

Abstract
The T cell receptor must translate modest, quantitative differences in ligand binding kinetics into the qualitatively distinct signals used to determine cell fate. Here, we use mice that express an endogenous T cell receptor (TCR) antagonist and an adoptive transfer system to examine the influence of TCR signal quality on the development of effector function. We show that activation of antigen-specific T cells in the presence of an antagonist results in a functional reprogramming of the primary immune response, marked by altered T cell homing, a failure to develop effector function, and ultimately clonal elimination by apoptosis. Importantly, antagonism does not block cell division, implying that the signals promoting clonal expansion and effector differentiation are distinct.
Keywords: immune tolerance, clonal deletion, lymphocyte activation, immunization, T lymphocyte effector
Introduction
Cell-autonomous instructional programs with sequentially acquired activation thresholds control all phases of primary and secondary T cell immune responses (1–5). For example, sustained TCR cross-linking by high affinity MHC/foreign peptide ligands on the surface of activated, professional APCs results in clonal expansion, followed by differentiation and effector function, clonal contraction, and ultimately memory formation. In contrast, shorter interactions between naive T cells and immature APCs presenting low doses of antigen induce unresponsiveness and tolerance (3, 6, 7). The outcome of T cell activation therefore depends largely on the overall strength and duration of the initial antigenic stimulation and the subsequent induction of the developmental programs that control heritable changes in gene expression (1, 8). Collectively, the continuum of differentiation stages after T cell stimulation characterizes the progressive model of T cell activation (1, 4, 6, 9).
Multiple variables control signal strength and timing, and therefore the capacity to induce full effector function. These include the affinity and binding kinetics of the TCR for cognate MHC/peptide ligands (10, 11), the presence or absence of costimulatory molecules (12, 13), the cell surface concentration of the relevant ligands/receptors (14, 15), the nature of the APCs (16, 17), and competition for limited niches in lymphatic tissue (18). These factors regulate the rate of TCR triggering, signal amplification, and ultimately the duration of the immunological synapse (19, 20). In the progressive model of T cell differentiation, total signal strength correlates with T cell “fitness,” or the capacity of T cells to resist cell death, develop effector function, and persist after primary stimulation in the absence of antigen (2, 3). Mechanisms that interrupt the progressive nature of this process are predicted to result in reduced survival capacity and clonal elimination.
One potent mechanism capable of limiting synapse formation and blocking T cell activation is TCR antagonism (21–25). Antagonists are both naturally occurring and synthetic ligands that fail to activate mature T cells (26–28). When present together with a stimulatory ligand, antagonists will inhibit the expected proliferative response. Both kinetic and membrane-proximal signaling mechanisms have been proposed for this class of ligands, based largely on data from in vitro experiments (10, 25, 29). Recently, TCR antagonists have been shown to tip the kinase/phosphatase balance at the CD3 complex in favor of recruitment of the activated phosphatase SHP-1, resulting in negative feedback and T cell unresponsiveness (30, 31). These results support the conclusion that TCR antagonists create a qualitatively distinct signal with a negative effect on T cell proliferation. The impact of this ubiquitous class of self-ligand on the programming of peripheral T cell development is largely unknown.
In previous work, we demonstrated that constitutive expression of a weak TCR antagonist ligand in vivo resulted in a 2.5-fold decrease in the number of antigen-specific T cells in the draining lymph nodes after adoptive transfer and primary immunization (32). Although this study was the first to demonstrate in vivo antagonism, its physiological significance was limited by the inability to distinguish between reduced proliferation and reduced survival as the mechanism. Given the emerging positive role for self-ligands in T cell activation (19, 33), it is important to determine if endogenous peptide–MHC complexes also have the capacity to negatively influence T cell programming and T cell survival, as predicted by studies of TCR antagonism that support a dominant negative signal model (34, 35).
We have examined this question by using a modification of our previous adoptive transfer system together with mice that constitutively express a strong TCR antagonist. Under the conditions of these experiments, other factors that contribute to the strength of TCR signaling are maximized and normally lead to a robust Th1 cell response. Here, we show that T cell activation in the presence of the antagonist results in a functional reprogramming of the primary immune response marked by clonal expansion, altered T cell homing, a failure to develop effector function, and ultimately clonal elimination by apoptosis. Although these data are consistent with the progressive model of T cell differentiation (1), they demonstrate the novel findings that the quality of T cell signaling has a potent impact on T cell fate, and that the developmental programs promoting T effector function and clonal expansion are distinct. They suggest a role for endogenous TCR antagonists in establishing peripheral tolerance and shaping the peripheral T cell repertoire.
Materials and Methods
Mice.
The generation and characterization of 3.L2 TCR transgenic mice and I72 and A72 transgenic mice have been described previously (36, 37). 3.L2 mice express a TCR specific for Hb(64–76)/I-Ek. I72 and A72 mice express a chimeric membrane protein containing an altered peptide ligand of Hb(64–76) in all APCs. In I72 mice, the normal asparagine at position 72 has been replaced by isoleucine and in A72 mice it has been replaced by an alanine. In hybridoma assays, these substitutions create peptides that function as 3.L2 TCR antagonists by inhibiting IL-2 production after in vitro stimulation (38). All experiments were performed with 6–8-wk-old mice that were heterozygous for the 3.L2 TCR, I72, and A72 transgenes. All transgenic mice were created in either the C57BL/6 or B6.AKR strain and maintained on the B6.AKR background by backcrossing for >30 generations. Where possible, transgene− littermates were used as controls. RAG-1−/− mice were maintained on the B6.AKR background as described above. The Animal Resource Committee at the Medical College of Wisconsin approved all animal experiments.
Antibodies.
Antibodies and secondary reagents were purchased from BD Biosciences and included the following: PE anti–mouse CD4; FITC anti–mouse CD8a; biotin anti–mouse CD19; purified anti–mouse CD16/CD32; biotin anti–mouse TCR-β chain; biotin anti–mouse CD45R/B220; FITC anti–mouse CD69; APC anti–mouse IL-2; APC anti–mouse IL-4; APC anti–mouse IFN-γ; APC anti–mouse TNF-α; and streptavidin-CyChrome. Biotinylated 3.L2 clonotypic antibody (CAb) was prepared as described previously (36).
Peptides.
The Hb(64–76), Hb(64–76)N72I (I72), and MCC (88–103) peptides were purified by HPLC and analyzed by mass spectroscopy (United Biochemical Research). The peptide sequences in single letter amino acid code are as follows: GKKVITAFNEGLK (Hb); GKKVITAFIEGLK (I72); and ANERADLIAYLKQATK (MCC).
Ex Vivo Antagonism Assay.
B6.AKR splenocytes were prepulsed with 10 μM Hb(64–76) peptide for 3 h at 37°C, 5% CO2, and washed three times to remove unbound peptide. CD4+ T cells were purified from the spleens of 3.L2 mice by magnetic sorting with anti-CD4 beads using the conditions specified by the manufacturer (Miltenyi Biotec), and the percent of CAb+ cells was determined by FACS. 5 × 105 prepulsed, washed splenocytes were added to 2.5 × 104 CAb+ cells in a 96-well plate, followed by titrated amounts of I72 or MCC(88–103) peptide. All cells and peptides were resuspended in cell culture media (RPMI 1640, 10% FBS, 5 × 10−5 M 2-ME, and 1 mM glutamax), and triplicate wells were set up for each peptide concentration. Cultures were incubated for 48 h at 37°C, 5% CO2, pulsed with 0.4 μCi/well [3H]thymidine for an additional 18 h, and then harvested onto fiber filtermats with a Micro96 harvester (Skatron) and counted.
T Cell Proliferation Assays.
CD4+ T cells were isolated from 3.L2 mice by magnetic bead sorting and the percentage of CAb+ cells was determined by FACS. 2.5 × 104 CD4+ CAb+ T cells were added to 5 × 105 splenocytes, and the T cells were stimulated with either Hb(64–76) or the I72 peptide as described previously (39). Results from several experiments were then averaged to obtain the data shown. In some experiments, the source of APCs was either I72 transgenic or B6.AKR (control) splenocytes. For the hybridoma assay, 5 × 105 splenocytes from either I72 or B6.AKR mice were used as APCs to present the MCC(88–103) peptide to 2.5 × 104 2B4 hybridoma cells in triplicate wells. The 2B4 hybridoma is specific for MCC(88–103)/I-Ek. After 24 h, 100 μl of culture supernatant was removed and the relative amount of IL-2 in the supernatant was determined using the IL-2–dependent cell line CTLL-2 as described previously (39).
Adoptive Transfer and Immunization.
Pooled donor splenocytes were isolated from 3.L2 or 3.L2 × RAG−/− TCR transgenic mice and filtered. The percent of CD4+ CAb+ T cells in the suspension was determined by FACS, and 106 CD4+ CAb+ cells were transferred intravenously into recipient mice by retro-orbital injection. After 24 h, transfer recipients were immunized subcutaneously with 2 or 20 nmol of Hb(64–76) peptide emulsified in CFA (100 μl per mouse). At various times after immunization, the draining lymph nodes and spleen were removed and analyzed by cell count and FACS.
5-(and -6)-Carboxyfluorescein Diacetate Succinimidyl Ester (CFSE) Staining.
Splenocytes were labeled with the intracellular fluorescent CFSE (Molecular Probes) before transfer. A single cell suspension containing 50 × 106 cells/ml in Dulbecco's PBS and 5μM CFSE was incubated at 37°C for 15 min. After this incubation, the cells were washed three times with ice-cold DPBS, filtered, and resuspended at a concentration of 20 × 106 CD4+ CAb+ cells/ml. Each transfer recipient received 5 × 106 CD4+ CAb+ cells (250 μL). Controls included recipients that were either not immunized or immunized with CFA alone.
Flow Cytometry.
Single cell suspensions of lymph node cells or splenocytes were stained as described previously (37). Samples were gated on live cells and a minimum of 100,000 live cell events per sample were collected.
Immunohistochemistry.
Popliteal lymph nodes were removed and sectioned as described previously (37). The sections were incubated with the primary antibody, a biotinylated anti–mouse CD45R/B220 (0.5 μg in 100 μL FACS buffer), for 1 h. Sections were washed four times with FACS buffer and incubated with Cy 3–conjugated streptavidin (Jackson ImmunoResearch Laboratories) for 30 min (1.8 μg streptavidin in 50 μL FACS buffer). Slides were then washed three times in FACS buffer and a 50% glycerol/PBS solution was applied, followed by a sealed coverslip. Immunofluorescence was visualized on an Olympus BX60 microscope. Digital images were captured by an Axio Vision 3.0 Carl Zeiss vision imaging system and processed using Adobe Photoshop and Deneba Canvas software.
Intracellular Cytokine Analysis.
CD4+ T cells were purified from the spleens and lymph nodes of day 5 transfer recipients. The percent of CAb+ lymphocytes was determined by FACS and 2.5 × 104 CAb+ cells were added to 4.75 × 105 B6.AKR splenocytes in a 96-well plate. The splenocytes were prepulsed with 10 μM Hb(64–76) peptide for 3 h at 37°C before the addition of the purified T cells. After the addition of the CAb+ cells, the culture was incubated at 37°C, 5% CO2, for 24 h. Brefeldin A was then added at a final concentration of 10 μg/ml and cells were incubated for an additional 12 h before analysis. Surface staining of cells was performed before intracellular staining using a modified FACS buffer containing 5 μg/ml brefeldin A. Cells were stained on ice for 30 min with the primary antibody, washed with the modified FACS buffer, and incubated for 20 min on ice with the streptavidin-conjugated fluorochrome. Cells were washed again with the modified FACS buffer and prepared for intracellular cytokine staining by adding 200 μl of a fixation and permeabilization solution (FACS buffer with 2% paraformaldehyde and 0.1% saponin), followed by a 20-min incubation at room temperature. After this incubation, permeabilization was continued by adding 3 ml of permeabilization wash (FACS buffer with 0.1% saponin) for 10 min at room temperature. Cells were collected by centrifugation and stained for IL-2, IL-4, IFN-γ, and TNF-α by adding APC-conjugated antibodies at 0.2 μg/ml in 100 μl of permeabilization wash for 1 h on ice. Unrelated antibodies of the same isotypes were used as controls. Cells were then washed in normal FACS buffer (without saponin) and then fixed with 1% paraformaldehyde in PBS-azide (0.1%). Cells were immediately subjected to FACS analysis.
Hb(64–76) Recall Responses.
Adoptive transfer recipients were immunized with 2 nmol Hb(64–76) peptide in CFA as described above. The mice were reimmunized 2 mo later with 20 nmol Hb(64–76) in IFA. 5 d after the secondary immunization, the draining lymph nodes and spleen were removed and single cell suspensions were prepared. An aliquot of this suspension was analyzed using flow cytometry. CD4+ T cells were isolated from these suspensions using anti-CD4 microbeads and Fc block (5 μg for 107 cells). Proliferation assays were performed in triplicate in 96-well flat-bottom plates using 3.5 × 105 purified CD4+ T cells, 6.5 × 105 B6.AKR splenocytes, and varying concentrations of Hb(64–76) peptide per well. After incubation for 48 h, cells were pulsed with 0.4 μCi/well [3H]thymidine and harvested 24 h later.
Online Supplemental Material.
Adoptively transferred 3.L2 T cells isolated from RAG-1−/− donors proliferate equally in I72+ and I72− recipients 48 h after immunization with Hb(64–76) peptide (Fig. S1). Adoptively transferred 3.L2 T cells proliferate at equal rates in A72+ recipients after immunization (Fig. S2). Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20041226/DC1.
Results
Naive T Cells Are Susceptible to Antagonism.
The I72 peptide is a strong antagonist for the 3.L2 T cell hybridoma, and we wished to establish that this peptide would also antagonize the proliferation of 3.L2 transgenic T cells (38). In this ex vivo antagonism assay, B6.AKR splenocytes were prepulsed with Hb(64–76) and used as APCs. When naive, freshly isolated 3.L2 transgenic T cells were added to the culture wells, they were activated and proliferated, as measured by [3H]thymidine incorporation. These conditions result in ∼17,000 cpm (Fig. 1 A). When increasing concentrations of I72 peptide were added, the 3.L2 response is reduced by ∼75% at 100 μM. These results demonstrate that naive transgenic 3.L2 T cells are susceptible to antagonism by the I72 peptide. The control peptide MCC(88–103), which also binds to I-Ek, failed to inhibit 3.L2 T cell proliferation, demonstrating the specificity of this inhibition. In ex vivo proliferation assays, the I72 peptide did not induce 3.L2 T cell proliferation (Fig. 1 B).
Figure 1.

The antagonist I72 inhibits the proliferation of naive 3.L2 T cells. (A) Ex vivo antagonism assay with purified naive 3.L2 T cells, splenocytes prepulsed with 10 μM Hb(64–76) peptide as APCs and no additional peptide (dotted line), increasing concentrations of the antagonist I72 peptide (•), or the MCC(88–103) control peptide (□). Data shown is the mean cpm from 12 independent experiments. (B) Proliferation assay with purified naive 3.L2 T cells and splenocytes as APCs, with increasing concentrations of Hb(64–76) (○) or I72 peptide (•). Data shown is the mean of seven experiments. (C) Proliferation assay with purified naive 3.L2 T cells stimulated by Hb(64–76) peptide and control splenocytes (□), or splenocytes from transgenic mice expressing the antagonist I72 in all MHC class II+ cells (•). Data shown is the mean of three experiments. (D) Hybridoma assay with the 2B4 T cell hybridoma, increasing concentrations of the MCC(88–103) peptide, and either control splenocytes (□) or I72 splenocytes as APCs (•). Data shown is the mean of two experiments.
The transgenic mouse line I72 expresses a chimeric membrane protein containing the I72 peptide as an epitope tag in all MHC class II+ cells (37). Previous data demonstrated the efficient processing and presentation of the I72 epitope in all APCs, including B cells and in the thymus. I72 mice express sufficient ligand to induce negative selection of 3.L2 transgenic T cells. Because the strength of the antagonism effect correlates with the concentration of I72 (Fig. 1 A), we wished to determine if APCs derived from I72 mice would also inhibit the naive 3.L2 T cell response. In this ex vivo assay, purified CD4+ 3.L2 T cells were mixed with either I72+ (transgenic) or I72− (B6.AKR) splenocytes in the presence of increasing concentrations of the Hb(64–76) peptide. Results show a 7–10-fold shift in the dose response curve at all peptide concentrations (Fig. 1 C), demonstrating that the antagonist effect produced by I72 splenocytes cannot be overcome at high doses of agonist peptide. In Fig. 1, a comparison of A with C demonstrates that the level of inhibition of proliferation achieved by I72+ splenocytes is, on average, equal to that achieved by the addition of 31.6–100 μM I72 peptide in the antagonism assay (50–75%).
Importantly, both I72 and B6.AKR splenocytes are equivalent in their ability to present MCC(88–103), an unrelated peptide that also binds to I-Ek and is recognized by the 2B4 hybridoma (Fig. 1 D). These data demonstrate that the inhibition seen in Fig. 1 C is not due to fewer available I-Ek binding sites, and led us to further examine the ability of I72 mice to inhibit both primary and secondary 3.L2 immune responses to Hb(64–76).
Antagonism Does Not Inhibit Clonal Expansion.
The effect of the I72 ligand on 3.L2 proliferation shortly after immunization was examined by labeling 3.L2 T cells with CFSE before adoptive transfer (5 × 106 cells). I72+ and I72− transfer recipients were immunized subcutaneously with 2 nmol Hb(64–76) 1 d after transfer. 2 d after immunization, the draining lymph nodes and spleen were examined by flow cytometry for 3.L2 T cell proliferation, as measured by the interval decreases in CFSE fluorescence. Both I72+ and I72− mice show proliferation of 3.L2 T cells in the lymph nodes and the spleen (Fig. 2, A and B, panels 3, 6, 9, and 12), and the kinetics of this early expansion are equivalent. In the absence of immunization, no proliferation of 3.L2 T cells is seen in the lymph nodes of either I72+ or I72− mice (Fig. 2, A and B, panels 1 and 4), demonstrating that the interaction of 3.L2 T cells with I72 in vivo does not result in T cell activation. 3.L2 T cells in the spleen also show no turnover in the absence of antigen-specific activation (Fig. 2, A and B, panels 7 and 10). When mice were immunized with CFA alone, no 3.L2 proliferation was observed in either the lymph node or spleen (Fig. 2, A and B, panels 2, 5, 8, and 11). Taken together, these results show that the presence of I72 does not inhibit 3.L2 T cells from entering cell cycle after immunization with Hb(64–76) peptide. A reduction in the proliferative response was certainly expected from the ex vivo antagonism data and the current models of peripheral T cell differentiation.
Figure 2.
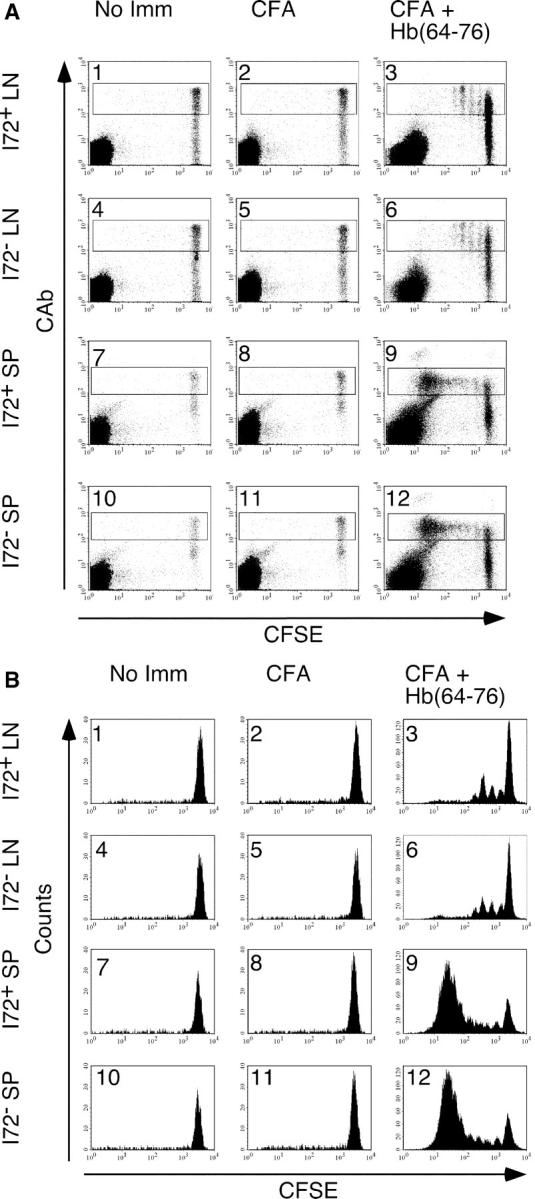
Adoptively transferred naive 3.L2 T cells expand equally in both I72+ and I72− mice after immunization with Hb(64–76) peptide. CFSE-labeled whole splenocytes containing 5 × 106 CAb+ cells were transferred intravenously into I72+ or I72− mice and immunized as indicated. The draining lymph nodes and spleens were harvested 48 h after immunization and analyzed by FACS. (A) Dot plot analysis of the CD4+ population for expression of the 3.L2 TCR and CFSE content. (B) Histogram analysis of the CD4+ CAb+ gate shown in A. This experiment is representative of seven similar experiments.
The CFSE data demonstrate that the interaction between 3.L2 T cells and the I72 ligand in the absence of immunization with Hb(64–76) does not result in cell division. These results also show that CFA-induced activation of APCs in the draining lymph nodes of I72 mice is not sufficient for 3.L2 T cell stimulation and does not result in 3.L2 bystander activation. Finally, 3.L2 T cells purified from unimmunized I72+ and I72− transfer recipients respond equally to ex vivo stimulation with Hb(64–76) peptide, suggesting that exposure to the endogenous antagonist I72 alone does not permanently alter TCR signaling (unpublished data).
In many TCR transgenic models, including 3.L2 mice, the transgenic β chain may pair with an endogenously rearranged α chain, generating a second TCR of unknown specificity. Although the data in Fig. 2 shows that all proliferation occurs in the CD4+ CAb+ population, we ruled out any potential contribution of a second TCR by repeating the experiment with 3.L2 T cells isolated from 3.L2 × RAG-1−/− mice. These data also show equal proliferation in both I72+ and I72− mice, confirming the results obtained with the transfer of whole splenocytes (Fig. S1, which is available at http://www.jem.org/cgi/content/full/jem.20041226/DC1).
When comparing lymph node with splenocyte CFSE staining, a greater number of divided 3.L2 T cells is observed in the spleens of both I72+ and I72− mice (Fig. 2, A and B, panels 9 and 12). The site of the initial activation of these cells is uncertain. After subcutaneous immunization, antigen-loaded, activated dendritic cells migrate to the draining lymph nodes, where the first T cell–APC interactions occur (17). However, by 48 h after immunization, it is also apparent that 3.L2 T cell division is initiated in the spleen. In our experiments, we cannot directly determine the number of 3.L2 T cell divisions that occur within the spleen or the number of 3.L2 T cells that were first stimulated in this lymphoid organ.
Altered Homing Properties of Antagonized T Cells.
Adoptive transfer and immunization were used to examine the primary 3.L2 immune response in the presence of the antagonist I72. In these experiments, whole splenocytes containing 106 unlabeled 3.L2 T cells were transferred intravenously, and the mice were immunized with two nmol Hb(64–76) in CFA 24 h later. Four-color analytical flow cytometry was used to examine the draining lymph nodes and spleen for cell surface expression of CD4, CD8, CD19, and the 3.L2 TCR at multiple time points after immunization.
A robust expansion of 3.L2 T cells is seen in the draining lymph nodes of I72− mice, with 2.8 × 106 3.L2 T cells per lymph node seen on day 5 after immunization (Fig. 3 A). Mice expressing the endogenous antagonist I72+ have a sevenfold reduction in 3.L2 T cell numbers at this same time point, with ∼0.4 × 106 3.L2 T cells per lymph node. The kinetics of T cell expansion are similar. The antigen-specific 3.L2 T cell response peaks in the lymph nodes on day 5 in both I72+ and I72− mice. 10 d after immunization, there are 0.5 × 106 3.L2 T cells per lymph node in I72− controls, whereas 3.L2 T cells in I72+ mice are barely detectable.
Figure 3.

Antagonism results in a redistribution of activated 3.L2 T cells. (A) Whole splenocytes containing 106 CAb+ cells were transferred intravenously into I72+ (•) and I72− (○) mice and immunized as described in Materials and Methods. The draining lymph nodes and spleens were removed 2–10 d after immunization. Lymph nodes were pooled and the total number of CD4+ CAb+ T cells per lymph node and spleen was determined by cell counting and FACS. Multiple experiments were averaged to obtain each data point, and the total number of mice used is as follows: day 2 (7 I72+, 6 I72−), day 3 (6 I72+, 5 I72−), day 4 (10 I72+, 9 I72−), day 5 (19 I72+, 16 I72−), day 7 (9 I72+, 9 I72−), and day 10 (6 I72+, 6 I72−). Error bars represent standard error of the mean. (B) Dual receptor T cells have no effect on the 3.L2 T cell proliferative response. A comparison of the number of 3.L2 T cells found in the lymph nodes and spleens of I72+ and I72− mice on day 5 after immunization using whole 3.L2 splenocytes (open bars) and splenocytes from 3.L2 × RAG-1−/− mice (closed bars). Only those experiments in which all four draining lymph nodes were available were used to generate this data, and the number of mice in each group is indicated. (C) There is no effect of antigen dose on the 3.L2 response. Transfer recipients were immunized with either 2 (light gray bars) or 20 nmol (dark gray bars) of Hb(64–76) peptide. The day 5 responses are shown, and the number of mice in each group is indicated.
Analysis of splenocytes shows a reversal of this effect, with preferential accumulation of 3.L2 T cells in I72+ spleens. In the I72+ mice, there are 16 × 106 3.L2 T cells per spleen at the peak response on day 5 (Fig. 3). In contrast, there are only 6 × 106 3.L2 T cells per spleen in I72− mice on day 5. This represents an ∼2.5-fold increase in the number of 3.L2 T cells in the spleens of I72+ mice at the peak of expansion. The numbers have equalized by day 10, when both I72+ and I72− mice have 4 × 106 3.L2 T cells per spleen. Importantly, the total number of T cells isolated from the four draining lymph nodes and spleen of I72+ and I72− mice at the peak of clonal expansion are nearly equivalent (Fig. 3 B; I72+ = 18.0 × 106/mouse and I72− = 18.3 × 106/mouse). This suggests that the magnitude of the proliferative response in both groups is the same.
A similar alteration in lymphocyte distribution was seen in adoptive transfer experiments performed with 3.L2 T cells isolated from 3.L2 × RAG−/− mice (Fig. 3 B) and with purified 3.L2 T cells (unpublished data). Also, results were the same when mice were immunized with 20 nmol Hb(64–76) peptide, rather than 2 nmol (Fig. 3 C). These control experiments demonstrate that neither the transfer of whole splenocytes or the dose of immunizing peptide influenced the results.
Furthermore, these results are consistent with our previous studies using 3.L2 T cells and mice expressing the weak antagonist A72 (32, 39). In these earlier studies, we observed a 2.5-fold decrease in the number of 3.L2 T cells in the draining lymph nodes, reflecting the weaker antagonism. The number of 3.L2 T cells in the spleen was not examined and we could not distinguish between reduced proliferation and reduced survival. Based on the data presented here, we labeled 3.L2 T cells with CFSE and performed adoptive transfer studies with mice expressing the A72 ligand as described in Fig. 2. Results confirm that after immunization with Hb(64–76) peptide, 3.L2 T cells in the draining lymph nodes and spleen divide at the same rate in both A72+ and I72+ mice, as expected (Fig. S2, which is available at http://www.jem.org/cgi/content/full/jem.20041226/DC1, and Fig. 2).
After adoptive transfer of 3.L2 T cells and immunization with Hb(64–76) peptide, B cell expansion is also seen in the draining lymph nodes of I72+ and I72− mice (Fig. 4 A). However, at the peak of the B cell response on day 7, there is a threefold decrease in the number of B cells in I72+ mice relative to I72− controls. These data suggest that activation of 3.L2 T cells in the presence of the I72 ligand results in poor or limited B cell help. Frozen sections of draining lymph nodes from I72+ and I72− stained for B cells confirm a decrease in secondary follicle formation on day 7 in I72+ mice (Fig. 4 B). There is a modest expansion of B cells in the spleens of I72+ and I72− mice, with no statistically significant difference between the two groups (Fig. 4 A; day 7, P = 0.23). It is important to note that the accelerated B cell response in the lymph nodes of I72− transfer recipients is due to the high precursor frequency of 3.L2 T cells. In normal mice (no transfer), the peak B cell response and the peak of secondary B cell follicle formation occur approximately 10 d after immunization and are the result of the response to CFA (unpublished data). It is therefore possible to distinguish the component of B cell expansion driven by activated 3.L2 T cells from that occurring in response to the adjuvant.
Figure 4.

T cell antagonism limits B cell expansion and secondary follicle formation. (A) From the experiments shown in Fig. 3, the total number of CD19+ cells per lymph node and spleen was also determined. (B) The location of B cells in draining lymph nodes was examined by immunohistochemistry. Frozen sections were stained with anti-B220 and visualized as described in Materials and Methods. The peak of B cell expansion and of secondary B cell follicle formation occurs 7 d after immunization and is absent in I72+ mice. No statistically significant difference in the number of splenic B cells was observed.
Antagonized T Cells Are Anergic.
The abortive immune response occurring in the draining lymph nodes of I72+ mice, marked by the large decrease in 3.L2 T cell numbers, suggested that the expanded population of 3.L2 T cells found in the spleens was nonfunctional. To evaluate this possibility, we purified CD4+ T cells from the lymph nodes and spleens of I72+ and I72− mice on day 5 after immunization. FACS was used to determine the number of 3.L2 T cells in each sample and 2.5 × 104 3.L2 T cells were mixed with 4.75 × 105 B6.AKR splenocytes per well in a 96-well plate. These cells were restimulated with 10 μM Hb(64–76) peptide for 18, 24, and 36 h, and were examined by FACS for the expression of CD69 and the intracellular cytokines IL-2, TNF-α, IFN-γ, and IL-4. The data in Fig. 5, A and B, are from one of three representative experiments analyzed at the time point that gives maximal cytokine expression (IL-2 18 h, all others 36 h). These data show that 3.L2 T cells isolated from the lymph nodes and spleens of I72+ mice are unable to make IL-2, IFN-γ, and TNF-α after restimulation ex vivo. This is in contrast to 3.L2 T cells isolated from the lymph nodes and spleens of I72− controls, which respond with a burst of IL-2, IFN-γ, and TNF-α production. The anergic 3.L2 T cells do up-regulate CD69, although at reduced levels compared with controls (Fig. 5 B). No T cells from these experiments produce IL-4 due to the strong Th1 cell bias created by immunizing with CFA (unpublished data). Together, these data demonstrate that 3.L2 T cells stimulated in the presence of the antagonist I72 fail to achieve full activation, resulting in antigen-specific clonal anergy.
Figure 5.
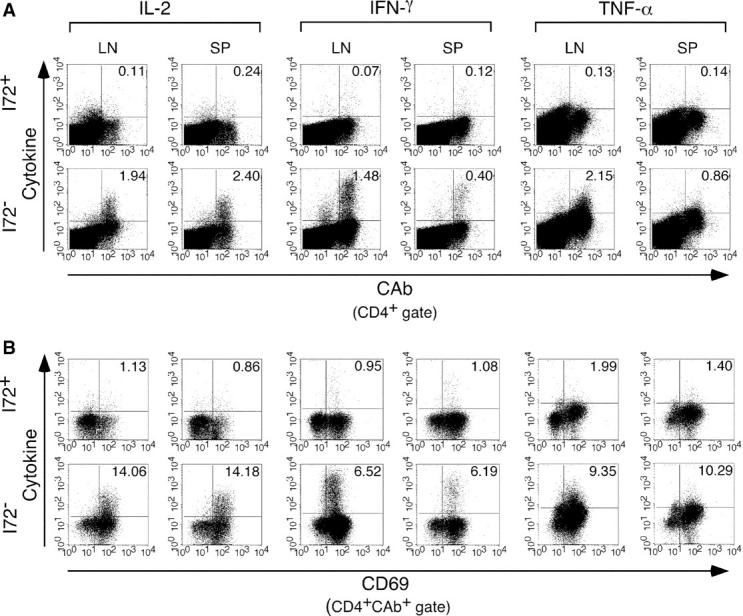
3.L2 T cells increase CD69 expression, but fail to develop effector function in the presence of the antagonist I72. CD4+ 3.L2 T cells were purified from the draining lymph nodes and spleens of I72+ and I72− mice on day 5 after immunization. Equal numbers of 3.L2 T cells were restimulated in culture with B6.AKR splenocytes prepulsed with Hb(64–76) peptide. After 24 h, the cells were removed and stained for cell surface markers and intracellular cytokines as described in Materials and Methods. Quadrants were established using isotype-matched control antibodies. (A) CD4+ cells were analyzed for the expression of the 3.L2 TCR and IL-2, IFN-γ, or TNF-α. No cytokine expression is seen in 3.L2 T cells isolated from I72+ mice. (B) CD4+ CAb+ cells were analyzed for the cell surface expression of CD69 and intracellular cytokines as described above. Many 3.L2 T cells isolated from I72+ mice have up-regulated CD69, yet fail to develop effector function. The data shown is representative of three experiments.
Antagonism Results in Activation of Caspase-3.
As shown above, 3.L2 T cells in immunized I72+ mice fail to acquire an effector phenotype. This lack of effector function together with the rapid loss of 3.L2 T cells from the spleens of I72+ mice seen by day 10 suggested that the proliferative program activated in these cells ultimately resulted in apoptosis. FACS experiments failed to show up-regulation of FasL or Fas on 3.L2 T cells from I72+ mice, making activation-induced cell death unlikely (unpublished data). Further examination by intracellular staining revealed activated caspase-3 in ∼30% of 3.L2 T cells from the spleens of I72+ mice. Activated caspase-3 was seen in only 1% of 3.L2 T cells isolated from I72− controls (Fig. 6). One plausible explanation, based on the lack of IL-2 production and the activation of caspase-3, is that the antagonized 3.L2 T cells fail to survive due to lack of a survival signal, leading to unopposed apoptotic cell death.
Figure 6.

Antagonized 3.L2 T cells have activated caspase-3. Lymph nodes and spleens from day 5 transfer recipients were analyzed by intracellular staining for activated caspase-3. The data was gated on CD4+ CAb+ cells (solid line) and the positive peak was established with an isotype-matched control antibody (dashed line). The percent of gated cells with activated caspase-3 is indicated.
T Cell Memory Is Aborted.
The loss of 3.L2 T cells from I72+ mice after immunization with Hb(64–76) peptide was rapid but not complete 10 d after primary immunization (Fig. 3). Given the altered distribution and lack of effector function seen in activated 3.L2 T cells in I72+ mice, it was possible that the development of 3.L2 T cell memory would also be affected. To test this hypothesis, I72+ and I72− littermate control mice were transferred and immunized as described in the experiments for Fig. 3. After 3, 4, 6, and 10 wk, the draining lymph nodes and spleen were removed and analyzed for the presence of 3.L2 T cells by FACS. The results show that 3.L2 T cells are clearly detectable in the lymph nodes of I72− mice at all time points, whereas no 3.L2 T cells are seen in the lymph nodes of I72+ mice at any time point. However, in the spleens of both I72+ and I72− mice, 3.L2 T cells can be visualized for 6 wk after primary immunization at approximately equal numbers (Fig. 7 A). In the absence of the antagonist, clonotype+ CD44hi cells could be detected in the spleens (5.6 × 103 cells/spleen) and lymph nodes (3.7 × 103 cells/lymph node) of I72− mice 10 wk after primary immunization (Fig. 7 B), consistent with the formation of 3.L2 T cell memory.
Figure 7.

3.L2 T cells fail to develop a memory response. (A) The fitness (survival) of 3.L2 T cells was determined by FACS analysis of draining lymph nodes and spleens at several time points after primary immunization. The data is gated on CD4+ cells, and the percent of lymphocytes that are CAb+ is indicated. In the far right panels, mice were reimmunized with 20 nmol Hb(64–76) peptide in IFA after 10 wk and analyzed 5 d later. The data are representative of three experiments and show persistence of 3.L2 T cells in I72− controls, but not mice that express the antagonist. (B) 10 wk after primary immunization, transfer recipients were analyzed for expression of the memory T cell marker CD44. Data show the percent of CAb+ cells that are CD44hi and CD44low, and are representative of three experiments. (C) Transfer recipients and controls were immunized as described in Materials and Methods. After 10 wk, CD4+ lymph node and spleen cells were purified from pooled samples and equal numbers of CD4+ cells were stimulated in a lymph node proliferation assay with fresh B6.AKR splenocytes as APCs. The data show a recall response in I72− transfer recipients (○), but not in I72+ mice (•), where the minimal response is less than that seen in I72− controls without adoptive transfer of 3.L2 T cells (□). I72+ mice without adoptive transfer (▪) have no recall response. The data are the mean cpm incorporated from three experiments.
This inability to detect 3.L2 T cells in I72+ mice by 6–8 wk after primary immunization was consistent with the hypothesis that the antagonism affects the development of T cell memory. To further evaluate this possibility, adoptive transfer recipients were reimmunized subcutaneously with 20 nmol Hb(64–76) peptide in IFA 10 wk after the primary immunization. 5 d later, the draining lymph nodes and spleen were removed. Cells were analyzed for surface expression of CD4, CD8, and the 3.L2 TCR (Fig. 7 A, far right panels). In I72+ mice, the number of CD4+ CAb+ cells in both lymph nodes and spleen did not exceed that seen with the isotype control antibody (isotype control values: 0.4 × 106 cells/spleen and 0.065 × 106 cells/lymph nodes; unpublished data), also suggesting a lack of 3.L2 T cell memory in these mice.
The suspected influence of the endogenous antagonist I72 on the formation of 3.L2 T cell memory was confirmed by proliferation assay. After secondary immunization, the CD4+ T cell population from lymph nodes and spleens was isolated by magnetic bead cell sorting to remove APCs containing I-Ek–Hb(64–76) or I-Ek–I72 complexes. Purified CD4+ T cells were then added to B6.AKR splenocytes in culture and stimulated with Hb(64–76) peptide. Results show a proliferative response to Hb(64–76) peptide in T cells isolated from I72− controls, but virtually no T cell response in the cells isolated from I72+ mice (Fig. 7 C). The small response seen in normal I72− mice (no adoptive 3.L2 transfer) immunized with Hb(64–76) peptide reflects the response of the endogenous T cell repertoire to immunization and boosting with Hb(64–76) peptide. In contrast, the proliferative response of I72+ transfer recipients and I72+ controls is approximately equal and barely detectable. This reduced reactivity might be due to negative selection of the Hb(64–76)-reactive repertoire in the thymus of I72+ mice. Taken together, these data demonstrate that activation of 3.L2 T cells in the presence of the endogenous antagonist I72 results in an abrogation of 3.L2 T cell memory.
Discussion
In this paper, we have used an adoptive transfer model to examine the effect of a TCR antagonist on an antigen-specific immune response. We find that activation of transferred CD4+ transgenic T cells in the presence of an endogenously expressed antagonist results in antigen-specific clonal expansion while altering T cell homing and eliminating the development of a Th1 cell phenotype. These experiments show a clear separation between the cellular programs that promote cell division and those that result in effector function and T cell memory. Furthermore, they illustrate one functional consequence of the inherent flexibility (40–42) in TCR–ligand interaction. The capability of T cells to simultaneously engage more than one peptide–MHC complex, in this case an endogenous antagonist and the cognate antigen, results in qualitative differences in TCR activation and modified developmental programs. These data demonstrate a role for the quality of TCR signaling in determining cell fate and the potency of endogenous peptide–MHC complexes in shaping the peripheral T cell repertoire.
In the progressive differentiation model for CD4+ and CD8+ T cells, the sequential acquisition of discrete functional capacities by each clone activated in a primary response depends upon the duration of TCR and cytokine stimulation (2, 3, 6). Therefore, outcome hinges upon the affinity of each TCR for antigen (11) and the ability of each clone to compete for activated APCs and cytokines (18). Less fit cells receiving insufficient stimulation will die by neglect, whereas cells receiving sufficient stimulation will sequentially become nonpolarized intermediates and polarized effectors. Those cells receiving excessive stimulation will die by activation-induced cell death. Memory cells are postulated to develop from both nonpolarized intermediates and effectors, and some intermediates may mediate specialized functions such as B cell help or suppression (1). In our system, the use of CFA and adequate doses of peptide assures that all 3.L2 T cells receive a strong activation signal from mature dendritic cells. However, the presence of the antagonist redirects this signal, and the result in vivo is a full proliferative response followed by altered homing, anergy, and elimination of the expanded clone. No 3.L2 T cell memory was observed, further demonstrating that nonpolarized intermediates or effector cells were not generated. We conclude that cell division is the first functional capacity to be acquired and that reaching this activation threshold does not automatically result in the development of effector function. Also, cell division in the absence of effector differentiation results in clonal elimination rather than clonal contraction, as no 3.L2 T cells remain after 6–10 wk in mice that express the antagonist. Thus, there is a clear separation between the instructional program of clonal expansion and those programs that result in effector function and T cell memory. This finding was not predicted by the many in vitro studies describing the effects of antagonist ligands, and it highlights the hierarchical, compartmental nature of an intact immune response.
Abortive proliferation as a mechanism of peripheral tolerance is also seen when immature dendritic cells cross-present self-antigen to CD8+ T cells (16). The deletion of these autoreactive T cells was inhibited by Bcl-2 overexpression and also blocked in Bim-deficient T cells, suggesting lack of a survival signal (43). Thus, low levels of T cell stimulation by immature dendritic cells might be sufficient for proliferation, but not survival or effector differentiation. In our experiments, we observe a similar phenotype, suggesting that antagonist-induced alterations in TCR signaling might be qualitatively similar to those mediated by immature dendritic cells.
The mechanism by which antagonists alter T cell activation is controversial. Affinity measurements between TCR and peptide–MHC class II complexes using surface plasmon resonance have shown that antagonist peptides have faster off rates than agonist peptides, leading to a reduced half-life (11, 44). When viewed in the context of the kinetic proofreading model, these data suggest that antagonist inhibition of T cell activation is due to a failure to fully induce the T cell signaling cascade. This model is supported by data demonstrating that antagonists elicit limited phosphorylation of the CD3-associated ζ chains (29, 45–47). Both the total amount of ζ phosphorylation and the ratio of the p23 to p21 kD isoforms are reduced by antagonist stimulation, possibly resulting in an inhibitory signal. In contrast, studies using mutant ζ chain hybridomas or transgenic mice have demonstrated that antagonism does not depend upon differential phosphorylation of either the p21 or the p23 ζ isoforms, suggesting that altered ζ phosphorylation is a consequence of antagonism (48, 49). In agreement with these later observations, antagonists rapidly recruit SHP-1 to the TCR signaling complex (34), and transfected dominant negative SHP-1 prevents antagonism (30). Lower affinity antagonist ligands trigger a negative feedback loop mediated by SHP-1 dephosphorylation of Lck. Stimulatory ligands activate a positive feedback mechanism that results in phosphorylation of Lck by Erk, thereby blocking SHP-1–Lck interactions (31). One caveat is that many studies involve an analysis of TCR signals induced by either agonist or antagonist ligands independently and in vitro. They do not examine T cell activation when both ligands are present together on an activated APC, as is the case in our experiments and is arguably the more physiologically relevant condition.
This body of evidence documents the relationship between ligand quality and the association of SHP-1 with the TCR signaling complex. When viewed in this context, one prediction based on our data is that upon transfer of 3.L2 T cells into I72 mice, SHP-1 would be rapidly recruited to the TCR signaling complex, resulting in a negative effect on subsequent T cell activation. This association is likely to require the continual presence of the antagonist ligand. The capacity of 3.L2 T cells purified from I72+ transfer recipients to be stimulated normally supports the transient, reversible nature of the antagonist effect and suggests that the full biological potential of TCR antagonism is only appreciated in the setting of inflammation.
It is important to point out that when the antagonist I72 is expressed in the thymus of 3.L2 mice, the result is the elimination of those cells with high levels of the transgenic TCR (37). In this adoptive transfer study, we circumvent thymic processes to focus on the peripheral effect of this class of self-ligand. This approach is warranted when one considers that not all self-proteins are expressed in the thymus. Furthermore, agonist ligands in low doses have been shown to promote positive selection (50–52) and differentiation into lineages with innate characteristics (53), suggesting that negative selection is not the only functional outcome of strong TCR–ligand interaction in the thymus. In the later example, the developing thymocytes are reprogrammed into a CD8αα lineage, demonstrating a divergent developmental pathway triggered by strong TCR–ligand interactions (53).
As a group, endogenous MHC ligands are important in T cell activation and contribute to T cell responses. They heighten sensitivity to foreign antigens (33) and augment clonal expansion after activation (54). During an immune response, the number of MHC complexes displaying a foreign peptide is small and endogenous peptide–MHC complexes contribute to synapse formation (19). This contribution to the total binding energy needed for stable synapse formation can be substantial, regardless of the peptide(s) bound by MHC molecules. For example, alanine scanning mutagenesis of the MHC class I–restricted 2C TCR, combined with binding data and structural analysis, revealed that more than half of the binding energy for the 2C–QL9-Ld complex can be assigned to 2C-Ld contacts (55). Taken together, these observations demonstrate that TCR–MHC contacts contribute to T cell signaling, even in the absence of TCR–peptide interactions.
Antagonists appear to represent a class of endogenous ligand with negative effects on peripheral T cell responses. In our system, the 3.L2–I72-I-Ek complexes have a half-life of 2–3 s, ∼25% of that seen for N72, the strongest stimulatory ligand (11). Based on binding energy alone, I72 would be predicted to enhance 3.L2 T cell activation. Our data demonstrate that this is not the case and support the hypothesis that antagonists alter T cell signal transduction, resulting in clonal elimination and T cell tolerance. Therefore, antagonists are a ubiquitous class of self-ligand that may have a significant effect on the shape of the peripheral T cell repertoire by eliminating clones with self-reactive TCRs that are activated during an immune response.
Acknowledgments
We thank Paul Allen for advice, the gift of the 3.L2 mice, and review of the manuscript; Jack Gorski, Bonnie Dittel, Dave Eckels, Manoj Mishra, Merav Lidar, and Carol Julin for review of the manuscript; and Jennifer Ziegelbauer for assistance with the mice.
This study was funded by Grants from the National Institutes of Health, March of Dimes, and the Children's Hospital of Wisconsin.
The authors have no conflicting financial interests.
Abbreviations used in this paper: CAb, clonotypic antibody; CFSE, 5-(and -6)-carboxyfluorescein diacetate succinimidyl ester.
References
- 1.Lanzavecchia, A., and F. Sallusto. 2000. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 290:92–97. [DOI] [PubMed] [Google Scholar]
- 2.van Stipdonk, M.J., G. Hardenberg, M.S. Bijker, E.E. Lemmens, N.M. Droin, D.R. Green, and S.P. Schoenberger. 2003. Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4:361–365. [DOI] [PubMed] [Google Scholar]
- 3.Gett, A.V., F. Sallusto, A. Lanzavecchia, and J. Geginat. 2003. T cell fitness determined by signal strength. Nat. Immunol. 4:355–360. [DOI] [PubMed] [Google Scholar]
- 4.Kaech, S.M., and R. Ahmed. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaech, S.M., E.J. Wherry, and R. Ahmed. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2:251–262. [DOI] [PubMed] [Google Scholar]
- 6.Iezzi, G., K. Karjalainen, and A. Lanzavecchia. 1998. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 8:89–95. [DOI] [PubMed] [Google Scholar]
- 7.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masopust, D., S.M. Kaech, and R. Ahmed. 2004. The role of programming in memory T-cell development. Curr. Opin. Immunol. 16:217–225. [DOI] [PubMed] [Google Scholar]
- 9.Reinhardt, R.L., A. Khoruts, R. Merica, T. Zell, and M.K. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 10.Alam, S.M., G.M. Davies, C.M. Lin, T. Zal, W. Nasholds, S.C. Jameson, K.A. Hogquist, N.R. Gascoigne, and P.J. Travers. 1999. Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity. 10:227–237. [DOI] [PubMed] [Google Scholar]
- 11.Kersh, G.J., E.N. Kersh, D.H. Fremont, and P.M. Allen. 1998. High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity. 9:817–826. [DOI] [PubMed] [Google Scholar]
- 12.Andres, P.G., K.C. Howland, A. Nirula, L.P. Kane, L. Barron, D. Dresnek, A. Sadra, J. Imboden, A. Weiss, and A.K. Abbas. 2004. Distinct regions in the CD28 cytoplasmic domain are required for T helper type 2 differentiation. Nat. Immunol. 5:435–442. [DOI] [PubMed] [Google Scholar]
- 13.Song, J., S. Salek-Ardakani, P.R. Rogers, M. Cheng, L. Van Parijs, and M. Croft. 2004. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat. Immunol. 5:150–158. [DOI] [PubMed] [Google Scholar]
- 14.Singh, N.J., and R.H. Schwartz. 2003. The strength of persistent antigenic stimulation modulates adaptive tolerance in peripheral CD4+ T cells. J. Exp. Med. 198:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith, K., B. Seddon, M.A. Purbhoo, R. Zamoyska, A.G. Fisher, and M. Merkenschlager. 2001. Sensory adaptation in naive peripheral CD4 T cells. J. Exp. Med. 194:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurts, C., H. Kosaka, F.R. Carbone, J.F. Miller, and W.R. Heath. 1997. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itano, A.A., and M.K. Jenkins. 2003. Antigen presentation to naive CD4 T cells in the lymph node. Nat. Immunol. 4:733–739. [DOI] [PubMed] [Google Scholar]
- 18.Kedl, R.M., B.C. Schaefer, J.W. Kappler, and P. Marrack. 2002. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat. Immunol. 3:27–32. [DOI] [PubMed] [Google Scholar]
- 19.Wulfing, C., C. Sumen, M.D. Sjaastad, L.C. Wu, M.L. Dustin, and M.M. Davis. 2002. Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat. Immunol. 3:42–47. [DOI] [PubMed] [Google Scholar]
- 20.Huppa, J.B., M. Gleimer, C. Sumen, and M.M. Davis. 2003. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat. Immunol. 4:749–755. [DOI] [PubMed] [Google Scholar]
- 21.Alexander, J., K. Snoke, J. Ruppert, J. Sidney, M. Wall, S. Southwood, C. Oseroff, T. Arrhenius, F.C. Gaeta, S.M. Colon, et al. 1993. Functional consequences of engagement of the T cell receptor by low affinity ligands. J. Immunol. 150:1–7. [PubMed] [Google Scholar]
- 22.De Magistris, M.T., J. Alexander, M. Coggeshall, A. Altman, F.C. Gaeta, H.M. Grey, and A. Sette. 1992. Antigen analog-major histocompatibility complexes act as antagonists of the T cell receptor. Cell. 68:625–634. [DOI] [PubMed] [Google Scholar]
- 23.Dittel, B.N., D.B. Sant'Angelo, and C.A. Janeway Jr. 1997. Peptide antagonists inhibit proliferation and the production of IL-4 and/or IFN-gamma in T helper 1, T helper 2, and T helper 0 clones bearing the same TCR. J. Immunol. 158:4065–4073. [PubMed] [Google Scholar]
- 24.Grakoui, A., S.K. Bromley, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science. 285:221–227. [DOI] [PubMed] [Google Scholar]
- 25.Zal, T., M.A. Zal, and N.R. Gascoigne. 2002. Inhibition of T cell receptor-coreceptor interactions by antagonist ligands visualized by live FRET imaging of the T-hybridoma immunological synapse. Immunity. 16:521–534. [DOI] [PubMed] [Google Scholar]
- 26.Plebanski, M., E.A. Lee, C.M. Hannan, K.L. Flanagan, S.C. Gilbert, M.B. Gravenor, and A.V. Hill. 1999. Altered peptide ligands narrow the repertoire of cellular immune responses by interfering with T-cell priming. Nat. Med. 5:565–571. [DOI] [PubMed] [Google Scholar]
- 27.Wang, J.H., T.J. Layden, and D.D. Eckels. 2003. Modulation of the peripheral T-cell response by CD4 mutants of hepatitis C virus: transition from a Th1 to a Th2 response. Hum. Immunol. 64:662–673. [DOI] [PubMed] [Google Scholar]
- 28.Dittel, B.N. 2001. Mechanisms of T cell receptor antagonism: implications in the treatment of disease. Curr. Mol. Med. 1:339–355. [DOI] [PubMed] [Google Scholar]
- 29.Kersh, E.N., G.J. Kersh, and P.M. Allen. 1999. Partially phosphorylated T cell receptor ζ molecules can inhibit T cell activation. J. Exp. Med. 190:1627–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilgore, N.E., J.D. Carter, U. Lorenz, and B.D. Evavold. 2003. Cutting edge: dependence of TCR antagonism on Src homology 2 domain-containing protein tyrosine phosphatase activity. J. Immunol. 170:4891–4895. [DOI] [PubMed] [Google Scholar]
- 31.Stefanova, I., B. Hemmer, M. Vergelli, R. Martin, W.E. Biddison, and R.N. Germain. 2003. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 4:248–254. [DOI] [PubMed] [Google Scholar]
- 32.Basu, D., C.B. Williams, and P.M. Allen. 1998. In vivo antagonism of a T cell response by an endogenously expressed ligand. Proc. Natl. Acad. Sci. USA. 95:14332–14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stefanova, I., J.R. Dorfman, and R.N. Germain. 2002. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 420:429–434. [DOI] [PubMed] [Google Scholar]
- 34.Dittel, B.N., I. Stefanova, R.N. Germain, and C.A. Janeway Jr. 1999. Cross-antagonism of a T cell clone expressing two distinct T cell receptors. Immunity. 11:289–298. [DOI] [PubMed] [Google Scholar]
- 35.Robertson, J.M., and B.D. Evavold. 1999. Cutting edge: dueling TCRs: peptide antagonism of CD4+ T cells with dual antigen specificities. J. Immunol. 163:1750–1754. [PubMed] [Google Scholar]
- 36.Kersh, G.J., D.L. Donermeyer, K.E. Frederick, J.M. White, B.L. Hsu, and P.M. Allen. 1998. TCR transgenic mice in which usage of transgenic α- and β-chains is highly dependent on the level of selecting ligand. J. Immunol. 161:585–593. [PubMed] [Google Scholar]
- 37.Williams, C.B., D.L. Engle, G.J. Kersh, J.M. White, and P.M. Allen. 1999. A kinetic threshold between positive and negative selection based on the longevity of the T cell receptor-ligand complex. J. Exp. Med. 189:1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kersh, G.J., and P.M. Allen. 1996. Structural basis for T cell recognition of altered peptide ligands: a single T cell receptor can productively recognize a large continuum of related ligands. J. Exp. Med. 184:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams, C.B., K. Vidal, D.L. Donermeyer, D.A. Peterson, J.M. White, and P.M. Allen. 1998. In vivo expression of a T cell receptor antagonist: T cells escape central tolerance but are antagonized in the periphery. J. Immunol. 161:128–137. [PubMed] [Google Scholar]
- 40.Kersh, G.J., and P.M. Allen. 1996. Essential flexibility in the T cell recognition of antigen. Nature. 222:495–498. [DOI] [PubMed] [Google Scholar]
- 41.Reiser, J.B., C. Darnault, C. Gregoire, T. Mosser, G. Mazza, A. Kearney, P.A. van der Merwe, J.C. Fontecilla-Camps, D. Housset, and B. Malissen. 2003. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat. Immunol. 4:241–247. [DOI] [PubMed] [Google Scholar]
- 42.Krogsgaard, M., N. Prado, E.J. Adams, X.L. He, D.C. Chow, D.B. Wilson, K.C. Garcia, and M.M. Davis. 2003. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol. Cell. 12:1367–1378. [DOI] [PubMed] [Google Scholar]
- 43.Davey, G.M., C. Kurts, J.F. Miller, P. Bouillet, A. Strasser, A.G. Brooks, F.R. Carbone, and W.R. Heath. 2002. Peripheral deletion of autoreactive CD8 T cells by cross presentation of self-antigen occurs by a Bcl-2–inhibitable pathway mediated by Bim. J. Exp. Med. 196:947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alam, S.M., P.J. Travers, J.L. Wung, W. Nasholds, S. Redpath, S.C. Jameson, and N.R.J. Gascoigne. 1996. T cell receptor affinity and thymocyte positive selection. Nature. 381:616–620. [DOI] [PubMed] [Google Scholar]
- 45.Sloan-Lancaster, J., A.S. Shaw, J.B. Rothbard, and P.M. Allen. 1994. Partial T cell signaling: altered phospho-zeta and lack of zap70 recruitment in APL-induced T cell anergy. Cell. 79:913–922. [DOI] [PubMed] [Google Scholar]
- 46.Madrenas, J., R.L. Wange, J.L. Wang, N. Isakov, L.E. Samelson, and R.N. Germain. 1995. Zeta phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science. 267:515–518. [DOI] [PubMed] [Google Scholar]
- 47.Kersh, E.N., A.S. Shaw, and P.M. Allen. 1998. Fidelity of T cell activation through multistep T cell receptor ζ phosphorylation. Science. 281:572–575. [DOI] [PubMed] [Google Scholar]
- 48.Liu, H., and D.A. Vignali. 1999. Differential CD3 zeta phosphorylation is not required for the induction of T cell antagonism by altered peptide ligands. J. Immunol. 163:599–602. [PubMed] [Google Scholar]
- 49.Pitcher, L.A., P.S. Ohashi, and N.S. van Oers. 2003. T cell antagonism is functionally uncoupled from the 21- and 23-kDa tyrosine-phosphorylated TCR zeta subunits. J. Immunol. 171:845–852. [DOI] [PubMed] [Google Scholar]
- 50.Sebzda, E., V.A. Wallace, J. Mayer, R.S.M. Yeung, T.W. Mak, and P.S. Ohashi. 1994. Positive and negative thymocyte selection induced by different concentrations of a single peptide. Science. 263:1615–1618. [DOI] [PubMed] [Google Scholar]
- 51.Wang, R., A. Nelson, K. Kimachi, H.M. Grey, and A.G. Farr. 1998. The role of peptides in thymic positive selection of class II major histocompatibility complex-restricted T cells. Proc. Natl. Acad. Sci. USA. 95:3804–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kraj, P., R. Pacholczyk, H. Ignatowicz, P. Kisielow, P. Jensen, and L. Ignatowicz. 2001. Positive selection of CD4+ T cells is induced in vivo by agonist and inhibited by antagonist peptides. J. Exp. Med. 194:407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamagata, T., D. Mathis, and C. Benoist. 2004. Self-reactivity in thymic double-positive cells commits cells to a CD8αα lineage with characteristics of innate immune cells. Nat. Immunol. 5:597–605. [DOI] [PubMed] [Google Scholar]
- 54.Stefanova, I., J.R. Dorfman, M. Tsukamoto, and R.N. Germain. 2003. On the role of self-recognition in T cell responses to foreign antigen. Immunol. Rev. 191:97–106. [DOI] [PubMed] [Google Scholar]
- 55.Manning, T.C., C.J. Schlueter, T.C. Brodnicki, E.A. Parke, J.A. Speir, K.C. Garcia, L. Teyton, I.A. Wilson, and D.M. Kranz. 1998. Alanine scanning mutagenesis of an alphabeta T cell receptor: mapping the energy of antigen recognition. Immunity. 8:413–425. [DOI] [PubMed] [Google Scholar]