; (red) oxygen; (blue) nitrogen; (orange) water. Potential hydrogen bonds are depicted as small purple spheres.
The B pockets of the Kbm8–peptide complexes are significantly altered compared with their Kb counterparts; the bm8 mutations allow the pocket to become much deeper and more neutral. A secondary effect of these mutations is the rearrangement of the Tyr45 side chain, causing a widening and further deepening of the pocket. This rearrangement is similar to the ones observed in other Kbm8 peptide structures (17). Accommodation of SerP2 by Kbm8 is very similar to what is observed in the Kb–HSV8 complex. However, the SerP2 rides higher in the Kbm8 B pocket than in the Kb B pocket (Fig. 4). This results in a shifting of Wat2 to allow for favorable interactions between SerP2 and pocket residues, and for the inclusion of an additional water, Wat4, behind it. In contrast with the Kb–H2E complex, the larger, and no longer negatively charged Kbm8 B pocket allows the GluP2 side chain to extend in an alternate conformation similar to the SerP2 side chains in the other complexes (Fig. 4). This extension displaces Wat2 and allows the GluP2 side chain to directly interact with Tyr45. Despite the differences in the peptide-binding grooves of Kb and Kbm8, and the B pocket in particular, the fit of both peptides is quantitatively similar, with SC coefficients ranging from 0.68 to 0.72 (Table I).
Comparison of pMHC Thermostability.
The rearrangements we have observed for the peptide P2 and MHC B pocket residues could potentially alter the stability of the respective pMHC and, thus, may explain our differential TCR recognition. Indeed, the sevenfold difference in thermostability observed for SEV9 and VSV8 peptides presented by Kb and Kbm1 has been implicated in their differential TCR recognition profiles (17). To address this issue, we measured the thermostability of each pMHC using CD. All pMHCs had Tm values >37°C, further supporting the observation that they are all stable under physiological conditions (Fig. 6). Reproducibly higher Tm values were observed for HSV8 complexes (∼49°C) when compared with those of the H2E complexes (∼44°C). Thus, the thermal stability of Kb and Kbm8 is dependent on which peptide is loaded, and the measured stability is in no way correlated with T cell recognition. Consistent with our CD experiments are previously reported data concerning the cell surface stability of our pMHCs on living cells as measured by conformation-sensitive antibodies (11, 14) and T cell–based assays (29).
Figure 6.

pMHC thermostability as measured by CD. CD ellipticity data at 218 nm corresponding to the first melting transition was normalized to a scale of 0 to 1. The Tm for each pMHC is noted in the legend. All pMHCs experienced a second transition at higher temperatures with a corresponding Tm of ∼69°C, which we interpret as the unfolding of mβ2m (reference 32).
Comparison of B Pocket Hydrogen-bonding Networks.
The diversity of our pMHCs and their TCR recognition does not seem to correlate with their apparent resting state surface properties, as resolved crystallographically, or with their relative cell surface and in vitro stabilities. However, this is not the case when the B pocket environment is examined. Analysis of B pocket hydrogen-bonding networks and chemical environments for the four pMHCs reveals an apparent correlation with T cell recognition profiles (Fig. 5). In the TCR-recognized Kb–HSV8 complex, SerP2 forms two distinct hydrogen bonds: one to Glu24 at the base of the pocket and another to Wat2, which mediates a linkage to Glu63. In contrast, this same peptide in complex with Kbm8 has an altered hydrogen-bonding pattern and distinct B pocket chemical environment. Specifically, the Glu24 to Ser24 bm8 mutation makes the pocket more neutral and causes Tyr45 to reposition to establish a water-mediated hydrogen bond with SerP2 of HSV8. Additionally, the repositioning of Wat2 allows Glu63 to hydrogen bond with the P2 main chain nitrogen. Although these B pocket alterations are small, they are clearly significant enough that the Kbm8–HSV8 complex does not activate Kb–HSV8-reactive T cells.
TCR recognition of Kbm8 presenting the dominant HSV-1 epitope is regained with the introduction of a compensating peptide mutation at the P2 secondary anchor position, SerP2 to GluP2. The resulting H2E peptide, in complex with Kbm8, is able to faithfully mimic the hydrogen-bonding network observed in the Kb–HSV8 complex (Fig. 5). The GluP2 side chain extends to displace Wat2 and place its carboxylate group in a position similar to the carboxylate of Glu24 in Kb. This similar positioning allows for both restoration of the pocket's negative charge and establishment of a hydrogen-bonding pattern analogous to Glu24 in Kb. However, TCR recognition is again lost when the H2E peptide is in complex with Kb. GluP2 assumes an alternate conformation, likely due to charge repulsion from Glu24, causing disruption of hydrogen bonding between Glu24 and Asn70; this allows Wat2 back into the pocket, and introduces a new hydrogen bond between its carboxylate and the P3 main chain nitrogen. This hydrogen bond complements part of its negative charge, likely helping to stabilize the Kb–H2E complex.
Thus, the results of our structural analysis would indicate that the designed peptide variant H2E is able to successfully form a similar packing geometry and chemistry of the B pocket when bound to Kbm8 as is otherwise formed only when HSV8 is complexed with Kb. In contrast, significant alterations to the B pocket hydrogen-bonding network are readily apparent in the other two pMHCs that fail to be recognized by Kb–HSV8-reactive T cells.
Discussion
In this paper, we have examined four distinct pMHCs whose sequence variations are localized to residues located in the B pocket of the peptide-binding groove. Our goal was to understand how a compensating mutation in a secondary anchor position of a dominant HSV-1 peptide was able to restore recognition by Kb-restricted, HSV-1–reactive T cells when in complex with an allelic variant of Kb, Kbm8. Thus, the second-site revertant Kbm8–H2E, like Kb–HSV8, elicits activating responses in T cell effector assays, whereas Kb–H2E and Kbm8–HSV8 do not. We have shown previously that neither impaired peptide binding nor presentation can explain this differential pMHC recognition (14). Our current results establish that the loading of HSV8 onto either Kb or Kbm8 results in pMHCs with similar thermal stabilities as measured by CD, whereas the loading of our designed H2E peptide results in systematically less thermally stable complexes.
Next, we turned to crystallography to elucidate structural details that may provide further insight into our functional observations. The crystal structures of all four pMHCs were determined and, as expected, the P2 residues of both HSV8 and H2E are sequestered from solvent in the B pocket, proximal to the allelic variant residues Tyr22/Phe22 and Glu24/Ser24. To our surprise, comparison of the TCR-unliganded structures did not reveal any concerted structural differences localized to the membrane-distal surfaces of the pMHCs otherwise considered important in TCR engagement. Furthermore, the small conformational differences of solvent-accessible residues observed in our work did not appear to correlate with the available TCR recognition data. However, comparison of the manner in which the P2 secondary anchor residue is engaged in the B pocket of the pMHCs revealed that the two recognized complexes have similar packing structures, whereas the B pocket interactions of the ignored complexes are distinct. We will now turn to considerations of how these results might be important for the TCR recognition process.
Peptide Anchor Residues and Their Role in the Immune Response.
There have been many studies directed at examining TCR–pMHC recognition, many of which have revolved around the role of solvent-exposed/TCR-contacting peptide residues. Using many different MHC class I and II systems, it has been shown that TCRs can be extremely sensitive to even conservative mutations at these solvent-exposed residues (9–11). These results have been supported by the growing number of TCR–pMHC complex structures available (6–8). It is clear from these structures that a limited number of solvent-exposed peptide residues interact extensively with CDR loops of the TCR, and that peptide anchor residues and their surrounding MHC environments are not major recognition determinants. Although anchor residues can unquestionably impact TCR recognition indirectly by modulating MHC binding affinity and selection, other mechanisms of action have been proposed (15, 42–44).
Our current results are similar to a paper we published several years ago in a class II model (13). We have shown that for I-Ek/Hb, a minor change in the peptide P6 secondary anchor residue results in both a distinct, nonoverlapping bulk T cell population as well as a 1,000-fold difference in specific T cell activation. Structure determination of these pMHCs revealed minor structural differences in solvent-exposed atoms and an altered hydrogen-bonding network in the P6 pocket. Similarly, our Kb/Kbm8 data demonstrate that perturbations of buried residues can have a significant impact on T cell repertoire generation, selection, and specificity. Indeed, although functional stability of Kb–Kbm8–HSV8 complexes was indistinguishable as measured by both Ab-based (14) and T cell–based assays (29), T cell repertoire selection in the thymus, T cell diversity mobilized in response to HSV-1 infection, and T cell–mediated resistance to lethal HSV-1 infection were all heavily impacted by the alterations in the B pocket of the aforementioned complexes (28, 29, 41). The majority of previous TCR recognition studies have focused on the dominant role solvent-accessible pMHC residues can play in T cell activation processes. Our results extend these studies and demonstrate that buried peptide and MHC pocket residues can also be significant determinants in an immune response. Thus, our data further highlight the exquisite structural sensitivity that governs pMHC recognition by TCRs.
Potential Impact of B Pocket Environment on TCR Recognition.
Analysis of our structural data reveals that the bm8 substitutions of B pocket residues and their interactions do not translate into significant positional changes in solvent-exposed residues. Furthermore, the small changes that we did observe failed to correlate with T cell activation data. Nevertheless, our differential T cell recognition data demonstrate that, even though on the surface they appear chemically similar, the various pMHCs are readily discriminated. Thus, it is apparent that the B pocket environment is modulating one or more properties on which TCR recognition and subsequent T cell activation is dependent.
One possible discriminatory mechanism could be that our B pocket mutations affect the way in which the pMHC is engaged by the TCR. The available structural data for TCR–pMHC complexes indicate that there is a significant amount of structural flexibility in the TCR, notably in the CDR3 loops, with less structural variation observed in pMHC residues upon complex formation (7, 8). Nevertheless, a recent set of structures has demonstrated that near-identical pMHCs, Kb/dEV8 and Kbm3/dEV8, result in two very distinct structures when in complex with a common TCR, 2C (45). The difference between the two pMHCs is a buried Asp77 to Ser77 mutation that is associated with a differential hydrogen-bonding pattern with the peptide. Specifically, the bm3 mutation is associated with altered presentation of the dEV8 peptide as well as an increase in overall TCR–pMHC contact and SC. Thus, an extremely minor change in hydrogen bonding and chemical environment can have substantial effects on the way a TCR engages nearly isostructural resting state pMHCs. In accordance with this data, we postulate that both our allelic and secondary anchor mutations need not be in direct contact with TCR to elicit profound changes in recognition. For example, one could speculate that the ability of Kb-restricted, HSV-1–reactive T cells to engage only Kb–HSV8 and Kbm8–H2E is a manifestation of each pMHC's unique ability to adopt a conformation consistent with stable complex formation, or alternatively the transition state energy barrier to achieve a stable complex is too high for the unrecognized pMHCs (46). However, irrespective of the discriminatory mechanism, our data clearly support the notion that MHC anchor pocket environments can be just as significant an antigenic determinant as solvent-exposed peptide side chains. Given that some of the most pronounced effects of the Kb/Kbm8 polymorphism exert themselves at the level of intrathymic positive selection of TCR repertoire (27, 28), it is likely that this environment can play a broad role, affecting all the biological consequences of the TCR–pMHC contact. Moreover, this also implies that the self-peptides that mediate differential positive selection of TCR when presented by either Kb or Kbm8 must themselves participate in analogous, variable B pocket interactions as does the dominant HSV-1 epitope, providing another level of similarity between antigenic and positively selecting peptides.
Acknowledgments
Authors would like to acknowledge P. Allen for peptide synthesis and helpful discussions.
This paper was supported by the United States Public Health Service grant nos. CA86803 (to J. Nikolich-Žugich and D.H. Fremont) and RR0163 (Oregon National Primate Research Center). Further support came from the Burroughs-Wellcome Fund and the Kilo Foundation. Atomic coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics.
The authors have no conflicting financial interests.
Abbreviations used in this paper: ASU, asymmetric unit; CD, circular dichroism; pMHC, MHC class I molecule in complex with bound peptide; SC, shape complementarity; Tm, midpoint of thermal denaturation.
References
- 1.Matsumura, M., D.H. Fremont, P.A. Peterson, and I.A. Wilson. 1992. Emerging principles for the recognition of peptide antigens by MHC class I molecules. Science. 257:927–934. [DOI] [PubMed] [Google Scholar]
- 2.Madden, D.R. 1995. The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 13:587–622. [DOI] [PubMed] [Google Scholar]
- 3.Bjorkman, P.J., M.A. Saper, B. Samraoui, W.S. Bennett, J.L. Strominger, and D.C. Wiley. 1987. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 329:512–518. [DOI] [PubMed] [Google Scholar]
- 4.Rammensee, H.G., K. Falk, and O. Rotzschke. 1993. Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 11:213–244. [DOI] [PubMed] [Google Scholar]
- 5.van der Merwe, P.A., and S.J. Davis. 2003. Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 21:659–684. [DOI] [PubMed] [Google Scholar]
- 6.Hennecke, J., and D.C. Wiley. 2001. T cell receptor-MHC interactions up close. Cell. 104:1–4. [DOI] [PubMed] [Google Scholar]
- 7.Garcia, K.C., L. Teyton, and I.A. Wilson. 1999. Structural basis of T cell recognition. Annu. Rev. Immunol. 17:369–397. [DOI] [PubMed] [Google Scholar]
- 8.Housset, D., and B. Malissen. 2003. What do TCR-pMHC crystal structures teach us about MHC restriction and alloreactivity? Trends Immunol. 24:429–437. [DOI] [PubMed] [Google Scholar]
- 9.Sloan-Lancaster, J., and P.M. Allen. 1996. Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu. Rev. Immunol. 14:1–27. [DOI] [PubMed] [Google Scholar]
- 10.Thomson, C.T., A.M. Kalergis, J.C. Sacchettini, and S.G. Nathenson. 2001. A structural difference limited to one residue of the antigenic peptide can profoundly alter the biological outcome of the TCR-peptide/MHC class I interaction. J. Immunol. 166:3994–3997. [DOI] [PubMed] [Google Scholar]
- 11.Huard, R., R. Dyall, and J. Nikolic-Zugic. 1997. The critical role of a solvent-exposed residue of an MHC class I-restricted peptide in MHC-peptide binding. Int. Immunol. 9:1701–1707. [DOI] [PubMed] [Google Scholar]
- 12.Wang, B., A. Sharma, R. Maile, M. Saad, E.J. Collins, and J.A. Frelinger. 2002. Peptidic termini play a significant role in TCR recognition. J. Immunol. 169:3137–3145. [DOI] [PubMed] [Google Scholar]
- 13.Kersh, G.J., M.J. Miley, C.A. Nelson, A. Grakoui, S. Horvath, D.L. Donermeyer, J. Kappler, P.M. Allen, and D.H. Fremont. 2001. Structural and functional consequences of altering a peptide MHC anchor residue. J. Immunol. 166:3345–3354. [DOI] [PubMed] [Google Scholar]
- 14.Dyall, R., D.H. Fremont, S.C. Jameson, and J. Nikolic-Zugic. 1996. T cell receptor (TCR) recognition of MHC class I variants: intermolecular second-site reversion provides evidence for peptide/MHC conformational variation. J. Exp. Med. 184:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saito, N.G., H.C. Chang, and Y. Paterson. 1999. Recognition of an MHC class I-restricted antigenic peptide can be modulated by para-substitution of its buried tyrosine residues in a TCR-specific manner. J. Immunol. 162:5998–6008. [PubMed] [Google Scholar]
- 16.Nathenson, S.G., J. Geliebter, G.M. Pfaffenbach, and R.A. Zeff. 1986. Murine major histocompatibility complex class-I mutants: molecular analysis and structure-function implications. Annu. Rev. Immunol. 4:471–502. [DOI] [PubMed] [Google Scholar]
- 17.Rudolph, M.G., J.A. Speir, A. Brunmark, N. Mattsson, M.R. Jackson, P.A. Peterson, L. Teyton, and I.A. Wilson. 2001. The crystal structures of K(bm1) and K(bm8) reveal that subtle changes in the peptide environment impact thermostability and alloreactivity. Immunity. 14:231–242. [DOI] [PubMed] [Google Scholar]
- 18.Pullen, J.K., H.D. Hunt, and L.R. Pease. 1991. Peptide interactions with the Kb antigen recognition site. J. Immunol. 146:2145–2151. [PubMed] [Google Scholar]
- 19.Fremont, D.H., M. Matsumura, E.A. Stura, P.A. Peterson, and I.A. Wilson. 1992. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science. 257:919–927. [DOI] [PubMed] [Google Scholar]
- 20.Saito, Y., P.A. Peterson, and M. Matsumura. 1993. Quantitation of peptide anchor residue contributions to class I major histocompatibility complex molecule binding. J. Biol. Chem. 268:21309–21317. [PubMed] [Google Scholar]
- 21.Fremont, D.H., E.A. Stura, M. Matsumura, P.A. Peterson, and I.A. Wilson. 1995. Crystal structure of an H-2Kb-ovalbumin peptide complex reveals the interplay of primary and secondary anchor positions in the major histocompatibility complex binding groove. Proc. Natl. Acad. Sci. USA. 92:2479–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohren, E.M., L.R. Pease, H.L. Ploegh, and T.N. Schumacher. 1993. Polymorphisms in pockets of major histocompatibility complex class I molecules influence peptide preference. J. Exp. Med. 177:1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Messaoudi, I., J. LeMaoult, and J. Nikolic-Zugic. 1999. The mode of ligand recognition by two peptide:MHC class I-specific monoclonal antibodies. J. Immunol. 163:3286–3294. [PubMed] [Google Scholar]
- 24.Chattopadhyay, S., M. Theobald, J. Biggs, and L.A. Sherman. 1994. Conformational differences in major histocompatibility complex–peptide complexes can result in alloreactivity. J. Exp. Med. 179:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yun, T.J., M.D. Tallquist, E.M. Rohren, J.M. Sheil, and L.R. Pease. 1994. Minor pocket B influences peptide binding, peptide presentation and alloantigenicity of H-2Kb. Int. Immunol. 6:1037–1047. [DOI] [PubMed] [Google Scholar]
- 26.Nikolic-Zugic, J., and F.R. Carbone. 1990. The effect of mutations in the MHC class I peptide binding groove on the cytotoxic T lymphocyte recognition of the Kb-restricted ovalbumin determinant. Eur. J. Immunol. 20:2431–2437. [DOI] [PubMed] [Google Scholar]
- 27.Nikolic-Zugic, J., and M.J. Bevan. 1990. Role of self-peptides in positively selecting the T-cell repertoire. Nature. 344:65–67. [DOI] [PubMed] [Google Scholar]
- 28.Dyall, R., I. Messaoudi, S. Janetzki, and J. Nikolic-Zugic. 2000. MHC polymorphism can enrich the T cell repertoire of the species by shifts in intrathymic selection. J. Immunol. 164:1695–1698. [DOI] [PubMed] [Google Scholar]
- 29.Messaoudi, I., J.A. Guevara Patino, R. Dyall, J. LeMaoult, and J. Nikolich-Žugich. 2002. Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science. 298:1797–1800. [DOI] [PubMed] [Google Scholar]
- 30.Busch, D.H., I.M. Pilip, S. Vijh, and E.G. Pamer. 1998. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 8:353–362. [DOI] [PubMed] [Google Scholar]
- 31.Batalia, M.A., T.J. Kirksey, A. Sharma, L. Jiang, J.P. Abastado, S. Yan, R. Zhao, and E.J. Collins. 2000. Class I MHC is stabilized against thermal denaturation by physiological concentrations of NaCl. Biochemistry. 39:9030–9038. [DOI] [PubMed] [Google Scholar]
- 32.Fahnestock, M.L., I. Tamir, L. Narhi, and P.J. Bjorkman. 1992. Thermal stability comparison of purified empty and peptide-filled forms of a class I MHC molecule. Science. 258:1658–1662. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski, Z., and W. Minor. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–344. [DOI] [PubMed] [Google Scholar]
- 34.Jones, T.A., J.Y. Zou, S.W. Cowan, and Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta. Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- 35.Brunger, A.T., P.D. Adams, G.M. Clore, W.L. DeLano, P. Gros, R.W. Grosse-Kunstleve, J.S. Jiang, J. Kuszewski, M. Nilges, N.S. Pannu, et al. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta. Crystallogr. D. Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- 36.Carson, M. 1987. Ribbon models of macromolecules. J. Mol. Graph. 5:103–106. [Google Scholar]
- 37.Lawrence, M.C., and P.M. Colman. 1993. Shape complemetarity at protein/protein interfaces. J. Mol. Biol. 234:946–950. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls, A., K.A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- 39.Sayle, R., M. Saqi, M. Weir, and A. Lyall. 1995. PdbAlign, PdbDist and DistAlign: tools to aid in relating sequence variability to structure. Comput. Appl. Biosci. 11:571–573. [DOI] [PubMed] [Google Scholar]
- 40.McDonald, I.K., and J.M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- 41.Messaoudi, I., J. LeMaoult, B.M. Metzner, M.J. Miley, D.H. Fremont, and J. Nikolich-Žugich. 2001. Functional evidence that conserved TCR CDR alpha 3 loop docking governs the cross-recognition of closely related peptide:class I complexes. J. Immunol. 167:836–843. [DOI] [PubMed] [Google Scholar]
- 42.Sharma, A.K., J.J. Kuhns, S. Yan, R.H. Friedline, B. Long, R. Tisch, and E.J. Collins. 2001. Class I major histocompatibility complex anchor substitutions alter the conformation of T cell receptor contacts. J. Biol. Chem. 276:21443–21449. [DOI] [PubMed] [Google Scholar]
- 43.Peterson, D.A., R.J. DiPaolo, O. Kanagawa, and E.R. Unanue. 2001. Cutting edge: a single MHC anchor residue alters the conformation of a peptide-MHC complex inducing T cells that survive negative selection. J. Immunol. 166:5874–5877. [DOI] [PubMed] [Google Scholar]
- 44.Denkberg, G., E. Klechevsky, and Y. Reiter. 2002. Modification of a tumor-derived peptide at an HLA-A2 anchor residue can alter the conformation of the MHC-peptide complex: probing with TCR-like recombinant antibodies. J. Immunol. 169:4399–4407. [DOI] [PubMed] [Google Scholar]
- 45.Luz, J.G., M. Huang, K.C. Garcia, M.G. Rudolph, V. Apostolopoulos, L. Teyton, and I.A. Wilson. 2002. Structural comparison of allogeneic and syngeneic T cell receptor–peptide–major histocompatibility complex complexes: a buried alloreactive mutation subtly alters peptide presentation substantially increasing Vβ interactions. J. Exp. Med. 195:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu, L.C., D.S. Tuot, D.S. Lyons, K.C. Garcia, and M.M. Davis. 2002. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature. 418:552–556. [DOI] [PubMed] [Google Scholar]