

Abstract
We previously reported that central–memory T cells (TCM cells), which express lymph node homing receptors CCR7 and CD62L, are largely devoid of effector functions but acquire characteristics of effector–memory T cells (TEM cells) (i.e., CCR7− T helper [Th]1 or Th2 cells) after stimulation with T cell receptor agonists or homeostatic cytokines. Here we show that three chemokine receptors identify functional subsets within the human CD4+ TCM cell pool. TCM cells expressing CXCR3 secreted low amounts of interferon γ, whereas CCR4+ TCM cells produced some interleukin (IL)-4, but not IL-5. In response to IL-7 and IL-15, CXCR3+ TCM and CCR4+ TCM cells invariably generated fully differentiated CCR7− Th1 and Th2 cells, respectively, suggesting that they represent pre-Th1 and pre-Th2 cells. Conversely, CXCR5+ TCM cells lacking CXCR3 and CCR4 remained nonpolarized and retained CCR7 and CD62L expression upon cytokine-driven expansion. Unlike naive cells, all memory subsets had a low T cell receptor rearrangement excision circle content, spontaneously incorporated bromodeoxyuridine ex vivo, and contained cells specific for tetanus toxoid. Conversely, recall responses to cytomegalovirus and vaccinia virus were largely restricted to CXCR3+ TCM and TEM cells. We conclude that antigen-specific memory T cells are distributed between TEM cells and different subsets of TCM cells. Our results also explain how the quality of primary T cell responses could be maintained by TCM cells in the absence of antigen.
Keywords: T cell subsets, memory maintenance, cytokines, differentiation, chemokine receptors
Introduction
Upon recognition of antigenic peptides on DCs, naive T lymphocytes proliferate and differentiate into a variety of effector cells depending on the stimulatory conditions and cytokine milieu (1, 2). Accumulating evidence indicates that during the T cell differentiation process, effector functions and homing potentials are coordinately regulated (3). For instance, developing Th1 cells acquire the capacity to produce IFN-γ and expression of chemokine receptors such as CCR5, CXCR3, and CXCR6 that drive them to sites of delayed-type hypersensitivity reactions. Conversely, developing Th2 cells acquire the capacity to produce IL-4 and express CCR3, CCR4, CCR8, and the prostaglandin D2 chemoattractant receptor CRTh2 (4–7), which are required to migrate at sites of allergic reactions (6, 8, 9).
Expression of the lymph node homing receptors CCR7 and CD62L (10, 11) has been used to define subsets of human memory T cells with distinct functional properties. T cells within the CCR7+ “central–memory” T cell (TCM cell) cell subset show hypo-acetylated cytokine genes and have no or low effector functions, but efficiently differentiate to Th1 or Th2 effector cells after TCR stimulation in the presence of IL-12 or IL-4, respectively (12–14). In contrast, T cells of the CCR7− “effector–memory” T cell (TEM cell) subset show polarized cytokine gene acetylation patterns in vivo and rapidly produce high amounts of IFN-γ and IL-4 upon antigenic stimulation (12–14). It has been proposed that in secondary immune responses, TCM cells generate new waves of effector cells in antigen-draining lymph nodes, whereas TEM cells provide immediate protection against invading pathogens in peripheral tissues (3, 14).
The maintenance of T cell memory is controlled by cytokines that promote cell survival and slow homeostatic proliferation (15). In particular, IL-7 and IL-15 have been shown to regulate mouse CD8+ memory T cell survival and self-renewal in the absence of antigen (16, 17), whereas naive and CD4+ memory cells require IL-7 and TCR ligands (16, 18, 19), but do not respond to IL-15 (17). Conversely, human CD4+ memory T cells proliferate in response to IL-15 in a TCR-independent fashion and with slow kinetics (20, 21), suggesting different roles for IL-15 in mouse and human CD4+ memory T cell homeostasis. Notably, TCM cells proliferating in response to IL-7 and IL-15 differentiate and generate Th1 and Th2 effector cells (21), but how uncommitted TCM cells proliferating in the absence of antigen could maintain the quality of the primary response remained unclear.
The existence of TCM and TEM cell subsets has also been documented in mice (22, 23). In this experimental system, it has been possible to directly examine the kinetics of memory cell generation and the capacity of effector and memory subsets to reconstitute long-term memory (24), and there is growing evidence that TCM cells have higher reconstitution potential (24, 25). In particular, effector Th1 cells, defined by their secretion of IFN-γ, were found to be short-lived and unable to reconstitute T cell memory. In contrast, a population of activated Th1 lineage cells, which did not secrete IFN-γ after primary antigenic stimulation, persisted for several months in vivo and developed the capacity to secrete IFN-γ upon subsequent stimulation (26).
Since the first description of TCM and TEM cells, it was evident that other chemokine receptors, as well as adhesion and costimulatory molecules, are expressed on different fractions of TCM cells (12, 14). Heterogeneity of human CD4+ TCM cells has further been documented using CXCR5, the receptor for CXCL13, a chemokine expressed in B cell follicles (27, 28). CXCR5+ TCM cells lacked effector functions and cells specific for tetanus toxoid (TT), but contained residual T cell receptor rearrangement excision circles (TRECs), suggesting that they represent recently activated cells (29–31). Conversely, other recent studies claimed that both TCM and TEM cells possess high levels of effector functions, and that consequently neither CCR7 nor CXCR5 expression identify nonpolarized CD4+ memory T cells (9, 32–35). Understanding TCM cell differentiation stage and potential is of importance for the homeostatic maintenance of memory T cells and for the conservation of T cell polarization in secondary responses.
Here we report that CXCR3 and CCR4 identify two novel subsets of pre-Th1 and pre-Th2 cells within TCM cells. These cells possessed low IFN-γ– or IL-4–producing capacities when compared with CXCR3+ and CCR4+ TEM cells and spontaneously differentiated to Th1 and Th2 effector cells in response to homeostatic cytokines IL-7 and IL-15 independently of conventional Th1 or Th2 cell–inducing stimuli. In contrast, TCM cells lacking CXCR3 or CCR4 and expressing CXCR5 were nonpolarized cells whose differentiation to Th1 or Th2 cells is dependent on TCR triggering and signaling by polarizing cytokines.
Materials and Methods
Cell Culture.
PBMCs were isolated from buffy-coated blood from healthy donors. Monocytes were depleted by adhesion for 30 min and CD4+ T cells were isolated by negative selection with magnetic beads using Automacs (Miltenyi Biotec). Memory T cells were isolated by further depletion of naive T cells with anti-CD45RA beads (Miltenyi Biotec). Memory T cell subpopulations were purified to >95% by cell sorting after five-color staining as follows: anti-CXCR5 (R&D Systems) followed by anti-IgG2b PE (Biosystems), anti-CCR7 (R&D Systems) followed by anti-IgG2a FITC (Biosystems), and anti-CXCR3 CyChrome, anti-CD45RA APC, and anti-CCR4 biotin followed by streptavidin-APC-Cy7 (BD Biosciences). Labeling of T cells with carboxyfluorescein succinimidyl ester (CFSE) was performed as described previously (21). Monocytes were purified by positive selection with anti-CD14 beads (Miltenyi Biotec). For DC differentiation, CD14+ cells were cultured for 4 d in complete medium (RPMI 1640 supplemented with 2 mM glutamine, 1% nonessential amino acids, 1% sodium pyruvate, 50 μg/ml kanamycin, 50 U/ml penicillin, and 50 μg/ml streptomycin; GIBCO BRL) containing 10% FCS (Hyclone), 50 ng/ml granulocyte/macrophage colony-stimulating factor (Novartis), and 1,000 U/ml IL-4. The DCs obtained were stimulated for 24 h with 100 ng/ml lipopolysaccharide (Salmonella abortus equi; Sigma-Aldrich) and pulsed for 30 min with 100 ng/ml toxic shock syndrome toxin (TSST). CFSE-labeled 5 × 104 T cells were cultured with TSST-pulsed DCs in flat-bottom wells at a 5:1 ratio, and recombinant cytokines were used at either 25 ng/ml (IL-7 and IL-15; R&D Systems), 10 ng/ml (TNF, IL-6, IL-10, IL-4, and IL-12; BD Biosciences), or 1,000 U/ml (IL-2; Roche), whereas neutralizing antibodies to IL-4 and IL-12 (BD Biosciences) were used at 2 μg/ml.
ELISA, Intracellular Cytokine Staining, and IFN-γ Secretion Assay.
Cytokine-producing capacity of FACS-purified subsets was assessed after stimulation of purified cell populations at 5 × 104/100 μl for 24 h with 50 nM phorbol-12-13-dibutyrate (PdBu) and 0.5 μg/ml ionomycin, or in wells coated with 2 μg/ml each of anti-CD3 (clone TR66) and anti-CD28 antibodies (BD Biosciences). Cytokine concentrations of supernatants were then assessed by ELISA according to a standard protocol and analyzed with the Softmax program. Intracellular IFN-γ was detected after stimulating cells in the presence of 10 μg/ml brefeldin A (Sigma-Aldrich) for the last 2 h and after fixation with paraformaldehyde and permeabilization with saponin. After saturation of nonspecific binding sites with 10% FCS, cells were incubated with APC-labeled antibody to IFN-γ and PE-labeled antibody to IL-2 or IL-4 (BD Biosciences), washed, and analyzed by flow cytometry on a FACSCalibur with CELLQuest software (Becton Dickinson). To sort live IFN-γ–producing cells, we stimulated cells for 60 h with 25 ng/ml cytokines (IL-7, IL-15, IL-12, TNF-α, and IL-18), and IFN-γ–producing cells were identified with an IFN-γ secretion assay kit (Miltenyi Biotec) and purified by cell sorting.
Recall Responses.
PBMCs from 50 ml of fresh blood from healthy volunteers were prepared, monocytes were isolated and either incubated for 16 h with a replication-deficient vaccinia virus (VV; provided by G. Sutter, Institute for Virology, Munich, Germany) or left untreated. 5 × 104 monocytes were then irradiated and incubated in the absence or presence of 1 μg/ml TT or 2.5 μg of an extract of CMV-derived proteins (provided by R. Campanelli, University of Pavia, Pavia, Italy) with purified T cell subsets at a 1:1 ratio in U-bottom wells in complete medium containing 5% human serum. On days 5 and 7, cells were stained for CD14 and CD4, and CFSE dilution of CD4+ CD14− viable cells was assessed by flow cytometry. On day 5, cells had not yet diluted CFSE completely, and the precursor frequency of antigen-specific cells could therefore be calculated as described previously (36). In some experiments, the presence of pathogen-specific cells was confirmed by restimulating cells with autologous monocytes treated as described above followed by assessment of cytokine production of proliferating T cells by intracellular staining.
Ex Vivo Bromodeoxyuridine (BrdU) Labeling.
The assay was performed as described previously (25). In brief, fresh PBMCs were cultured with 10 μg/ml BrdU (Sigma-Aldrich) for 16 h. CD4+ cells were then positively selected with anti-CD4 magnetic beads and stained for CD45RA and chemokine receptor expression. Cells were then fixed, permeabilized, treated with DNase (Boehringer), stained with FITC-labeled anti-BrdU antibody (Becton Dickinson), and analyzed by flow cytometry.
Quantitative PCR of TRECs.
Memory CD4+ T cells were isolated by MACS as described above, stained for CD4, CD45RO, CCR7, and CXCR5, and sorted to >99.9% purity. Quantification of signal joint TRECs in sorted CD4+ T cell subsets was performed by real-time quantitative PCR with the 5′ nuclease (TaqMan) assay using an ABI 7700 sequence detector (Applied Biosystems). As described previously (37), 1–2 × 105 cells were lysed in 10 mM Tris, pH8, containing 100 μg/ml of proteinase K (GIBCO BRL) for 2 h at 56°C, and then for 15 min at 95°C. PCR reaction of lysates was performed with 500 nM of primers (CACATCCCTTTCAACCATGCT and GCCAGCTGCAGGGTTTAGG) and 125 nM of probe FAM-ACACCTCTGGTTTTTGTAAAGGTGCCCACT-TAMRA. PCR conditions were as follows: 1 cycle of 2 min at 50°C, 1 cycle of 10 min at 95°C, followed by 40 cycles of 30 s at 95°C, and 1 min at 65°C. Levels of DNA were standardized by normalizing with 18S rRNA sequences.
Results
Subsets of Human CD4+ TCM Cells Identified by Expression of CXCR5, CXCR3, and CCR4.
Purified human CD4+ T cells were analyzed for chemokine receptor expression by five-color staining. CD45RA+ cells expressed CCR7, but were largely negative for the other chemokine receptors, consistent with the view that they are predominantly antigen-inexperienced “naive” T cells (not depicted). Conversely, the following three main subsets could be identified in CD45RA− cells according to CCR7 and CXCR5 expression: CXCR5+ CCR7+ cells (CXCR5+ TCM), CXCR5− CCR7+ cells (CXCR5− TCM), and CXCR5− CCR7− cells (TEM; Fig. 1 A). Within these main subsets, staining with antibodies to CXCR3 and CCR4 revealed further heterogeneity (Fig. 1 B). CXCR3 and CCR4 were expressed on different populations of TEM cells, which contain Th1 and Th2 effector cells (4, 9, 38). However, CXCR3 and CCR4 were also expressed on some TCM cells, especially within the CXCR5− subset (Fig. 1 B). Thus, the following four major subsets of TCM cells were identified: (a) CXCR3− CCR4− CXCR5+ TCM cells (“CXCR5+ TCM”), (b) CXCR3− CCR4− CXCR5− TCM cells (“−/− TCM”), (c) CXCR5− CXCR3+ CCR4− TCM cells (“CXCR3+ TCM”), and (d) CXCR5− CCR4+ CXCR3− TCM cells (“CCR4+ TCM”). Mean values ± standard deviations of the four subsets in four healthy donors were 11 ± 5%, 18 ± 10%, 17 ± 8%, and 17 ± 12%, respectively.
Figure 1.

Expression of chemokine receptors defines CD4+ TCM cell subsets with distinct replicative potentials. Purified CD4+ T cells were stained with antibodies specific for CXCR5, CCR7, CXCR3, CCR4, and CD45RA, and analyzed by five-color flow cytometry. (A) CCR7 and CXCR5 expression of CD45RA− cells of one representative donor out of four. (B) CXCR5+ TCM, CXCR5− TCM, and TEM cells were analyzed for CXCR3 and CCR4 expression. Percentages indicate the mean frequency of subsets in the memory pool of five healthy donors. (C) Purified, CFSE-labeled CD4+ T cell subsets were stimulated with IL-7 and IL-15, or TSST-loaded DCs for 7 d. CFSE profiles of viable (propidium iodide−) cells were analyzed by flow cytometry. Numbers in the top row indicate the percentage of dividing cells and numbers in parenthesis indicate the mean division number. Numbers in the bottom row indicate the fraction of TSST-responsive TCR Vβ2+ cells. One representative donor out of four is shown.
Because CXCR3 and CCR4 have been associated with differentiated Th1 and Th2 cells (7, 38), we analyzed expression of other surface markers that are acquired or lost with T cell differentiation (12, 17, 39; Table I). As expected, naive T cells expressed uniformly CD27 and CD62L, but not the IL-2/15Rβ chain (CD122), whereas most TEM cells had lost CD27 and CD62L expression, but were CD122+. CXCR5+ TCM cells had a phenotype similar to naive cells, whereas CXCR5− TCM cells expressed intermediate levels of CD27, CD62L, and CD122. In particular, CXCR3+ TCM cells were CD122+ and had partially lost CD62L expression, consistent with a more differentiated phenotype. The differences in CD122 expression were functionally relevant because they closely correlated with proliferation in response to IL-7 and IL-15 (Fig. 1 C, top), being low in naive cells and CXCR5+ TCM cells, intermediate in CCR4+ TCM cells and −/−TCM cells, and high in CXCR3+ TCM and TEM cells.
Table I.
Expression of CD27, CD62L, and IL-2/IL-15Rβ Chain (CD122) on Naive T Cells and Memory T Cell Subsets a
| TCM
|
||||||
|---|---|---|---|---|---|---|
| CXCR5+ | CXCR5− | |||||
| TN | −/− | CXCR3 | −/− | CCR4+ | TEM | |
| CD27+ (%)b | 99 ± 1 | 98 ± 2 | 76 ± 9 | 83 ± 9 | 72 ± 17 | 40 ± 8 |
| CD62Lhi (%)b | 99 ± 1 | 89 ± 5 | 55 ± 15 | 84 ± 8 | 79 ± 9 | 23 ± 12 |
| CD122 (MFI)c | 1 ± 1 | 3 ± 2 | 8 ± 3 | 6 ± 2 | 5 ± 2 | 10 ± 3 |
| PI+ (%)d | 3 ± 2 | 7 ± 4 | 17 ± 9 | 9 ± 5 | 15 ± 8 | 25 ± 10 |
Mean ± standard deviation of four healthy donors.
Percent of positive cells.
Mean fluorescence intensity.
Propidium iodide+ cells after stimulation for 3 d with DC+ TSST.
We then compared the expansion potential of purified CFSE-labeled CD4+ naive and memory T cell subsets after TCR stimulation with TSST-loaded DCs because replicative capacity diminishes with T cell differentiation (25, 40). Proliferation and accumulation was high in naive cells, CXCR5+ TCM cells, and −/−TCM cells, intermediate in CCR4+ and CXCR3+ TCM cells, and low in TEM cells (Fig. 1 C, bottom). As reported for the CD8 compartment (25), the reduced accumulation of TEM cells was associated with a high rate of apoptosis (Table I). Similar results were obtained upon stimulation with anti-CD3 and anti-CD28 antibodies (not depicted).
Together, these results show that subsets of CD4+ TCM cells identified by CXCR5, CXCR3, and CCR4 expression differ in their proliferative response to cytokines and TCR ligands, and suggest that CXCR5+ TCM cells and −/−TCM cells are at an early stage of memory cell differentiation, whereas CCR4+ and CXCR3+ TCM cells have characteristics of more mature cell types.
CXCR3 and CCR4 Identify TCM Cells with Low IFN-γ– and IL-4–producing Capacities.
Next, we analyzed effector cytokine–producing capacities of TCM and TEM cell subsets. Total TCM and TEM cell populations were sorted for CXCR3 and CCR4 expression, stimulated with PdBu and ionomycin, and secreted cytokines were quantified by ELISA (Fig. 2 A). TCM cells lacking CXCR3 and CCR4 failed to produce IFN-γ, IL-4, and IL-5, whereas double negative cells in the TEM subset produced all three cytokines. Consistent with the role of CXCR3 and CCR4 as Th1 and Th2 cell markers, CXCR3-expressing cells produced predominantly IFN-γ, whereas CCR4+ cells produced mainly type 2 cytokines. However, CXCR3+ TCM and CCR4+ TCM cells produced less effector cytokines than the corresponding TEM cell subset, and IL-5 production was entirely restricted to TEM cells. Similar results were obtained when CXCR5+ TCM and CXCR5− TCM cell subsets were analyzed separately (not depicted). Because CXCR3+ TCM cells contained a considerable fraction of CD62L− cells (Table I), we further analyzed IFN-γ–producing capacity of CXCR3+ TCM and TEM cells according to CD62L expression (Fig. 2 B). CXCR3+ TCM cells expressing CD62L produced only low amounts of IFN-γ, whereas CXCR3+ TCM cells lacking CD62L produced high levels of IFN-γ comparable to TEM cells. Thus, IFN-γ production among CCR7+ cells is largely restricted to a TEM cell–like subset of CD62L− CXCR3+ cells. Collectively, these results show that CXCR3 and CCR4 identify cells in the TCM cell pool that are nonpolarized or produce low levels of IFN-γ or IL-4.
Figure 2.

Ex vivo cytokine-producing capacities of CD4+ memory T cell subsets. (A) Purified CD4+ TCM and TEM cell subsets were stimulated with PdBu and ionomycin for 24 h and supernatants were analyzed for IFN-γ (diluted 1:4, white bars), IL-4 (black bars), and IL-5 (gray bars) by ELISA. Stimulation with anti-CD3 and anti-CD28 antibodies gave similar results (not depicted). Shown is the mean of four experiments with cells from different donors. (B) CXCR3+ CD4+ T cells were sorted for CCR7 and CD62L expression as indicated and IFN-γ production was assessed as described above. The mean of three independent experiments with three different donors is shown.
Cytokine-stimulated CXCR3+ and CCR4+ TCM Cells Differentiate to Th1 and Th2 Cells.
We previously showed that some cytokine-stimulated TCM cells spontaneously differentiate to Th1 or Th2 cells, whereas naive cells require TCR ligands or inflammatory cytokines for differentiation (21, 41). To understand whether the subsets defined by CXCR5, CXCR3, and CCR4 could discriminate cells with predetermined fates, we induced proliferation of purified CD4+ T cell subsets by either TSST-loaded DCs or IL-7 and IL-15 (Fig. 3 A). Because memory subsets showed different proliferative responses to IL-7 and IL-15 (Fig. 1 C), and acquisition of effector cytokine–producing capacities progressively increases with division number (42, 43), analysis was performed by gating on cells that had performed the same number of divisions (cytokines: 4; TSST: >7). Under both conditions of stimulation, CXCR5+ TCM cells that lacked CXCR3 and CCR4 expression remained nonpolarized, whereas −/−TCM cells generated some Th1 and Th2 cells and acquired CXCR3 and CCR4 on a fraction of cells at the same time (Fig. 3 C). Cytokine-stimulated CXCR5+ TCM cells progressively lost CXCR5 expression, but homogeneously maintained high levels of CCR7 and CD62L, whereas a fraction of CXCR5− TCM cells progressively lost CCR7 and CD62L expression, thus acquiring the phenotype of TEM cells (Fig. 3 B). Notably, TEM cells remained CCR7− and maintained high effector functions under these conditions.
Figure 3.
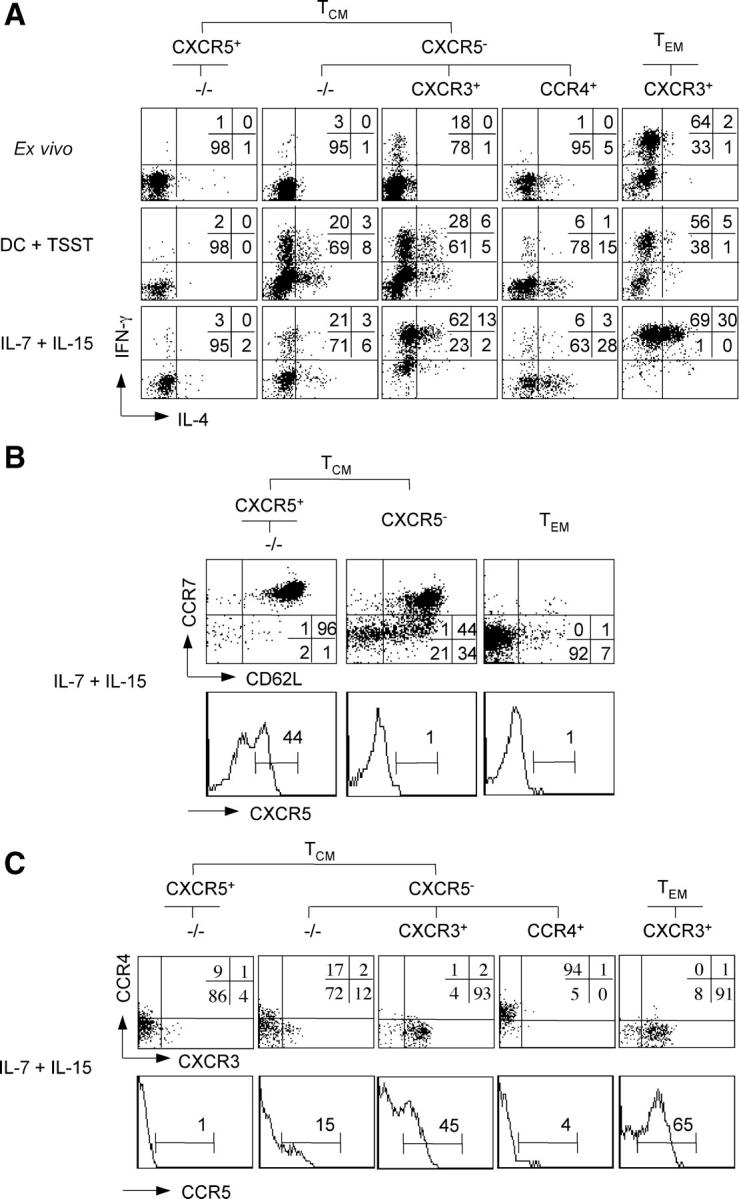
(A) Differentiation of TCM cell subsets in response to TCR or cytokine stimulation. Purified CD4+ T cell subsets were CFSE labeled and stimulated with TSST-loaded DCs or with IL-7 and IL-15 in the presence of neutralizing anti–IL-4 and anti–IL-12 antibodies. After 7 d, cells were stimulated with PdBu and ionomycin, stained with APC-labeled anti–IFN-γ and PE-labeled anti–IL-4 antibodies, and cells of the same division number were analyzed by flow cytometry. Unstimulated T cell subsets were also analyzed ex vivo as control. Numbers indicate the percentage of cells producing IFN-γ or IL-4. One respresentative experiment out of five is shown. (B and C) Modulation of homing receptor expression by cytokine-stimulated CD4+ memory T cell subsets. Purified CFSE-labeled CD4+ T cell subsets were stimulated with IL-7 and IL-15. TCM and TEM cells in B were sorted for CXCR5, CCR7, and CD62L expression, whereas in C they were sorted for CXCR5, CCR7, CXCR3, and CCR4 expression as indicated. After 7 d, cells in division four were analyzed for the expression of CXCR5, CCR7, and CD62L (B), or CXCR3, CCR4, and CCR5 (C). One representative donor out of four is shown.
When CXCR3+ TCM and CCR4+ TCM cells were expanded with homeostatic cytokines, they maintained CXCR3 and CCR4 expression (Fig. 3 C) and spontaneously differentiated into Th1 and Th2 cells, respectively (Fig. 3 A). Thus, CCR4+ TCM cells produced high levels of IL-4 and also secreted IL-5 (not depicted), a type 2 cytokine produced exclusively by TEM cells (Fig. 2 A). Moreover, the amount of IFN-γ produced by CXCR3+ TCM cells stimulated with IL-7 and IL-15 was comparable to that produced by CXCR3+ TEM cells ex vivo, and a fraction of CXCR3+ TCM cells acquired expression of CCR5 (Fig. 3 C), a receptor for inflammatory chemokines expressed on Th1 effector cells (5). Together, these results demonstrate that TCM cells that lack CXCR3 and CCR4 expression are nonpolarized precursors, whereas CXCR3+ and CCR4+ TCM cells represent pre-Th1 and pre-Th2 cells that become fully differentiated Th1 and Th2 effector cells in response to homeostatic cytokines.
To exclude a selective outgrowth of preexisting IFN-γ–producing cells from CXCR3+ TCM cells, we wished to deplete cells with IFN-γ–producing capacity from cytokine-stimulated cultures. To this aim it was necessary to induce IFN-γ production of Th1 cell–polarized memory cells without activating TCR-dependent signaling. It is well established that TCR-independent IFN-γ production of activated Th1 cells can be induced by inflammatory cytokines IL-12 and IL-18 (44, 45). We found that production of IFN-γ by resting CD4+ memory cells in response to IL-12 and IL-18 required activation by IL-7 and IL-15, was boosted by TNF-α, and occurred with delayed kinetics (Fig. 4 A). In contrast, IL-2 production was restricted to TCR-stimulated cells (not depicted), confirming that IFN-γ production by cytokine-stimulated cells is TCR independent. Notably, TCR and cytokine stimulation induced a similar fraction of cells to secrete IFN-γ before cell division (Fig. 4 A, note empty/filled symbols), and IFN-γ production was in both cases restricted to CXCR3+ TCM and TEM cells (Fig. 2 and not depicted). Thus, mature Th1 cell–polarized memory cells can be identified by IFN-γ secretion after either TCR or optimal cytokine activation.
Figure 4.

(A) Kinetics and requirements of TCR- and cytokine-induced IFN-γ production. CFSE-labeled CD4+ memory T cells were stimulated for the indicated times with either anti-CD3 and anti-CD28 antibodies (squares), or with TNF-α, IL-12, and IL-18 in the absence (circles) or presence (triangles) of IL-7 and IL-15. IFN-γ production was analyzed by intracellular staining. Empty symbols indicate conditions with undivided cells, whereas filled symbols indicate conditions with dividing cells. The mean percentage of IFN-γ+ cells of three independent experiments is plotted. (B) Cytokine-stimulated CXCR3+ TCM cells lacking IFN-γ–producing capacity become Th1 cell effector cells. Purified CFSE-labeled CXCR3+ TCM and CCR4+ TCM cells were stimulated with IL-7, IL-15, TNF-α, IL-12, and IL-18 for 60 h, and IFN-γ–secreting cells were purified by cell sorting. IFN-γ+ and IFN-γ− cells were then expanded for an additional 5 d with IL-7 and IL-15, briefly stimulated with PdBu and ionomycin, and analyzed for IL-4 and IFN-γ production by intracellular staining. One representative donor out of three is shown.
Next, we induced IFN-γ production by purified CFSE-labeled CXCR3+ TCM cells with cytokines, sorted undivided IFN-γ cells after 60 h, expanded them with IL-7 and IL-15, and analyzed effector cytokine–producing capacities of proliferating cells (Fig. 4 B). A large fraction of IFN-γ− CXCR3+ TCM cells differentiated under these conditions and acquired the capacity to produce high levels of IFN-γ. Sorting IFN-γ cells after 72 h gave similar results (not depicted). In contrast, CCR4+ TCM cells secreted IL-4 under the same conditions, whereas IFN-γ+ CXCR3+ TCM cells maintained high IFN-γ production. We conclude that CXCR3+ TCM cells lacking IFN-γ–producing capacity become Th1 effector cells after TCR-independent proliferation induced by cytokines.
We then analyzed the effects of polarizing cytokines on T cell differentiation induced by TCR agonists or homeostatic cytokines. IL-4 and IL-12 induced differentiation of TCR-stimulated CXCR5+ and especially −/−TCM cells into IL-4– and IFN-γ–producing cells, respectively, whereas they failed to modulate T cell differentiation in IL-7 plus IL-15–activated T cells (Fig. 5, A and B). Under the same conditions of TCR stimulation, IL-4 induced CXCR3+ T cells to produce the opposite cytokine IL-4 and promoted CCR4 expression, whereas IL-12 induced CCR4+ cells to produce IFN-γ (Fig. 5 A) and up-regulate CXCR3 (not depicted). Again, polarizing cytokines had little effect on the extent of T cell differentiation in IL-7 plus IL-15–stimulated cells (compare Figs. 3 A and 5 B). Similar results were obtained when IL-2 substituted for IL-7 and IL-15, and in the absence or presence of TNF, IL-6 and IL-10, DC-derived cytokines that strongly boost proliferation of TCM cells in response to IL-7 and IL-15 (not depicted; reference 21). Together, these findings suggest that flexibility of cytokine gene expression of human memory T cells requires TCR triggering and polarizing cytokines (13).
Figure 5.

Effects of polarizing cytokines on TCR- and cytokine-induced differentiation. Purified CFSE-labeled CD4+ T cell subsets were stimulated with DC plus TSST or IL-7 plus IL-15 in the absence or presence of IL-12 and neutralizing anti–IL-4 antibody (Th1-condition) or IL-4 and neutralizing anti–IL-12 antibody (Th2-condition). After 7 d, cells were stimulated with PdBu and ionomycin and cells of the same division number were analyzed for IFN-γ and IL-4 production by intracellular staining. The percentages of INF-γ+ cells (white bars), IL-4+ cells (black bars), and of cells producing both cytokines (gray bars) are represented. One representative experiment out of five with different donors is shown.
Proliferation History, In Vivo Turnover, and Recall Responses of CD4+ Memory T Cell Subsets.
The proliferation history and in vivo turnover of the different memory T cell subsets was then assessed by measuring the amounts of TRECs and the spontaneous BrdU incorporation of ex vivo–isolated cells. TRECs carrying a particular signal joint sequence (37) were quantified by TaqMan PCR in T cell subsets from five healthy donors (Fig. 6 A). As expected, CD4+ CD45RA+ naive T cells contained high levels of TRECs, whereas B cells and T cell clones were negative (not depicted). Compared with naive T cells, CXCR5+ TCM and CXCR5− TCM cells contained much lower amounts of TRECs, whereas TEM cells contained the lowest amount. Although there were considerable quantitative differences among individual donors, these data indicate that the different subsets of TCM cells have divided to a similar extent.
Figure 6.

Proliferation history, in vivo turnover, and recall responses of CD4+ memory T cell subsets. (A) Total CXCR5+ TCM, CXCR5− TCM, and TEM cells were analyzed for their single joint TREC content and compared with naive cells from the same donor (naive cells: 100%). Bars indicate the mean TREC levels in memory subsets of five different donors. (B) Freshly isolated PBMCs were incubated with BrdU, CD4+ T cells were isolated, and BrdU incorporation was analyzed as a function of CD45RA and chemokine receptor expression by flow cytometry. The mean percentage of BrdU+ cells in a given subset of four donors is shown. (C) CD4+ naive and memory subsets were sorted, labeled with CFSE, and incubated with autologous monocytes that had either been infected with VV or incubated with TT or an extract of CMV-derived proteins. CFSE profiles of viable CD4+ CD14− cells on day 7 of one representative donor are shown. (D) Recall responses of memory subsets to TT of eight different donors were assessed as described above, and the frequency of TT-specific cells was calculated after 5 d. The frequency of TT-specific cells in the indicted memory subsets of seven different TT-responsive donors was plotted against the time of the last boost.
To measure the spontaneous BrdU incorporation, freshly isolated PBMCs were incubated with BrdU. CD4+ T cells were then purified and T cell subsets analyzed by intracellular staining with anti-BrdU antibodies. As shown in Fig. 6 B, CXCR5+ TCM, CXCR3+ TCM, and CCR4+ TCM cells spontaneously incorporated BrdU to a similar extent, whereas TEM and especially −/−TCM cells had a higher proliferation rate and naive cells were below the detection limit. These data indicate that memory T cells in different subsets slowly turn over under steady-state conditions in vivo.
To investigate whether the different memory subsets contained cells specific for recall antigens, T cell populations were isolated from smallpox- and/or tetanus-vaccinated donors and stimulated for 7 d with autologous monocytes that were incubated with TT or with an extract of CMV-derived proteins. To assess memory against smallpox, monocytes were infected with a replication-deficient VV. In these experiments, purified CFSE-labeled T cell subsets and CFSE dilution were used to read out proliferation of pathogen-specific T cells. From eight donors analyzed, seven responded strongly to TT, whereas one donor that had been boosted 20 yr ago had a low response (not depicted). Five donors responded strongly to CMV, and four of six donors that had also been vaccinated against smallpox had a detectable response to VV. Responses to autologous monocytes alone were undetectable or very low (not depicted).
We analyzed the distribution of TT-, CMV-, and VV-specific T cells among different memory subsets (one donor responding to all three pathogens is shown in Fig. 6 C). In all cases, pathogen-specific cells were detected in both the TCM and TEM cell pools. TT-specific cells were undetectable among naive cells, but present in all memory subsets in six of seven responsive donors. Conversely, CMV-specific cells were largely restricted to CXCR3+ TCM and TEM cells in all five responsive donors, consistent with the notion that CMV infection promotes a Th1 cell response (46). Consistent with previous reports, VV-specific cells were less frequent (47, 48), but were detectable in CXCR3+ TCM and TEM cells and, interestingly, in three of four donors in CXCR5+ TCM cells. Together, these results show that all memory subsets contain cells specific for recall antigens, and that the distribution of antigen-specific cells within TCM cell subsets varies for different pathogens.
Next, we compared precursor frequencies of TT-specific cells in different subsets in donors that had been boosted recently or several years ago (Fig. 6 D). TT-specific cells were relatively frequent among TEM cells (>1:500) in all donors. Conversely, CXCR5+ TCM cells were less frequent (<1:500), especially in donors that had not been boosted for several years, possibly explaining the failure of previous studies to detect TT-specific cells in this subset using thymidine incorporation (29, 31). Interestingly, the relative distribution among CXCR3+ TCM and CCR4+ TCM cells was highly variable, with some donors having higher numbers of CCR4+ TCM cells and others containing predominantly CXCR3+ TCM cells. These results indicate that TT-specific T cells are present in high frequency in TEM cells, even several years after vaccination, and are distributed in different subsets of TCM cells.
Discussion
We have shown that the human CD4+ TCM cell pool can be subdivided into subsets of nonpolarized cells and pre-Th1 and pre-Th2 cells based on chemokine receptor expression. These subsets have extensively divided in vivo and contain cells specific for recall antigens and with self-renewal capacity. Upon TCR-independent proliferation induced by homeostatic cytokines, TCM cell subsets are committed for different fates and become Th1, Th2, or remain nonpolarized cells, explaining how the quality of the primary immune response could be maintained by TCM cells in the absence of antigen.
Th cells expressing CXCR5 comprise CCR7− CD57+ follicular Th cells in tonsils (29, 30, 49), and nonpolarized circulating CXCR5+ TCM cells of unknown function and specificity that might have a recent activation history (31). Our results show that nonpolarized cells are present in both CXCR5+ and CXCR5− TCM cell subsets and lack CXCR3 and CCR4 expression. Using CFSE dilution we were further able to show that CXCR5+ TCM cells contained low numbers of TT-specific cells even several years after vaccination. Moreover, three of four smallpox-vaccinated, responsive donors contained VV-specific cells at low frequency in the CXCR5+ TCM cell subset. Using quantitative PCR, we found that CXCR5+ TCM and CXCR5− TCM cells contained comparable amounts of residual TRECs, whereas TEM cells had slightly lower levels, suggesting that TCM cell subsets had divided to a comparable extent (approximately seven times). However, because naive and memory cells were identified by CD45 isoform expression that is not a stable marker (25, 50), the number of divisions performed by memory cells might be underestimated by our analysis. Spontaneous BrdU uptake indicated that CXCR5+ TCM cells have an in vivo proliferation rate that is comparable to that of other memory subsets. The relative small cell size and the absence of CD69 on BrdU+ cells suggest that this proliferation is driven by homeostatic mechanisms rather than by antigen. Why −/−TCM cells have a higher turnover than other memory subsets is currently unclear. They might be particularly fit because they combine a relatively high cytokine responsiveness with a low susceptibility to apoptosis (51). Alternatively, they might be preferentially located in cytokine-rich microenvironments or in proximity to DCs that boost proliferation in response to IL-7 and IL-15 (21). In any case, these results show that all CD45RA− subsets, including CXCR5+ TCM cells, are memory cells that have extensively divided, slowly turnover in vivo, and contain cells specific for recall antigens.
Different viruses are known to induce CD8+ memory cells belonging preferentially to different subsets (52, 53). Here we showed that although pathogen-specific CD4+ T cells are present in both the TCM and TEM cell pools, they have characteristic distributions in TCM cell subsets, reflecting the Th1/Th2 cell polarization induced by the pathogens or vaccinations. Thus, TT-specific cells were detected in all subsets, consistent with the notion that vaccination against tetanus induces a mixed Th1/Th2 cell response (54). Conversely, CMV and VV promote Th1 cell polarization (46, 47), and virus-specific cells were consequently detected in CXCR3+ TCM cells but not in CCR4+TCM cells. In one donor, we were able to show that VV-specific TEM cells were also CXCR3+ (not depicted). Collectively, these results suggest that immune responses generate heterogeneous populations of memory cells that belong to different subsets and comprise a broad spectrum of differentiation stages. The distribution between CXCR3+ and CCR4+ subsets in the TCM and TEM cell pools might be useful to monitor the quality of the memory response to different pathogens.
Human memory T cells can be subdivided into CCR7+ TCM and CCR7− TEM cells with different effector functions and homing potentials, suggesting a division of labor between these two subsets (12). However, several recent reports showed that antigen-experienced CCR7+ cells possess immediate effector functions (9, 32–35). Although we identified here CCR7+ memory cells with IFN-γ– and IL-4–producing capacities as CXCR3+ TCM and CCR4+ TCM cells, respectively, the following lines of evidence suggest that these cells are not fully differentiated effectors: (a) they had a higher expansion potential than TEM cells and most cells had retained CD27 and CD62L expression; (b) IFN-γ production by CXCR3+ TCM cells was low and largely restricted to unconventional CCR7+ CD62L− cells; (c) although CCR4+ TCM cells produced some IL-4, production of IL-5, which acts on eosinophils at peripheral sites of allergic inflammation, was limited to TEM cells; (d) many CXCR3+ TCM and CCR4+ TCM cells had retained flexibility to differentiate to Th2 and Th1 cells, respectively, upon antigenic stimulation in the presence of appropriate polarizing cytokines; and (e) upon cytokine stimulation, they further differentiated, losing CCR7 and CD62L and acquiring nonlymphoid homing potential and high levels of effector functions. Together, these findings are consistent with the notion that nonlymphoid tissue homing potential and effector cytokine–producing capacities are progressively acquired upon T cell differentiation and reside predominantly in the TEM cell subset of the human CD4+ memory cell pool (3). However, because effector functions and nonlymphoid homing potentials are acquired in a stochastic manner (55), some cells have characteristics that are intermediate between TCM and TEM cells.
We previously proposed that cytokine-driven differentiation of TCM cells might be a mechanism to replenish short-lived TEM cells in the absence of antigen (21), but how nonpolarized TCM cells could faithfully maintain polarized Th1 or Th2 effector cell populations remained unclear. A recent report showed that CD4 T cell priming upon viral infection generated both short-lived effector cells and long-lived precursors that lacked effector functions, but spontaneously acquired IFN-γ–producing capacity when transferred into antigen-free hosts (26). We showed here that CXCR3+ TCM and CCR4+ TCM cells invariably differentiated to CCR7− Th1 or Th2 effector cells in an antigen-independent fashion, whereas CXCR5+ TCM cells remained nonpolarized and CCR7+. CXCR3 and CCR4 are preferentially induced under type 1 and type 2 priming conditions, respectively (6, 7), and CXCR3+ TCM and CCR4+ TCM cells might therefore represent committed precursors of the Th1 and Th2 cell lineage with the capacity to generate effector cells for extended periods in the absence of antigen. Unlike cytokines, TCR ligands can still instruct TCM cells to become Th1 or Th2 cells in the presence of appropriate polarizing cytokines (13). This differential flexibility might allow the human immune system to mount qualitatively different responses in the case of cross-reactive antigens (56), or alternatively, to maintain the quality of the primary response under homeostatic conditions.
Acknowledgments
We thank I. Giacchetto for technical assistance and Elisabetta Traggiai and Stefan Wirths for critical reading and comments.
This work has been supported in part by the European Community (contract no. QLK-CT-201-0105 to F. Sallusto), the NIH (grant no. U19AI057266-01 to A. Lanzavecchia), and by the Swiss National Science Foundation (grant no. 3100A0-104168 to J. Geginat and no. 3100A0-101962 to F. Sallusto). L. Rivino is supported by the Fondazione per la Ricerca sulla Trasfusione e i Trapianti.
The authors have no conflicting financial interests.
L. Rivino, M. Messi, and D. Jarrossay contributed equally to this work.
Abbreviations used in this paper: BrdU, bromodeoxyuridine; CFSE, carboxyfluorescein succinimidyl ester; PdBu, phorbol-12-13-dibutyrate; TCM cell, central–memory T cell; TEM cell, effector–memory T cell; TREC, T cell receptor rearrangement excision circle; TSST, toxic shock syndrome toxin; TT, tetanus toxoid; VV, vaccinia virus.
References
- 1.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 2.Mosmann, T.R., and R.L. Coffman. 1989. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 7:145–173. [DOI] [PubMed] [Google Scholar]
- 3.Lanzavecchia, A., and F. Sallusto. 2000. Dynamics of T lymphocyte responses: intermediates, effectors and memory cells. Science. 290:92–97. [DOI] [PubMed] [Google Scholar]
- 4.Sallusto, F., A. Lanzavecchia, and C.R. Mackay. 1998. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today. 19:568–574. [DOI] [PubMed] [Google Scholar]
- 5.Qin, S., J.B. Rottman, P. Myers, N. Kassam, M. Weinblatt, M. Loetscher, A.E. Koch, B. Moser, and C.R. Mackay. 1998. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Invest. 101:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Ambrosio, D., A. Iellem, R. Bonecchi, D. Mazzeo, S. Sozzani, A. Mantovani, and F. Sinigaglia. 1998. Selective up-regulation of chemokine receptors CCR4 and CCR8 upon activation of polarized human type 2 Th cells. J. Immunol. 161:5111–5115. [PubMed] [Google Scholar]
- 7.Sallusto, F., D. Lenig, C.R. Mackay, and A. Lanzavecchia. 1998. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 187:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langenkamp, A., K. Nagata, K. Murphy, L. Wu, A. Lanzavecchia, and F. Sallusto. 2003. Kinetics and expression patterns of chemokine receptors in human CD4+ T lymphocytes primed by myeloid or plasmacytoid dendritic cells. Eur. J. Immunol. 33:474–482. [DOI] [PubMed] [Google Scholar]
- 9.Kim, C.H., L. Rott, E.J. Kunkel, M.C. Genovese, D.P. Andrew, L. Wu, and E.C. Butcher. 2001. Rules of chemokine receptor association with T cell polarization in vivo. J. Clin. Invest. 108:1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forster, R., A. Schubel, D. Breitfeld, E. Kremmer, I. Renner-Muller, E. Wolf, and M. Lipp. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 99:23–33. [DOI] [PubMed] [Google Scholar]
- 11.Bradley, L.M., S.R. Watson, and S.L. Swain. 1994. Entry of naive CD4 T cells into peripheral lymph nodes requires L-selectin. J. Exp. Med. 180:2401–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sallusto, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 401:708–712. [DOI] [PubMed] [Google Scholar]
- 13.Messi, M., I. Giacchetto, K. Nagata, A. Lanzavecchia, G. Natoli, and F. Sallusto. 2003. Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat. Immunol. 4:78–86. [DOI] [PubMed] [Google Scholar]
- 14.Sallusto, F., J. Geginat, and A. Lanzavecchia. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22:745–763. [DOI] [PubMed] [Google Scholar]
- 15.Sprent, J., D.F. Tough, and S. Sun. 1997. Factors controlling the turnover of T memory cells. Immunol. Rev. 156:79–85. [DOI] [PubMed] [Google Scholar]
- 16.Schluns, K.S., W.C. Kieper, S.C. Jameson, and L. Lefrancois. 2000. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 1:426–432. [DOI] [PubMed] [Google Scholar]
- 17.Zhang, X., S. Sun, I. Hwang, D.F. Tough, and J. Sprent. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 8:591–599. [DOI] [PubMed] [Google Scholar]
- 18.Seddon, B., and R. Zamoyska. 2002. TCR and IL-7 receptor signals can operate independently or synergize to promote lymphopenia-induced expansion of naive T cells. J. Immunol. 169:3752–3759. [DOI] [PubMed] [Google Scholar]
- 19.Seddon, B, P. Tomlinson, and R. Zamoyska. 2003. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 4:680–686. [DOI] [PubMed] [Google Scholar]
- 20.Kanegane, H., and G. Tosato. 1996. Activation of naive and memory T cells by interleukin-15. Blood. 88:230–235. [PubMed] [Google Scholar]
- 21.Geginat, J., F. Sallusto, and A. Lanzavecchia. 2001. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4+ T cells. J. Exp. Med. 194:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reinhardt, R.L., A. Khoruts, R. Merica, T. Zell, and M.K. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 23.Roman, E., E. Miller, A. Harmsen, J. Wiley, U.H. von Andrian, G. Huston, and S.L. Swain. 2002. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J. Exp. Med. 196:957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wherry, E.J., V. Teichgraber, T.C. Becker, D. Masopust, S.M. Kaech, R. Antia, U.H. von Andrian, and R. Ahmed. 2003. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 4:225–234. [DOI] [PubMed] [Google Scholar]
- 25.Geginat, J., A. Lanzavecchia, and F. Sallusto. 2003. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 101:4260–4266. [DOI] [PubMed] [Google Scholar]
- 26.Wu, C.Y., J.R. Kirman, M.J. Rotte, D.F. Davey, S.P. Perfetto, E.G. Rhee, B.L. Freidag, B.J. Hill, D.C. Douek, and R.A. Seder. 2002. Distinct lineages of T(H)1 cells have differential capacities for memory cell generation in vivo. Nat. Immunol. 3:852–858. [DOI] [PubMed] [Google Scholar]
- 27.Cyster, J.G. 1999. Chemokines and cell migration in secondary lymphoid organs. Science. 286:2098–2102. [DOI] [PubMed] [Google Scholar]
- 28.Baggiolini, M. 1998. Chemokines and leukocyte traffic. Nature. 392:565–568. [DOI] [PubMed] [Google Scholar]
- 29.Breitfeld, D., L. Ohl, E. Kremmer, J. Ellwart, F. Sallusto, M. Lipp, and R. Forster. 2000. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192:1545–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaerli, P., K. Willimann, A.B. Lang, M. Lipp, P. Loetscher, and B. Moser. 2000. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192:1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaerli, P., P. Loetscher, and B. Moser. 2001. Cutting edge: induction of follicular homing precedes effector Th cell development. J. Immunol. 167:6082–6086. [DOI] [PubMed] [Google Scholar]
- 32.Unsoeld, H., S. Krautwald, D. Voehringer, U. Kunzendorf, and H. Pircher. 2002. Cutting edge: CCR7+ and CCR7− memory T cells do not differ in immediate effector cell function. J. Immunol. 169:638–641. [DOI] [PubMed] [Google Scholar]
- 33.Debes, G.F., U.E. Hopken, and A. Hamann. 2002. In vivo differentiated cytokine-producing CD4(+) T cells express functional CCR7. J. Immunol. 168:5441–5447. [DOI] [PubMed] [Google Scholar]
- 34.Ravkov, E.V., C.M. Myrick, and J.D. Altman. 2003. Immediate early effector functions of virus-specific CD8+ CCR7+ memory cells in humans defined by HLA and CC chemokine ligand 19 tetramers. J. Immunol. 170:2461–2468. [DOI] [PubMed] [Google Scholar]
- 35.Wolint, P., M.R. Betts, R.A. Koup, and A. Oxenius. 2004. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J. Exp. Med. 199:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gett, A.V., and P.D. Hodgkin. 2000. A cellular calculus for signal integration by T cells. Nat. Immunol. 1:239–244. [DOI] [PubMed] [Google Scholar]
- 37.Douek, D.C., R.D. McFarland, P.H. Keiser, E.A. Gage, J.M. Massey, B.F. Haynes, M.A. Polis, A.T. Haase, M.B. Feinberg, J.L. Sullivan, et al. 1998. Changes in thymic function with age and during the treatment of HIV infection. Nature. 396:690–695. [DOI] [PubMed] [Google Scholar]
- 38.Bonecchi, R., G. Bianchi, P.P. Bordignon, D. D'Ambrosio, R. Lang, A. Borsatti, S. Sozzani, P. Allavena, P.A. Gray, A. Mantovani, and F. Sinigaglia. 1998. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J. Exp. Med. 187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamann, D., P.A. Baars, M.H. Rep, B. Hooibrink, S.R. Kerkhof-Garde, M.R. Klein, and R.A. van Lier. 1997. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 186:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weng, N.P., B.L. Levine, C.H. June, and R.J. Hodes. 1995. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc. Natl. Acad. Sci. USA. 92:11091–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murali-Krishna, K., and R. Ahmed. 2000. Cutting edge: naive T cells masquerading as memory cells. J. Immunol. 165:1733–1737. [DOI] [PubMed] [Google Scholar]
- 42.Gett, A.V., and P.D. Hodgkin. 1998. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc. Natl. Acad. Sci. USA. 95:9488–9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bird, J.J., D.R. Brown, A.C. Mullen, N.H. Moskowitz, M.A. Mahowald, J.R. Sider, T.F. Gajewski, C.R. Wang, and S.L. Reiner. 1998. Helper T cell differentiation is controlled by the cell cycle. Immunity. 9:229–237. [DOI] [PubMed] [Google Scholar]
- 44.Okamura, H., H. Tsutsi, T. Komatsu, M. Yutsudo, A. Hakura, T. Tanimoto, K. Torigoe, T. Okura, Y. Nukada, K. Hattori, et al. 1995. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 378:88–91. [DOI] [PubMed] [Google Scholar]
- 45.Yang, J., T.L. Murphy, W. Ouyang, and K.M. Murphy. 1999. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur. J. Immunol. 29:548–555. [DOI] [PubMed] [Google Scholar]
- 46.Rentenaar, R.J., L.E. Gamadia, N. van DerHoek, F.N. van Diepen, R. Boom, J.F. Weel, P.M. Wertheim-van Dillen, R.A. van Lier, and I.J. ten Berge. 2000. Development of virus-specific CD4(+) T cells during primary cytomegalovirus infection. J. Clin. Invest. 105:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Combadiere, B., A. Boissonnas, G. Carcelain, E. Lefranc, A. Samri, F. Bricaire, P. Debre, and B. Autran. 2004. Distinct time effects of vaccination on long-term proliferative and IFN-γ–producing T cell memory to smallpox in humans. J. Exp. Med. 199:1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hammarlund, E., M.W. Lewis, S.G. Hansen, L.I. Strelow, J.A. Nelson, G.J. Sexton, J.M. Hanifin, and M.K. Slifka. 2003. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 9:1131–1137. [DOI] [PubMed] [Google Scholar]
- 49.Kim, C.H., L.S. Rott, I. Clark Lewis, D.J. Campbell, L. Wu, and E.C. Butcher. 2001. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center–localized subset of CXCR5+ T cells. J. Exp. Med. 193:1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bell, E.B., and S.M. Sparshott. 1990. Interconversion of CD45R subsets of CD4 T cells in vivo. Nature. 348:163–166. [DOI] [PubMed] [Google Scholar]
- 51.Gett, A.V., F. Sallusto, A. Lanzavecchia, and J. Geginat. 2003. T cell fitness determined by signal strength. Nat. Immunol. 4:355–360. [DOI] [PubMed] [Google Scholar]
- 52.Champagne, P., G.S. Ogg, A.S. King, C. Knabenhans, K. Ellefsen, M. Nobile, V. Appay, G.P. Rizzardi, S. Fleury, M. Lipp, et al. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 410:106–111. [DOI] [PubMed] [Google Scholar]
- 53.Appay, V., P.R. Dunbar, M. Callan, P. Klenerman, G.M. Gillespie, L. Papagno, G.S. Ogg, A. King, F. Lechner, C.A. Spina, et al. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8:379–385. [DOI] [PubMed] [Google Scholar]
- 54.Rowe, J., C. Macaubas, T. Monger, B.J. Holt, J. Harvey, J.T. Poolman, R. Loh, P.D. Sly, and P.G. Holt. 2001. Heterogeneity in diphtheria-tetanus-acellular pertussis vaccine-specific cellular immunity during infancy: relationship to variations in the kinetics of postnatal maturation of systemic th1 function. J. Infect. Dis. 184:80–88. [DOI] [PubMed] [Google Scholar]
- 55.Reiner, S.L., and R.A. Seder. 1999. Dealing from the evolutionary pawnshop: how lymphocytes make decisions. Immunity. 11:1–10. [DOI] [PubMed] [Google Scholar]
- 56.Welsh, R.M., and L.K. Selin. 2002. No one is naive: the significance of heterologous T-cell immunity. Nat Rev. Immunol. 2:417–426. [DOI] [PubMed] [Google Scholar]