

Abstract
Virus-specific CD8+ T cells are associated with declining viremia in acute human immunodeficiency virus (HIV)1 infection, but do not correlate with control of viremia in chronic infection, suggesting a progressive functional defect not measured by interferon γ assays presently used. Here, we demonstrate that HIV-1–specific CD8+ T cells proliferate rapidly upon encounter with cognate antigen in acute infection, but lose this capacity with ongoing viral replication. This functional defect can be induced in vitro by depletion of CD4+ T cells or addition of interleukin 2–neutralizing antibodies, and can be corrected in chronic infection in vitro by addition of autologous CD4+ T cells isolated during acute infection and in vivo by vaccine-mediated induction of HIV-1–specific CD4+ T helper cell responses. These data demonstrate a loss of HIV-1–specific CD8+ T cell function that not only correlates with progressive infection, but also can be restored in chronic infection by augmentation of HIV-1–specific T helper cell function. This identification of a reversible defect in cell-mediated immunity in chronic HIV-1 infection has important implications for immunotherapeutic interventions.
Keywords: HIV-1, CD8+ T cells, CD4+ T cells, vaccine, protective immunity
Introduction
Acute HIV-1 infection is characterized by high level plasma viremia, which leads to an activation of the cellular immune system and the rapid expansion of HIV-1–specific CD8+ T cells (1). The first appearance of these cells in the peripheral blood is followed by a rapid and dramatic decline of HIV-1 plasma viremia, probably reflecting the strong antiviral activities of these cells (2, 3). The HIV-1–specific CD8+ T cell responses detected during acute HIV-1 infection are typically low in magnitude and narrowly directed against a paucity of viral epitopes (4–6). Thus, the apparent antiviral activity of these cells in acute infection constitutes a striking contrast to chronic HIV-1 infection, where high levels of viral replication occur in the presence of strong, polyclonal and broadly diversified HIV-1–specific CD8+ T cell responses, as determined by the assessment of antigen-specific interferon γ secretion (7–9). These data suggest a progressive functional defect of HIV-1–specific CD8+ T cells in chronic infection that is not measured by assays quantifying solely antigen-specific interferon γ production of T cells.
Recent data demonstrated that HIV-1–specific CD8+ T cells in individuals with long-term nonprogressive infection have a strong HIV-1–specific ex vivo proliferative capacity, whereas this effector function seems to be absent in individuals with high level viremia (10). A similar loss of HIV-1–specific ex vivo proliferation was also observed for HIV-1–specific CD4+ T cells, which show strong proliferative capacities in acute and long-term nonprogressive infection (11–13), but no detectable ex vivo proliferation in the presence of ongoing viral replication. However, a direct functional connection between these two cell subsets has not been revealed and functional interactions between HIV-1–specific CD4+ and CD8+ T cells are currently insufficiently understood.
In this study, we demonstrate that HIV-1–specific CD4+ and CD8+ T cells in acute HIV-1 infection have strong ex vivo proliferative capacities, which are rapidly lost in the presence of continuing viral replication, but partially preserved by early institution of antiretroviral therapy. HIV-1–specific proliferation of CD8+ T cells critically depended on the presence of IL-2–secreting antigen-specific CD4+ T cells, as it was restored in CD8+ T cells from the phase of chronic infection in vitro by the addition of autologous HIV-specific CD4+ T cells isolated during acute infection and more importantly, in vivo by the induction of HIV-1–specific CD4+ T helper cell responses using an HIV-1 immunogen. Overall, these data suggest that the proliferative impairment of HIV-1–specific CD8+ T cells during chronic infection is not primarily due to an intrinsic functional defect of these cells, but rather represents a direct consequence of the progressive loss of IL-2–secreting, HIV-1–specific CD4+ T cells.
Materials and Methods
Study Individuals.
A total of 35 HIV-1–infected individuals recruited from the Massachusetts General Hospital, the Fenway Community Health Care Center, or the Lemuel Shattuck Hospital were included in this study. 18 persons had primary HIV-1 infection, defined by negative or incompletely positive HIV-1–specific ELISA and/or Western blot reactions in the presence of detectable viral load or HIV-1 seroconversion within 6 mo before study enrollment (4). Seven additional subjects with long-term nonprogressive disease courses (CD4+ T cell count of >500/ml and viral load of <1,000 copies/ml for at least 5 yr in the absence of antiretroviral therapy) and 10 individuals with chronic progressive HIV-1 infection (viral load of >30,000 copies/ml or CD4+ T cell count of <300 cell/μl) were also included. The clinical and demographic characteristics of the study individuals are summarized in Table I. In addition, cryopreserved PBMCs from 10 study individuals who had previously participated in a clinical pilot trial using an HIV-1 immunogen (14) were analyzed in this study. The study was approved by the respective institutional review boards and was conducted in accordance with human experimentation guidelines of the Massachusetts General Hospital.
Table I.
Demographical and Clinical Characteristics of the Study Persons
| CD4+ T cell count (median, range) |
HIV-1 RNA (median, range) |
||||||
|---|---|---|---|---|---|---|---|
| Study population |
Age (median, range) |
Sex(m/f ratio) | Ethnicity | Baseline | 1 yr follow-up |
Baseline | 1 yr follow-up |
| Subjects with long-term nonprogressing HIV-1 infection (n = 7) |
47 (34–51) |
5:2 | 6 Caucasians, 1 Hispanic |
805 cells/μl (566–1,118) |
n/a | 555 copies/ml (<50–1,000) |
n/a |
| Subjects with primary HIV-1 infection (n = 18) |
46.4 (32–57) |
17:1 | 16 Caucasians, 1 African American, 1 Haitian |
562 cells/μl (307–1,141) |
untreated:
490 cells/μl (342–1,050) treated: 753 cells/μl (434–948) |
105,000 copies/ml (216–2,500,000) |
untreated:
26,300 copies/ml (4,231–63,900) treated: <50 copies/ml (all patients) |
| Subjects with chronic progressive HIV-1 infection (n = 10) |
42.5 (37–56) |
9:1 | 5 Caucasians, 2 African Americans, 2 Hispanics |
177 cells/μl (20–460) |
n/a | 42,344 copies/ml (7,247–232,000) |
n/a |
HLA Typing.
High and intermediate resolution HLA class I typing was performed at a commercial laboratory (Dynal) by sequence-specific PCRs according to standard procedures. DNA for typing was extracted using the Purgene DNA Isolation kit for whole blood samples (Gentra Systems).
Lymphocyte Separation and Culture.
Blood specimens were drawn in ACD tubes (Becton Dickinson). Fresh PBMCs were separated from whole blood by Ficoll-Hypaque (Histopaque 1077; Sigma-Aldrich) density gradient centrifugation. PBMCs were cultured in RPMI 1640 medium (Sigma-Aldrich) supplemented with 2 mM l-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 10 mM Hepes, and 10% heat-inactivated FCS. Recombinant human IL-2 (provided by the AIDS Research & Reference Reagent Repository, National Institutes of Health) was added in some assays at a concentration of 50I U/ml.
Synthetic Peptides.
410 synthetic 17–19 amino acid peptides, overlapping by 10 amino acids and spanning the entire HIV-1 clade B 2001 consensus sequence (http://hiv-web.lanl.gov), and peptides corresponding to described optimal HIV-1 CD8+ T cell epitopes (15), were synthesized at the Massachusetts General Hospital Peptide Core Facility on an automated peptide synthesizer using F-moc technology. 14 CMV-specific peptides that were selected on the basis of their capacity to bind to HLA class II DRB1*0401 in binding assays (16) were similarly synthesized.
ELISPOT Assay.
ELISPOT assays were performed as described previously (4). In brief, PBMCs were plated in 96-well polyvinylidene plates that had been precoated with 0.5 μg/ml of an anti–human interferon γ mAb (Mabtech). PBMCs were added at a concentration of 50,000–100,000 cells per well in a volume of 100 μl RPMI 1640 medium supplemented with 10% FCS, 10 mM Hepes buffer, 2 mM l-glutamine, and 50 U/ml penicillin-streptomycin. The final concentration of the peptides in every single well was 14 μg/ml. Plates were incubated overnight at 37°C, 5% CO2, and developed on the next day as described elsewhere (17). Wells containing PBMCs and medium with phytohemagglutinin or without any peptide were used as positive or negative controls and run in triplicate on each plate. To calculate the number of specific T cells, the number of spots in the negative control wells was subtracted from the counted number of spots in each well. Responses were considered positive if there were >50 spot-forming cells (SFCs)/106 PBMCs and at least three times the mean number of SFCs of the three control wells. CD8+ T cell dependence of responses was determined by depletion of CD4+ T cells using the MiniMACS cell depletion system (Miltenyi Biotec).
Ex Vivo Proliferation Assay.
PBMCs were first suspended at 106/ml in PBS and incubated at 37°C for 7 min with 0.25 μM carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes). After the addition of serum and washes with PBS, cells were suspended at 106/ml in medium (RPMI 1640 supplemented with glutamine, 10% human FCS, penicillin, and streptomycin). No exogenous cytokines were added to the medium unless otherwise indicated. Pools of overlapping HIV-1–specific peptides representing the entire amino acid sequence of either Gag, Nef, Pol, Env, a combined pool of Tat, Rev, Vif, Vpr, Vpu, or tetanus toxoid (Aventis Behring) were then added at a concentration of 20 ng/ml per peptide. On day 6, cells were harvested, washed with PBS, and stained with mAbs (CD4 PE, CD8 APC, and CD3 PerCP; BD Biosciences). Cells were then washed and fixed in 1% paraformaldehyde and subjected to flow cytometric analysis. Where indicated, IL-2–neutralizing antibodies (clone MQ1-17H12; BD Biosciences) or isotype control antibodies (clone A4A; Neomarkers) were added at 10 μg/ml. In some experiments, 106 autologous CD4+ T cells isolated and cryopreserved during acute or chronic HIV-1 infection were thawed and added to 5 × 106 PBMCs from chronic HIV-1 infection. Flow cytometric data (100,000 nongated events) were acquired on a FACSCalibur four-color flow cytometer using CELLQuest software or an LSR II flow cytometer using the FACS DiVa software (all instruments and software from BD Biosciences). The mean background proliferation was calculated based on the proliferating fractions in media alone. The antigen-specific proportion of proliferating cells was calculated by subtracting the proportion of proliferating cells in unstimulated samples from the proliferating fraction in response to antigen.
Multiparameter Flow Cytometric Analysis.
We used a panel of five different monoclonal surface antibodies (CD25 [IL-2Rα chain] PE-Cy7, CD8 APC-Cy7, CD4 APC-Cy5.5, IL-7Rα chain PE, and IL-15Rα chain Alexa 430; all from BD Biosciences) in addition to staining with APC-labeled MHC class I–peptide tetramer complexes (Beckman Coulter) and CFSE. For intracellular cytokine stainings, cells were initially stained with surface antibodies. After fixation and permeabilization, cells were stained with intracellular antibodies as described previously (17). Samples were acquired on an LSR II flow cytometric device (BD Biosciences), using the FACS DiVa software (BD Biosciences) according to the manufacturer's instructions. Data analysis was performed with the FlowJo software package (TreeStar).
Depletion and Enrichment of Selective Cellular Subsets.
Isolation of CD8+ and CD4+ T cells as well as depletion of CD3+ T cells from whole blood samples was performed by use of the respective Rosette Sep cell separation kits (StemCell Technologies Inc.). Depletion of CD4+ from isolated PBMCs was performed using magnetic anti-CD4+ beads and the MACS cell separation system (Miltenyi Biotec). All cell enrichment and depletion procedures were conducted by negative selection to ensure that isolated cells were not labeled with antibodies.
Statistical Analysis.
Results are given as means or medians with ranges. Statistical analysis was based on Student's t tests. A p-value of <0.05 was considered significant. When two adjacent peptides were recognized in the ELISPOT assays, we deleted the weaker of the two responses. In the case of three adjacent peptides eliciting a response, the weakest of all three peptides was deleted and the responses were counted as two epitopic regions. Statistical analysis and graphical presentation was performed by use of the GraphPad Prism software package.
Results
Strong Lymphoproliferative Responses of HIV-1–specific CD8+ T Cells in Primary HIV-1 Infection.
A number of recent studies have analyzed the magnitude, breadth, and protein specificity of HIV-1–specific CD8+ T cell responses in primary HIV-1 infection by interferon γ ELISPOT technology or intracellular cytokine staining (4–6, 18). Here, we extended these studies and used a flow cytometric proliferation assay based on the sequential loss of CFSE labeling in dividing cells to determine the ex vivo proliferative capacity of HIV-1–specific CD8+ T cells in a total of 18 different subjects with primary HIV-1 infection. In addition, 10 individuals with untreated chronic progressive HIV-1 infection and 7 individuals with untreated long-term nonprogressive HIV-1 infection were included as reference populations. The demographic and clinical characteristics of the study subjects are summarized in Table I.
Fig. 1 shows representative experimental results from individuals with long-term nonprogressive HIV-1 infection (Fig. 1 A), chronic progressive HIV-1 infection (Fig. 1 B), and primary HIV-1 infection (Fig. 1 C). Although similar frequencies of dividing CD8+ T cells were observed in the three study subjects after stimulation with PHA, populations of proliferating antigen-specific CD8+ T cells were only seen in subjects with primary or long-term nonprogressive HIV-1 infection after stimulation of cells with a pool of overlapping peptides spanning the HIV-1 Nef protein. These proliferating CD8+ T cells were specific for HIV-1 Nef, as demonstrated by the specific staining with MHC class I tetramers refolded with HIV-1 Nef peptides (Fig. 1, D and E). Overall, the proportion of CD8+ T cells proliferating in response to stimulation with viral peptides spanning the entire HIV-1 proteome reached a median of 10.9% (range: 3.5–22%) and 23.6% (range: 11.8–59.9%) in individuals with primary or long-term nonprogressing HIV-1 infection, respectively. In contrast, essentially no HIV-1–specific CD8+ T cell ex vivo proliferation was observed in study persons with chronic progressive HIV-1 infection (Fig. 1 F; reference 10), whereas the proportion of proliferating CMV-specific CD8+ T cells was not reduced in a subset of individuals with chronic HIV-1 infection when compared with persons with acute infection (median of 7% [range: 4–17%] vs. 9.1% [range: 7.5–23%]; P = 0.5).
Figure 1.

Cross-sectional assessment of CD8+ T cell proliferation after stimulation with HIV-1 peptide pools in individuals with primary, chronic progressive, and chronic long-term nonprogressive HIV-1 infection. (A–C) Dot plots showing the flow cytometric analysis of HIV-1–specific CD8+ T cell proliferation after stimulation of PBMCs with no stimulus, phytohemagglutinin (PHA), or a pool of overlapping peptides spanning the entire HIV-1 Nef protein in subjects with long-term nonprogressive (A), chronic progressive (B), or primary (C) HIV-1 infection. Values in top left corner of dot plots indicate the proportion of CFSElow CD8+ T cells. (D and E) Corresponding antigen specificity of proliferation cells. 34% of proliferating cells in the study individual in A were binding to the HLA-A3-QVPLRPMTYK (QK10) tetramer (D), whereas 82% of the CD8+ T cells proliferating in the study person in C were specific for the HLA-B8-FLKEKGGL (FL8) tetramer (E). (F and G) Comparative analysis of proliferation and interferon γ secretion by CD8+ T cells in response to stimulation with overlapping peptides spanning the entire HIV-1 proteome. Data from study subjects with chronic progressive HIV-1 infection (CPHI; n = 10), chronic long-term nonprogressive HIV-1 infection (CNPHI; n = 7), and primary HIV-1 infection (PHI; n = 18) are shown.
The lymphoproliferative CD8+ T cell responses in primary HIV-1 infection targeted multiple viral regions, with a median proportion of 2.5% (range: 0–16.4%), 1.6% (range: 0–15%), 0.3% (range: 0–5.3%), 0.25% (range: 0–11%), or 0.1% (range: 0–6.48%) of CD8+ T cells responding to stimulation with Nef, Gag, Pol, Env, or the remaining regulatory and accessory HIV-1 proteins (Vpr, Vpu, Vif, Rev, and Tat), respectively. Notably, the observed pattern of lymphoproliferative HIV-1–specific CD8+ T cells in the three study groups was strikingly different from the corresponding HIV-1–specific CD8+ T cell responses measured using an interferon γ ELISPOT assay. As described previously (4, 7), the total magnitude of interferon γ–secreting, HIV-1–specific CD8+ T cells in individuals with primary HIV-1 infection was significantly lower than in individuals with chronic infection, whereas no difference in the total HIV-1–specific CD8+ T cell magnitude was found between individuals with progressive and long-term nonprogressive chronic infection (Fig. 1 G). Taken together, these data show that HIV-1–specific CD8+ T cells in primary and long-term nonprogressive HIV-1 infection have strong ex vivo proliferative capacities, whereas this effector function is absent in chronic progressive HIV-1 infection.
Parallel Evolution of Lymphoproliferative HIV-1–specific CD4+ and CD8+ T Cell Responses after Primary HIV-1 Infection.
To more closely determine the fate of HIV-1–specific CD8+ T cell lymphoproliferative responses mounted during primary HIV-1 infection, we longitudinally followed the evolution of these responses during the ensuing disease process, using the CFSE-based proliferation assay in conjunction with an interferon γ ELISPOT assay. In line with previous findings (4, 19), we observed that the total magnitude of HIV-1–specific interferon γ–secreting CD8+ T cells increased by a median of threefold over a 1-yr period in individuals with ongoing viral replication in the absence of antiretroviral combination therapy (Fig. 2 A). In contrast, the proliferative capacity of these cells diminished dramatically during the same time period in antiretroviral therapy–naive individuals, with almost no CD8+ T cells proliferating in response to HIV-1 antigen after 1 yr of follow-up. In individuals with rapid institution of antiretroviral therapy during primary HIV-1 infection (Fig. 2 B), we observed a stable magnitude of interferon γ–secreting, HIV-1–specific CD8+ T cell responses over a 1-yr study period (a median of 1,420 SFCs/106 PBMCs vs. a median of 1,985 SFCs/106 PBMCs). Interestingly, the corresponding proportions of CD8+ T cells proliferating in response to HIV-1 antigen declined substantially after primary HIV-1 infection, but were maintained at clearly detectable levels and significantly exceeded the proportion of antigen-specific proliferating CD8+ T cells in persons with continuing viral replication (a median of 2.2% [range: 0.7–14.25%] vs. a median of 0.4% [range: 0.3–1.2%], respectively; P = 0.03), although there was no statistically significant difference between these two study cohorts at baseline during primary infection (P = 0.5). In line with previous reports (12, 20), a partial conservation of lymphoproliferative responses by initiation of antiretroviral therapy during primary HIV-1 infection was also observed for HIV-1–specific CD4+ T cells, whereas the HIV-1–specific proliferation of CD4+ T cells was essentially lost 1 yr after acute HIV-1 infection in individuals with continuously ongoing viral replication (Fig. 2, A and B). Overall, we observed a strong correlation between the total proportion of proliferating HIV-1–specific CD4+ and CD8+ cells (Fig. 2 C). In contrast, no correlation was seen between HIV-1–specific CD8+ T cell lymphoproliferative responses and the corresponding magnitude of HIV-1–specific, interferon γ–secreting CD8+ T cells (Fig. 2 D). Taken together, these data indicate a parallel evolution of lymphoproliferative HIV-1–specific CD8+ and CD4+ T cell responses, and suggest a potential link between the ex vivo proliferative responses of these two antigen-specific T cell populations.
Figure 2.

Longitudinal evolution of HIV-1–specific CD8+ T cell proliferative responses after primary HIV-1 infection. (A and B) Simultaneous assessment of antigen-specific proliferation and interferon γ secretion of CD8+ and CD4+ T cells after stimulation with overlapping peptides spanning the entire HIV-1 proteome at baseline and after 1 yr of follow-up in study persons with untreated (A) and treated (B) primary HIV-1 infection. (C and D) Correlation between HIV-1–specific CD8+ and CD4+ T cell lymphoproliferative responses. Proportions of CD8+ cells proliferating after exposure to overlapping peptides spanning the HIV-1 proteome were plotted against the corresponding proportion of CD4+ T cells (C) and against the corresponding magnitude of CD8+ T cell–mediated SFCs/106 PBMCs using an interferon γ ELISPOT assay (D). Data from the cross-sectional and longitudinal analysis were included. Dashed lines indicate the 95% confidence interval of the regression line.
Ex Vivo Proliferation of HIV-1–specific CD8+ Cells Critically Depends on IL-2.
Previous studies indicate a decisive role of IL-2 for maintaining the ex vivo proliferative activity of HIV-1–specific CD4+ T helper cells (21). Here, we conducted a series of experiments to elucidate the relevance of IL-2 for also sustaining ex vivo proliferative capacities of HIV-1–specific CD8+ T cells. Overall, the neutralization of IL-2 by IL-2–specific mAbs resulted in an almost complete abrogation of ex vivo proliferative activities of HIV-1–specific CD8+ T cells from study subjects with acute HIV-1 infection, whereas control antibodies did not yield similar effects (Fig. 3, A–D). This was the case both for the entire population of CD8+ T cells dividing after stimulation with HIV-1 peptides (Fig. 3, A and C), as well as for subsets of proliferating CD8+ T cells specific for certain defined HIV-1 CD8+ T cell epitopes, as determined by staining with HIV-1 epitope-specific MHC class I tetramers (Fig. 3, B and D). IL-2–neutralizing mABs similarly abrogated the proliferative capacity of CMV-specific CD8+ T cells (not depicted), indicating that IL-2 dependence was not confined to HIV-1–specific CD8+ T cells. In addition, CD8+ T cells proliferating in response to HIV-1 peptides significantly up-regulated the surface expression of the IL-2Rα chain. In contrast, these dividing CD8+ T cells down-regulated or maintained constant cell surface expression levels of the α receptor chains for the homeostatic cytokines IL-7 and IL-15 (Fig. 3, E and F). Taken together, these results indicate a critical relevance of IL-2 for the ex vivo proliferative capacity of HIV-1–specific CD8+ T cells.
Figure 3.

Antigen-specific ex vivo proliferation of HIV-1–specific CD8+ T cells critically depends on IL-2. (A and B) Dot plots (A) or histograms (B) showing the flow cytometric analysis of the proportion of CD8+ T cells (A) or HLA-B8-FLKEKGGL (FL8) tetramer–specific CD8+ T cells (B) proliferating in response to stimulation with a pool of overlapping peptides spanning HIV-1 Nef in the presence or absence of anti–IL-2 mAb. Values in top left corner of dot plots indicate the proportion of CFSElow CD8+ T cells. (C and D) Proportion of CD8+ T cells (C) or B8-FL8 tetramer–specific CD8+ T cells (D) proliferating after exposure to a pool of overlapping Nef peptides in the presence or absence of IL-2 antibodies. Mean and standard deviation of four experiments in four different study persons are shown. (E) Flow cytometric analysis of the surface expression of the α chain of the IL-2, IL-7, and IL-15 receptor in CD8+ T cells proliferating after stimulation with overlapping HIV-1 Nef peptides. (F) Median fluorescence of antibodies directed against the α chain of the IL-2, IL-7, and IL-15 receptor in CD8+ T cells proliferating (black bars) or nonproliferating (white bars) after stimulation with HIV-1 Nef peptides. Data reflect the mean and standard deviation of five independent experiments in four different study subjects.
IL-2 Production by CD4+ T Cells Supports HIV-1–specific CD8+ T Cell Proliferation.
To more closely determine cell populations supporting the ex vivo proliferative activity of HIV-1–specific CD8+ T cells in individuals with primary HIV-1 infection, we assessed HIV-1–specific CD8+ T cell lymphoproliferative responses after the selective ex vivo removal of distinct leukocyte subsets (Fig. 4, A–C). Interestingly, HIV-1–specific CD8+ T cell proliferation was almost entirely blocked when lymphocyte samples had been depleted of CD4+ cells before the addition of HIV-1 peptides (Fig. 4, A and B). In contrast, HIV-1–specific proliferation was restored after coincubation of isolated CD8+ T cells with isolated CD4+ T cells, indicating that CD4+ T cells are essential for the ex vivo proliferative activities of HIV-1–specific CD8+ T cells (Fig. 4, A and B). The HIV-1–specific proliferative capacity of CD8+ T cells that was lost after depletion of autologous CD4+ cells was also restored by the addition of exogenous IL-2 (Fig. 4, D–G), indicating that IL-2 can directly support HIV-1–specific CD8+ T cells in the absence of CD4+ T helper cells. In addition, exogenous IL-2 restored the ex vivo proliferative capacity of a subset of HIV-1–specific CD8+ T cells in individuals with chronic progressive HIV-1 infection that was otherwise lost (Fig. 4, H and I). Thus, these data show that the ex vivo proliferative activity of HIV-1–specific CD8+ T cells critically depends on IL-2 secreted by CD4+ T cells.
Figure 4.
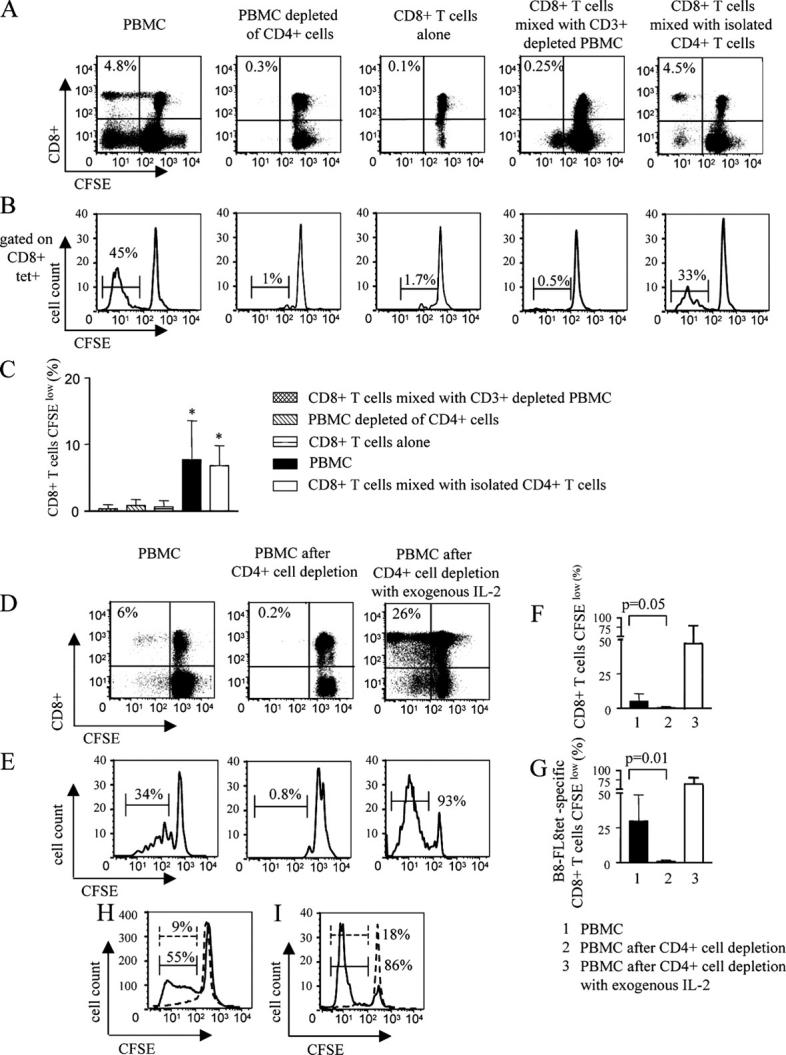
Ex vivo proliferation of HIV-1–specific CD8+ T cells is supported by CD4+ T cells. (A–C) Dot plots (A) and histograms (B) indicating the flow cytometric assessment of CD8+ T cells (A) or B8-FL8–specific CD8+ T cells (B) proliferating after stimulation with a Nef peptide pool in the presence or absence of indicated leukocellular subsets. Cells were gated according to forward scatter (FSC)/side scatter (SSC) characteristics of the lymphocyte population in A. In B, lymphocytes were additionally gated according to CD8+ expression and tetramer binding. Values in top left corner of dot plots indicate the proportion of CFSElow CD8+ T cells. A and B show one representative experiment and C indicates the mean and standard deviation of the proportion of CFSElow CD8+ T cells in four independent experiments (*, P < 0.05). (D–G) Dot plots (D) and histograms (E) showing the flow cytometric analysis of the proliferation of CD8+ T cells (D) or B8-FL8–specific CD8+ T cells (E) in responses to stimulation with a Nef peptide pool in whole PBMC samples, CD4+ cell–depleted PBMC samples, and in CD4+ cell–depleted PBMC samples that were supplemented with exogenous IL-2. Gating was performed as described for A and B. D and E show one representative experiment and F and G give the mean and standard deviation from ten independent experiments for bulk CD8+ T cells (F) and three different experiments for tetramer-specific cells (G), respectively. (F and G) Left black bars represent proliferating cells in whole PBMC samples, middle bars show PBMC samples depleted of CD4+ cells, and right white bars indicate PBMC samples depleted of CD4+ cells, but supplemented with exogenous IL-2. (H and I) Rescue of HIV-1–specific CD8+ T cell proliferation by exogenous IL-2 in chronic replicative HIV-1 infection. (H) CD8+ T cell proliferation in the presence of exogenous IL-2 after stimulation of PBMC samples from chronic HIV-1 infection with Nef pool peptides (solid line) or no antigenic stimulation (dashed line). Gating was performed according to FSC/SSC characteristics and CD8+ expression. (I) Proliferation of HLA-A3-QK10 tetramer–specific CD8+ T cells in the presence of IL-2 after stimulation of PBMC samples from chronic HIV-1 infection with Nef pool peptides (solid line) or no antigenic stimulation (dashed line). Cells were gated according to FSC/SSC characteristics, as well as CD8 expression and HLA-A3 QK10 tetramer binding. (H and I) Percentages indicate the proportion of CFSElow CD8+ T cells. One representative example of four different experiments is shown.
Antigenic Stimulation of CD4+ T Cells Significantly Enhances the Antigen-specific Proliferative Activity of HIV-1–specific CD8+ T Cells.
In our previous experiments, the ex vivo proliferation capacity of HIV-1–specific CD8+ T cells was assessed after stimulation of PBMCs with pools of overlapping peptides spanning HIV-1 proteins, which simultaneously elicited lymphoproliferative CD4+ and CD8+ T cell responses, but did not allow for the analysis of the lymphoproliferative activity of HIV-1–specific CD8+ T cells in the absence of concurrent CD4+ T cell proliferative immune responses. We subsequently tested the ex vivo proliferative capacity of HIV-1–specific CD8+ T cells that had been stimulated with defined optimal HIV-1–specific CD8+ T cell epitopic peptides in the presence or absence of a simultaneous stimulus for HIV-1–specific CD4+ T cell proliferative responses. Fig. 5, A and B, shows results from the HLA-B8–expressing study individual AC-31. Only a limited proportion of CD8+ T cells specific for the HLA-B8–restricted Nef epitope FLKEKGGL (B8-FL8) and virtually no CD4+ T cells proliferated when PBMCs samples were stimulated with the HLA-B8–restricted CD8+ T cell epitopic peptide B8-FL8 alone. In contrast, a dramatically stronger HIV-1–specific CD8+ T cell proliferative response was observed after stimulation of PBMCs with the B8-FL8 peptide and an additional HIV-1 Nef peptide (PEKEVLVWKFDSRLAFHH) that, when used alone, elicited a selective CD4+ T cell–mediated lymphoproliferative response, but no significant CD8+ T cell proliferation (Fig. 5, A, B, D, and E). The enhancement of the ex vivo proliferation of HIV-1–specific CD8+ T cells by synchronized stimulation of HIV-1–specific CD4+ T cells was almost entirely blocked by adding IL-2–neutralizing antibodies (Fig. 5 C). Finally, we observed that the ex vivo proliferation of HIV-1–specific CD8+ T cells can also be enhanced by simultaneous stimulation of CD4+ T cells specific for CMV or tetanus toxoid (Fig. 5, D–F). Thus, these data illustrate that antigen-specific lymphoproliferative CD4+ T cell responses significantly enhance the ex vivo proliferative activity of HIV-1–specific CD8+ T cells in an IL-2–dependent fashion.
Figure 5.

Ex vivo proliferative activities of HIV-1–specific CD8+ T cells are dramatically enhanced by simultaneous stimulation of antigen-specific CD4+ T cells. (A) CD8+ and CD4+ T cell proliferation after stimulation with the HIV-1 Nef CD8+ T cell epitope B8-FL8 or with the overlapping HIV-1 Nef peptide PEKEVLVWKFDSRLAFHH, or both peptides together. Dot plots of one representative flow cytometric experiment are shown. Cells were gated according to FSC/SSC characteristics of the lymphocyte population. (B) B8-FL8 tetramer–specific CD8+ T cells proliferating after stimulation with B8-FL8 peptide only (top) or in conjunction with the overlapping Nef peptide PEKEVLVWKFDSRLAFHH (bottom). (C) CD8+ T cell proliferation after simultaneous stimulation of PBMC samples with the CD8+ T cell epitope B8-FL8 and the HIV-1 Nef peptide PEKEVLVWKFDSRLAFHH in the presence of IL-2 antibodies. Cells were gated according to FSC/SSC characteristics of the lymphocyte population. (D and E) Proportion of CD8+ T cells (D), B8-FL8 tetramer–specific CD8+ T cells (E), or CD4+ T cells (F) proliferating after stimulation with the B8-FL8 epitopic peptide in the presence or absence of concomitant stimulation with tetanus toxoid, a CMV peptide, or the overlapping HIV-1 Nef peptide PEKEVLVWKFDSRLAFHH. Mean and standard deviation from three independent experiments in three different study subjects are shown.
Autologous HIV-1–specific CD4+ T Cells Isolated in Acute Infection Can Reconstitute the Proliferative Activity of HIV-1–specific CD8+ T Cells in Chronic Infection.
The above data demonstrate that HIV-1–specific CD8+ T cell proliferation critically depends on IL-2 produced by antigen-specific CD4+ T cells. Next, we tested whether CD4+ T cells harvested during acute HIV-1 infection, when triggered by HIV-1, could rescue the ex vivo proliferative activity of HIV-1–specific CD8+ T cells in chronic, untreated HIV-1 infection. Fig. 6 A shows data from study individual AC-98. Strong CD8+ T cell–mediated lymphoproliferative immune responses were observed in acute HIV-1 infection after PBMC stimulation with a pool of overlapping HIV-1 Nef peptides, with a significant proportion of these proliferating cells being specific for the HLA-A3–restricted Nef epitope A3-QK10. These lymphoproliferative responses were almost completely lost during chronic HIV-1 infection, despite the physical preservation of QK10-specific CD8+ T cells, as determined by staining with QK10-specific MHC class I tetramers (Fig. 6, A and B). Yet, the addition of autologous CD4+ T cells isolated during acute HIV-1 infection to the PBMC sample from chronic infection rescued the proliferation of a subset of CD8+ T cells specific for the QK10 tetramer after stimulation with a pool of overlapping Nef peptides, whereas the addition of the same number of autologous CD4+ T cells isolated during chronic HIV-1 infection did not increase the proliferative activity of HIV-1–specific CD8+ T cells in chronic infection. The added autologous CD4+ T cells from acute HIV-1 infection exhibited strong HIV-1 Nef-specific lymphoproliferative capacities and IL-2 secretion (Fig. 6, A and C), whereas CD4+ T cells from the chronic disease phase were neither able to secrete IL-2 nor proliferate in an antigen-specific manner (not depicted). In two additional study subjects, we observed that HLA-A2–restricted CD8+ T cells specific for the p17 Gag epitope SLYNTVATL (SL9) had almost entirely lost their antigen-specific proliferative capacity in chronic infection, despite being detectable at high frequencies, as determined by specific staining of CD8+ T cells with SL9–MHC class I tetramer complexes. Yet, after addition of autologous CD4+ T cells harvested during acute HIV-1 infection, the ex vivo proliferative capacities of a subset of these cells were rescued (Fig. 6, D and E). Again, we found that the CD4+ T cells harvested during acute infection included HIV-1–specific CD4+ T cells with strong capacities for ex vivo proliferation and IL-2 secretion after encounter with HIV-1 peptides (not depicted). Moreover, the enhancement of HIV-1–specific CD8+ T cell lymphoproliferative responses by CD4+ T cells isolated during acute HIV-1 infection was almost entirely abrogated by IL-2–neutralizing antibodies (not depicted). Taken together, these results show that CD4+ T cells isolated during acute HIV-1 infection can rescue the ex vivo proliferative capacity of HIV-1–specific CD8+ T cells in chronic HIV-1 infection by an IL-2–dependent mechanism.
Figure 6.

Autologous CD4+ T cells harvested during primary HIV-1 infection can restore the ex vivo proliferative activity of HIV-1–specific CD8+ T cells in chronic HIV-1 infection. (A) Flow cytometric analysis of CD8+ and CD4+ T cell proliferation after stimulation with a pool of overlapping Nef peptides in study subject AC-98 during primary and chronic HIV-1 infection and in chronic HIV-1 infection after the addition of isolated autologous CD4+ T cells harvested during acute HIV-1 infection. Values in top left corner of dot plots indicate the proportion of CFSElow CD8+ or CD4+ T cells, respectively. (B) Corresponding histograms indicating the proportion of proliferating A3-QK10–specific CD8+ T cells. (C) Intracellular cytokine staining of CD4+ T cells from acute HIV infection in study subject AC-98 after stimulation with Nef pool peptides. Cells were gated according to FSC/SSC characteristics. Value in top right corner indicates proportion of IL-2+ CD4+ T cells. (D and E) Proportion of CD8+ T cells (D) and HIV-1 tetramer–specific CD8+ T cells (E) proliferating in chronic HIV-1 infection after stimulation with a pool of HIV-1–specific Nef peptides in the presence (white bars) or absence (black bars) of added autologous CD4+ T cells from acute HIV-1 infection. Data indicate the mean and standard deviation from three study individuals described in Results.
In Vivo Reconstitution of the Proliferative Activity of HIV-1–specific CD8+ T Cells by Vaccine-induced, IL-2–secreting, HIV-1–specific CD4+ T Cells.
A number of studies have indicated that the administration of inactivated gp120-depleted HIV-1 can result in the induction of strong HIV-1–specific CD4+ T cell lymphoproliferative responses (22–25). We showed recently in a placebo-controlled phase II clinical trial in 10 individuals (5 receiving HIV vaccine and 5 receiving adjuvant alone), that the vaccine could elicit vigorous HIV-1–specific CD4+ T cell–mediated lymphoproliferative immune responses in chronically infected HIV-1 persons treated with highly active antiretroviral therapy, but did not increase the magnitude of HIV-1–specific CD8+ T cell responses when measured by an interferon γ ELISPOT assay (14). We predicted, based on the in vitro data described above, that the in vivo induction of HIV-1–specific CD4+ T cell proliferative responses in these individuals should result in the simultaneous development of HIV-1–specific CD8+ T cell proliferative responses, and used cryopreserved samples from this randomized trial to further characterize the quality of HIV-1–specific CD4+ T cell responses induced by the vaccine, as well as their impact on HIV-1–specific CD8+ T cell responses.
After five consecutive immunizations with gp120-depleted, inactivated HIV, a median of 0.6% (range: 0.4–1.9%) of CD4+ T cells secreted IL-2 in response to stimulation with overlapping peptides spanning HIV-1, whereas IL-2 secretion by CD4+ T cells was minimal in five recipients of placebo (median proportion of 0.1% [range: 0–0.2%]; P = 0.05). We subsequently assessed the impact of immunization on HIV-1–specific proliferative T cell responses. In line with our previous data, HIV-1–specific lymphoproliferative activities of CD4+ T cells were negligible at baseline in all 10 individuals with chronic infection. After immunization, HIV-1–specific CD4+ T cells in vaccinees but not in control individuals developed strong proliferative capacities (Fig. 7, A and C), as described previously using a standard tritium-incorporation assay (14). In addition, strong lymphoproliferative activities were also observed in HIV-1–specific CD8+ T cells from vaccine recipients, but virtually no CD8+ T cell–mediated lymphoproliferative activities were observed in control individuals (Fig. 7, B and D). CD4+ and CD8+ T cell–mediated lymphoproliferative responses in vaccinees were confined to those antigens contained in the immunogen, with no induction of responses to envelope proteins that were removed during the preparation of the antigen (26). Thus, these data indicate that the in vivo augmentation of virus-specific CD4+ T cell responses can lead to the reconstitution of HIV-1–specific CD8+ T cell lymphoproliferative immune responses in vivo.
Figure 7.

In vivo reconstitution of HIV-1–specific CD8+ T cell lymphoproliferative activities by vaccine-mediated induction of IL-2–secreting, HIV-1–specific CD4+ T cells. (A and B) Dot plots reflecting the lymphoproliferative activity of HIV-1–specific CD4+ (A) and CD8+ T (B) cells after stimulation with a pool of overlapping Gag peptides before and after five consecutive administrations of an Env-depleted immunogen or placebo. Percentages indicate the proportion of CFSElow CD8+ and CD4+ T cells. (C and D) Proportions of CD4+ (C) and CD8+ (D) T cells proliferating in response to stimulation with HIV-1 peptides spanning the entire HIV-1 proteome in five recipients of placebo and the vaccine. Data from baseline and after five consecutive administrations of the immunogen/placebo are shown.
Discussion
HIV-1–specific CD8+ T cells play a critical role in the initial control of viral replication in acute infection (27, 28). Yet, the functional correlates for CD8+ T cell–mediated HIV-1 immune control are not well understood. Here, we show that HIV-1–specific CD8+ T cells in acute HIV-1 infection exhibit strong ex vivo proliferative capacities, whereas this effector function is rapidly lost in the presence of ongoing viral replication. Moreover, our data demonstrate that lymphoproliferative CD4+ T cell responses enhanced HIV-1–specific CD8+ T cell proliferation in an IL-2–dependent fashion, whereas no HIV-1–specific CD8+ T cell proliferation was observed in individuals with acute infection after in vitro depletion of CD4+ T cells. Finally, the proliferative defect of HIV-1–specific CD8+ T cell responses in chronic infection was partially corrected in vitro by adding autologous IL-2–secreting CD4+ T cells isolated during acute infection and in vivo by the induction of HIV-1–specific CD4+ T cells using an Env-depleted immunogen. Thus, these data demonstrate a progressive loss of HIV-1–specific CD8+ T cell function that is closely linked to the loss of HIV-1–specific, IL-2–secreting CD4+ T cells, but can be rescued in vitro and more importantly in vivo by reconstituting HIV-1–specific CD4+ T cell help.
Recent data have demonstrated that HIV-1–specific CD8+ T cell responses measured by their ability of antigen-specific interferon γ secretion do not differ in individuals with progressive and long-term nonprogressive HIV-1 infection and are not directly associated with the level of viral replication (7, 9). In contrast, HIV-1–specific CD8+ T cells in individuals with long-term nonprogressive infection exhibit strong antigen-dependent ex vivo proliferative capacities, whereas HIV-1–specific CD8+ T cells in subjects with progressive disease courses lose their abilities to proliferate ex vivo in an antigen-specific manner (10). Here, we extend these findings, demonstrating that strong HIV-1–specific CD8+ T cell–mediated lymphoproliferative immune responses are present in acute HIV-1 infection, when high level plasma viremia declines after the first appearance of cellular immune responses against HIV-1. HIV-1–specific CD8+ T cell proliferation was rapidly lost in individuals with ongoing viral replication, despite the persistence of or an increase in the number of interferon γ–secreting, HIV-1–specific CD8+ T cells. In contrast, early suppression of viral replication by antiretroviral therapy preserved these proliferative responses. These data demonstrate that the ability of HIV-1–specific CD8+ T cells to proliferate in response to antigenic stimulation ex vivo can be conserved in individuals with suppressed HIV-1 viremia, but is lost rapidly after acute infection in the presence of ongoing viral replication.
It has recently been shown that the lack of proliferative capacity of HIV-1–specific CD4+ T cells in chronic HIV-1 infection is associated with diminished IL-2 secretion by these cells (13, 21), suggesting a potential relevance of autocrine IL-2 secretion for maintaining HIV-1–specific CD4+ T cell lymphoproliferative responses. In addition, antigen-dependent IL-2 secretion of HIV-1–specific CD4+ T cells is present in acute HIV-1 infection, but sequentially lost during the ensuing disease process, which is closely paralleled by the loss of CD4+ T cell lymphoproliferative immune responses (13, 20). Here, we show that the loss of the proliferative capacity of HIV-1–specific CD4+ T cells is similarly paralleled by a loss of lymphoproliferative HIV-1–specific CD8+ T cell responses, suggesting a mutual functional interaction of these T cell subsets. Moreover, our in vitro data demonstrate that HIV-1–specific CD4+ T cells isolated from acute HIV-1 infection partially restored lymphoproliferative capacities of HIV-1–specific CD8+ T cells in chronic HIV-1 infection by an IL-2–dependent mechanism, whereas no CD8+ T cell proliferation was seen in the presence of CD4+ T cells from chronic HIV-1 infection, which had lost both their antigen-specific lymphoproliferative activity and their ability to secrete IL-2. More importantly, therapeutic immunization aimed at induction of HIV-1–specific CD4+ T cells was able to repair the proliferation deficiency of HIV-1–specific CD8+ cells in vivo in persons with chronic infection. In fact, the administration of a CD4+ T cell–targeted vaccine resulted in HIV-1–specific CD8+ T cell responses with similar lymphoproliferative capacities, as in individuals with acute or long-term nonprogressive HIV-1 infection. Thus, our data suggest that the minimal proliferative capacities of HIV-1–specific CD8+ T cells in chronic HIV-1 infection are not primarily due to a functional defect of these cells, but are rather related to insufficient support by HIV-1–specific CD4+ T helper cells. Nevertheless, even after providing IL-2–secreting CD4+ T helper cells, not all HIV-1–specific CD8+ T cells proliferated, suggesting that within the entire HIV-1–specific CD8+ T cell compartment, populations with different thresholds for antigen-specific proliferation exist.
Our data identify IL-2 secretion as the most prominent mechanism used by CD4+ T helper cells to support HIV-1–specific CD8+ T cell lymohproliferative responses, as IL-2–neutralizing antibodies fully abrogated the proliferative enhancement mediated by CD4+ T cells. This observation is in line with previous data showing that exogenous IL-2 can correct cell cycle perturbations, normalize the overall intracellular protein turnover, and restore the phase-specific pattern of the expression of cell cycle–dependent proteins of lymphocytes in HIV-1–infected individuals (29, 30). However, although IL-2 administration together with highly active antiretroviral therapy has been shown to result in a significant increase in CD4+ T cell counts, it was not associated with enhancement of HIV-1–specific T cell responses or immune-mediated control of HIV-1 infection (31, 32). These data suggest that the HIV-1–specific immune response depends less on systemic levels of IL-2, but rather on the levels of IL-2 provided in the microenvironment of the antigen-specific interaction between antigen-presenting cells, virus-specific CD4+ T cells, and virus-specific CD8+ T cells. This is further supported by our in vivo data demonstrating increased HIV-1–specific CD8+ T cell proliferation after the induction of IL-2–secreting, HIV-1–specific CD4+ T cell responses, strengthening the conclusion that IL-2 secretion in an antigen-specific manner appears to be more relevant for the maintenance of HIV-1–specific proliferative CD8+ T cell responses than systemic levels of IL-2.
Taken together, these data demonstrate a parallel impairment of both HIV-1–specific CD4+ and CD8+ T cell proliferative responses after acute HIV-1 infection in the presence of ongoing viral replication, and suggest a critical role of IL-2–secreting CD4+ T helper cells for sustaining the proliferative capacity of these HIV-1–specific T cell responses both in vitro and in vivo. These results provide evidence for direct functional linkage of HIV-1–specific CD4+ and CD8+ T cell responses and contribute to the understanding of key molecular events contributing to the immunopathogenesis of HIV-1 infection.
Acknowledgments
This study was supported by the National Institutes of Health (to M. Altfeld, E.S. Rosenberg, and B.D. Walker), the Doris Duke Charitable Foundation (to M. Altfeld, E.S. Rosenberg, and B.D. Walker), the Howard Hughes Medical Institute (to B.D. Walker), the Foundation for AIDS and Immunology Research (to X.G. Yu), the Schweizerische Stiftung fuer medizinisch-biologische Stipendien (to D.E. Kaufmann), and the Deutsche Forschungsgemeinschaft (to M. Lichterfeld).
The authors have no conflicting financial interests.
Abbreviations used in this paper: CFSE, carboxyfluorescein succinimidyl ester; FSC, forward scatter; SFC, spot-forming cell; SSC, side scatter.
References
- 1.Kahn, J.O., and B.D. Walker. 1998. Acute human immunodeficiency virus type 1 infection. N. Engl. J. Med. 339:33–39. [DOI] [PubMed] [Google Scholar]
- 2.Borrow, P., H. Lewicki, B.H. Hahn, G.M. Shaw, and M.B. Oldstone. 1994. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68:6103–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koup, R.A., J.T. Safrit, Y. Cao, C.A. Andrews, G. McLeod, W. Borkowsky, C. Farthing, and D.D. Ho. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altfeld, M., E.S. Rosenberg, R. Shankarappa, J.S. Mukherjee, F.M. Hecht, R.L. Eldridge, M.M. Addo, S.H. Poon, M.N. Phillips, G.K. Robbins, et al. 2001. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J. Exp. Med. 193:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalod, M., M. Dupuis, J.C. Deschemin, C. Goujard, C. Deveau, L. Meyer, N. Ngo, C. Rouzioux, J.G. Guillet, J.F. Delfraissy, et al. 1999. Weak anti-HIV CD8(+) T-cell effector activity in HIV primary infection. J. Clin. Invest. 104:1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao, J., J. McNevin, S. Holte, L. Fink, L. Corey, and M.J. McElrath. 2003. Comprehensive analysis of human immunodeficiency virus type 1 (HIV-1)-specific gamma interferon-secreting CD8+ T cells in primary HIV-1 infection. J. Virol. 77:6867–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addo, M.M., X.G. Yu, A. Rathod, D. Cohen, R.L. Eldridge, D. Strick, M.N. Johnston, C. Corcoran, A.G. Wurcel, C.A. Fitzpatrick, et al. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 77:2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Draenert, R., C.L. Verrill, Y. Tang, T.M. Allen, A.G. Wurcel, M. Boczanowski, A. Lechner, A.Y. Kim, T. Suscovich, N.V. Brown, et al. 2004. Persistent recognition of autologous virus by high-avidity CD8 T cells in chronic, progressive human immunodeficiency virus type 1 infection. J. Virol. 78:630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Betts, M.R., D.R. Ambrozak, D.C. Douek, S. Bonhoeffer, J.M. Brenchley, J.P. Casazza, R.A. Koup, and L.J. Picker. 2001. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection. J. Virol. 75:11983–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Migueles, S.A., A.C. Laborico, W.L. Shupert, M.S. Sabbaghian, R. Rabin, C.W. Hallahan, D. Van Baarle, S. Kostense, F. Miedema, M. McLaughlin, et al. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 3:1061–1068. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg, E.S., M. Altfeld, S.H. Poon, M.N. Phillips, B.M. Wilkes, R.L. Eldridge, G.K. Robbins, R.T. D'Aquila, P.J. Goulder, and B.D. Walker. 2000. Immune control of HIV-1 after early treatment of acute infection. Nature. 407:523–526. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg, E.S., J.M. Billingsley, A.M. Caliendo, S.L. Boswell, P.E. Sax, S.A. Kalams, and B.D. Walker. 1997. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 278:1447–1450. [DOI] [PubMed] [Google Scholar]
- 13.Younes, S.A., B. Yassine-Diab, A.R. Dumont, M.R. Boulassel, Z. Grossman, J.P. Routy, and R.P. Sekaly. 2003. HIV-1 viremia prevents the establishment of interleukin 2–producing HIV-specific memory CD4+ T cells endowed with proliferative capacity. J. Exp. Med. 198:1909–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbins, G.K., M.M. Addo, H. Troung, A. Rathod, K. Habeeb, B. Davis, H. Heller, N. Basgoz, B.D. Walker, and E.S. Rosenberg. 2003. Augmentation of HIV-1-specific T helper cell responses in chronic HIV-1 infection by therapeutic immunization. AIDS. 17:1121–1126. [DOI] [PubMed] [Google Scholar]
- 15.Brander, C., and P. Goulder. 2000. The evolving field of HIV CRL epitope mapping: new approaches for the identification of novel epitopes. HIV Molecular Database. B.T.M. Korber, C. Brander, B.D. Walker, R.A. Koup, J. Moore, B. Haynes, and G. Meyer, editors. Los Alamos National Laboratory. Los Alamos, New Mexico. 11–120.
- 16.Sidney, J., S. Southwood, C. Oseroff, M.-F. Del Guercio, and A. Sette. 1998. Measurement of MHC/peptide interactions by gel filtration. Curr. Prot. Immunol. 18:18.3.2–18.3.19. [DOI] [PubMed] [Google Scholar]
- 17.Altfeld, M., M.M. Addo, R.L. Eldridge, X.G. Yu, S. Thomas, A. Khatri, D. Strick, M.N. Phillips, G.B. Cohen, S.A. Islam, et al. 2001. Vpr is preferentially targeted by CTL during HIV-1 infection. J. Immunol. 167:2743–2752. [DOI] [PubMed] [Google Scholar]
- 18.Alter, G., G. Hatzakis, C.M. Tsoukas, K. Pelley, D. Rouleau, R. LeBlanc, J.G. Baril, H. Dion, E. Lefebvre, R. Thomas, et al. 2003. Longitudinal assessment of changes in HIV-specific effector activity in HIV-infected patients starting highly active antiretroviral therapy in primary infection. J. Immunol. 171:477–488. [DOI] [PubMed] [Google Scholar]
- 19.Lacabaratz-Porret, C., A. Urrutia, J.M. Doisne, C. Goujard, C. Deveau, M. Dalod, L. Meyer, C. Rouzioux, J.F. Delfraissy, A. Venet, and M. Sinet. 2003. Impact of antiretroviral therapy and changes in virus load on human immunodeficiency virus (HIV)-specific T cell responses in primary HIV infection. J. Infect. Dis. 187:748–757. [DOI] [PubMed] [Google Scholar]
- 20.Harari, A., S. Petitpierre, F. Vallelian, and G. Pantaleo. 2004. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood. 103:966–972. [DOI] [PubMed] [Google Scholar]
- 21.Iyasere, C., J.C. Tilton, A.J. Johnson, S. Younes, B. Yassine-Diab, R.P. Sekaly, W.W. Kwok, S.A. Migueles, A.C. Laborico, W.L. Shupert, et al. 2003. Diminished proliferation of human immunodeficiency virus-specific CD4+ T cells is associated with diminished interleukin-2 (IL-2) production and is recovered by exogenous IL-2. J. Virol. 77:10900–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moss, R.B., C. Brandt, W.K. Giermakowska, J.R. Savary, G. Theofan, M. Zanetti, D.J. Carlo, and M.R. Wallace. 2003. HIV-specific immunity during structured antiviral drug treatment interruption. Vaccine. 21:1066–1071. [DOI] [PubMed] [Google Scholar]
- 23.Moss, R.B., E. Webb, W.K. Giermakowska, F.C. Jensen, J.R. Savary, M.R. Wallace, and D.J. Carlo. 2000. HIV-1-specific CD4 helper function in persons with chronic HIV-1 infection on antiviral drug therapy as measured by ELISPOT after treatment with an inactivated, gp120-depleted HIV-1 in incomplete Freund's adjuvant. J. Acquir. Immune Defic. Syndr. 24:264–269. [DOI] [PubMed] [Google Scholar]
- 24.Moss, R.B., M.R. Wallace, P. Lanza, W. Giermakowska, F.C. Jensen, G. Theofan, C. Chamberlin, S.P. Richieri, and D.J. Carlo. 1998. In vitro p24 antigen-stimulated lymphocyte proliferation and beta-chemokine production in human immunodeficiency virus type 1 (HIV-1)-seropositive subjects after immunization with an inactivated gp120-depleted HIV-1 immunogen (Remune). Clin. Diagn. Lab. Immunol. 5:308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moss, R.B., W.K. Giermakowska, M.R. Wallace, J.R. Savary, F.C. Jensen, and D.J. Carlo. 2000. Cell-mediated immune responses to autologous virus in HIV-1-seropositive individuals after treatment with an HIV-1 immunogen. AIDS. 14:2475–2478. [DOI] [PubMed] [Google Scholar]
- 26.Turner, J.L., R.J. Trauger, A.E. Daigle, and D.J. Carlo. 1994. HIV-1 immunogen induction of HIV-1-specific delayed-type hypersensitivity: results of a double-blind, adjuvant-controlled, dose-ranging trial. AIDS. 8:1429–1435. [PubMed] [Google Scholar]
- 27.Schmitz, J.E., M.J. Kuroda, S. Santra, V.G. Sasseville, M.A. Simon, M.A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B.J. Scallon, et al. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 283:857–860. [DOI] [PubMed] [Google Scholar]
- 28.Jin, X., D.E. Bauer, S.E. Tuttleton, S. Lewin, A. Gettie, J. Blanchard, C.E. Irwin, J.T. Safrit, J. Mittler, L. Weinberger, et al. 1999. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus–infected macaques. J. Exp. Med. 189:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paiardini, M., D. Galati, B. Cervasi, G. Cannavo, L. Galluzzi, M. Montroni, D. Guetard, M. Magnani, G. Piedimonte, and G. Silvestri. 2001. Exogenous interleukin-2 administration corrects the cell cycle perturbation of lymphocytes from human immunodeficiency virus-infected individuals. J. Virol. 75:10843–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankar, P., M. Russo, B. Harnisch, M. Patterson, P. Skolnik, and J. Lieberman. 2000. Impaired function of circulating HIV-specific CD8(+) T cells in chronic human immunodeficiency virus infection. Blood. 96:3094–3101. [PubMed] [Google Scholar]
- 31.The AIDS Clinical Trials Group 248 Study Team. 2004. Daily low-dose subcutaneous interleukin-2 added to single- or dual-nucleoside therapy in HIV infection does not protect against CD4+ T-cell decline or improve other indices of immune function: results of a randomized controlled clinical trial (ACTG 248). J. Acquir. Immune Defic. Syndr. 36:576–587. [DOI] [PubMed]
- 32.Dybul, M., B. Hidalgo, T.W. Chun, M. Belson, S.A. Migueles, J.S. Justement, B. Herpin, C. Perry, C.W. Hallahan, R.T. Davey, et al. 2002. Pilot study of the effects of intermittent interleukin-2 on human immunodeficiency virus (HIV)-specific immune responses in patients treated during recently acquired HIV infection. J. Infect. Dis. 185:61–68. [DOI] [PubMed] [Google Scholar]