

Abstract
HACS1 is a Src homology 3 and sterile alpha motif domain–containing adaptor that is preferentially expressed in normal hematopoietic tissues and malignancies including myeloid leukemia, lymphoma, and myeloma. Microarray data showed HACS1 expression is up-regulated in activated human B cells treated with interleukin (IL)-4, CD40L, and anti–immunoglobulin (Ig)M and clustered with genes involved in signaling, including TNF receptor–associated protein 1, signaling lymphocytic activation molecule, IL-6, and DEC205. Immunoblot analysis demonstrated that HACS1 is up-regulated by IL-4, IL-13, anti-IgM, and anti-CD40 in human peripheral blood B cells. In murine spleen B cells, Hacs1 can also be up-regulated by lipopolysaccharide but not IL-13. Induction of Hacs1 by IL-4 is dependent on Stat6 signaling and can also be impaired by inhibitors of phosphatidylinositol 3-kinase, protein kinase C, and nuclear factor κB. HACS1 associates with tyrosine-phosphorylated proteins after B cell activation and binds in vitro to the inhibitory molecule paired Ig-like receptor B. Overexpression of HACS1 in murine spleen B cells resulted in a down-regulation of the activation marker CD23 and enhancement of CD138 expression, IgM secretion, and Xbp-1 expression. Knock down of HACS1 in a human B lymphoma cell line by small interfering ribonucleic acid did not significantly change IL-4–stimulated B cell proliferation. Our study demonstrates that HACS1 is up-regulated by B cell activation signals and is a participant in B cell activation and differentiation.
Keywords: B lymphocytes, interleukin-4, signaling, gene expression, adaptor protein
Introduction
HACS1 is an adaptor protein identified in our transcriptional study of genes expressed in multiple myeloma (1). The gene encoding HACS1 maps to human chromosome 21q11.2, a region frequently disrupted by translocation events in hematopoietic disorders. Our previous work demonstrated that this gene is highly expressed as a 2.2-kb transcript in some hematopoietic malignancies including acute myeloid leukemia, myeloma, and lymphoma, and in normal bone marrow, lymph node, spleen and thymus. HACS1 mRNA expression is also observed at a low level in heart, brain, placenta, and lung. HACS1 encodes a 441–amino acid protein containing a Src homology 3 (SH3) domain in the middle of the protein and a sterile alpha motif (SAM) domain at the COOH terminus. It contains three putative nuclear localization signals, suggesting that it can be transported to the nucleus. The SH3 motif of the HACS1 protein shares 39% homology with CRK, an adaptor protein that binds to several tyrosine-phosphorylated proteins. HACS1 also shares 50% amino acid homology with HACS2/SLY, a highly homologous gene on the X chromosome (2). In addition, an NH2-terminal splice variant, SAMSN1, encodes a 373–amino acid protein that preserves both the SH3 and SAM domains. Human HACS1/SAMSN1 is part of a large segment of chromosome 21 genes conserved on mouse chromosome 16. The orthologous murine Hacs1/Samsn1 encodes a highly conserved protein (87%) of 364 amino acids. A recent study of expression profiles of mouse orthologs to human chromosome 21 genes by a combination of large scale mRNA in situ hybridization and in silico mining of expressed sequence tags (ESTs) suggests that Hacs1/Samsn1 might be associated with blood vessel formation during embryogenesis (3). Despite this information, the gene regulation and function of the HACS1 protein remains unknown.
Here, we demonstrate that HACS1 gene expression is up-regulated by treatment of B cells with IL-4 and other B cell activators. By dissecting IL-4 receptor signaling pathways, we further demonstrate that up-regulation of HACS1 gene expression by IL-4 requires STAT6 and activation of phosphatidylinositol (PI) 3-kinase, protein kinase C (PKC), and NF-κB. Functional assays further indicate that HACS1 likely mediates its effects in the signaling cascades that regulate B cell activation and differentiation.
Materials and Methods
Cytokines, Antibodies, and Reagents.
Cytokines including hIL-4, mIL-4, mIL-2, mIL-3, mIL-6, mIL-7, mIL-10, hIL-13, mIL-13, and TNF-α were from R&D Systems. Anti-CD19 and anti-B220 microbeads were from Miltenyi Biotec Canada. Anti-CD40 and goat F(ab′)2 anti–human or mouse IgM were from Southern Biotechnology Associates, Inc. Anti-HACS1 is a rabbit polyclonal antibody raised against a glutathione S-transferase fusion containing the SAM domain and COOH-terminal region of the human HACS1 protein. The HACS1 antibody recognizes both mouse and human forms of HACS1 and was used for both immunoprecipitation and Western blotting. Anti-nPKCζ and anti–pNF-κB p50 were from Santa Cruz Biotechnology, Inc. Wortmannin and pyrrolidinedithiocarbamate (PDTC) were from Sigma-Aldrich. Rapamycin (FRAP/mTOR inhibitor), PD98059 (MEK1 inhibitor), anti–phospho-Akt, anti-Akt, anti–phospho-PKCζ, and anti-STAT6 were from Cell Signaling Technology. Bisindolylmaleimide I (Bis I) and Bay11-7082 were from Calbiochem-Novabiochem. The antiphosphotyrosine antibody 4G10 was from Upstate Biotechnology, anti–paired Ig-like receptor B (PIR-B) polyclonal anti-sera was a gift from Dr. B. Neel (American Red Cross, Bethesda, MD), and anti-HA antibody was from Roche Diagnostic. HRP-conjugated anti–rabbit or anti–mouse secondary antibodies and ECL reagent were from Amersham Bioscience. Stat6 knockout mouse was purchased from Jackson Laboratory.
Bioinformatics.
Microarray data from the analysis of diffuse large B cell lymphoma (4) was chosen primarily because HACS1 was among the genes spotted on the cDNA microarray (NIH “Lymphochip”). First, the “Lymphochip” was Blast searched (http://llmpp.nih.gov/lymphoma/search.shtml/) using HACS1 (sequence data available from GenBank/EMBL/DDBJ under accession no. AF218085) as the query sequence. Alignment results showed two significant EST hits: IMAGE: 1269200 (sequence data available from GenBank/EMBL/DDBJ under accession no. AA747998) and IMAGE:1356357 (sequence data available from GenBank/EMBL/DDBJ under accession no. AA831672). Further analysis was conducted using the Stanford Online Universal Resource for Clones and ESTs (SOURCE) (http://source.stanford.edu) retrieval system (5). The SOURCE database showed cDNA clone IMAGE: 1269200 alone has microarray data available.
B Cell Purification and Stimulation.
Human peripheral blood B cells and murine splenic B cells were selected by magnetic cell purification using anti-CD19 microbeads and anti-B220 microbeads, respectively, to 95% purity, as confirmed by flow cytometry analysis. Cells were cultured (2.0 × 106 cells/ml) in 6-well plates in RPMI 1640 containing 10% FCS and stimulated with different cytokines or antibodies. Cytokines added to these cultures included IL-4, IL-2, IL-3, IL-6, IL-7, IL-10, and IL-13 (all 10 ng/ml). Other stimuli were anti-IgM, anti-CD40 (both 5 μg/ml), and LPS (5 μg/ml). Cells were also pretreated with various signaling inhibitors (0.1 μM wortmannin; 10 μM PD 98059; 20 nM rapamycin; 5 μm Bis I; 2, 5, and 10 μm Bay11-7082; and 1 and 5 mM PDTC) for 15 min, followed with treatment of 10 ng/ml IL-4 or 5 μg/ml anti-CD40 for 8 h or overnight.
BJAB cells were cultured at 37°C with 5% CO2 in RPMI 1640 supplemented with 10% FBS. For transient transfections, BJAB cells were electroporated with 20 μg of pEF(HA)2PIR-B (cytoplasmic tail) in RPMI 1640 using a GENEPulser (Bio-Rad Laboratories) set at 250 V and 960 μF. Cells were cultured for 24 h, resuspended in 500 μL RPMI 1640 media, prewarmed for 5 min at 37°C, and then stimulated with or without goat anti–human IgM (Zymed Laboratories) for 5 min. The stimulations were halted using phosphatase inhibitor buffer (100 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO4, PBS, pH 7.4), collected by centrifugation, and lysed in 0.2% TNE (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.2% NP-40) lysis buffer containing protease inhibitors and 1 mM Na3VO4.
RT-PCR.
RT-PCR was performed using standard methods. The primers used to amplify HACS1 were: forward, 5′-TATTGACATGGCCACCAAGA-3′ and reverse, 5′-GGCCAATTGGAAAATCAGTG-3′; HACS2/SLY forward, 5′-TCCAGCAGCTTCAAGGATTT-3′ and reverse, 5′-CATCTTGCCCA-TCTTCCTGT-3′; Xbp-1 forward, 5′-CCTTGTGGTTGAG-AACCAGG-3′ and reverse, 5′-CTAGAGGCTTGGTGTA-TAC-3′.
Confocal Immunofluorescence Microscopy of B Cells.
Primary splenic murine B cells were treated with or without 10 ng/ml IL-4 for the indicated times and seeded onto glass coverslips that had been pretreated with 0.01% polyornithine (Sigma-Aldrich). Coverslips were washed twice with PBS plus Ca2+ or Mg2+. Cells were fixed with 4% paraformaldehyde for 30 min at room temperature and permeabilized with 0.2% Triton X-100 in PBS plus Ca2+ and Mg2+ for 10 min. After incubation in blocking buffer (5% BSA in PBS plus Ca2+ and Mg2+), the permeabilized B cells were incubated with anti-HACS1 antibody (1:1,000) and subsequently with Alexa 488–labeled (green fluorescence) anti–rabbit secondary antibody (1:500) at 37°C for 30 min. Cell nuclei were stained with propidium iodide (red fluorescence). Coverslips were mounted with Dako Fluorescent Mounting medium and viewed with a Leica 4D confocal microscope.
Yeast Two-Hybrid and cDNA Library Screening.
To identify binding partners for HACS1, a yeast two-hybrid screen was conducted using the CytoTrap system (Stratagene) which is based on the reestablishment of the Ras signaling pathway to detect in vivo protein–protein interactions in the cytoplasm. The full-length human cDNA of HACS1 was cloned in frame into pSos vector that was used as the bait. A mouse spleen cDNA library was screened after cotransformation of the bait plasmid and library into cdc25H yeast. The yeast colonies were selected based on growth at 37°C from which plasmid DNA was isolated for sequence analysis.
Generation of Expression Constructs and Retroviruses.
The retrovirus vector miev was originally from Dr. Robert Hawley (Harvard Institues of Medicine, Boston, MA). The cDNAs encoding HACS1 protein were subcloned into this vector followed by sequencing. Generation of ecotropic retrovirus was performed as described (6). Briefly, 10 μg of DNA was used to transfect the GP+E86 packaging cell line, and after 1 wk of transfection, EGFP+ cells were sorted using a Coulter Elite cell sorter. The sorted GP+E86 cells were then expanded and analyzed for HACS1 expression by Western Blot. Finally, the retroviral supernatant was harvested from packaging cells.
Retroviral Transduction of B Cells and B Cell Activation Analysis.
Transduction of splenic B lymphocytes was performed using purified splenic B220+ cells. Briefly, B220+ cells were stimulated with 5 μg/ml LPS (Sigma-Aldrich) overnight and cocultured with viral packaging cells (miev, HACS1/miev) in the presence of 4 μg/ml polybrene for 2 d. GFP+ B cells were sorted and counted. 2.5 × 105 of cells were analyzed by Western blot or RT-PCR to test expression of HACS1 or Xbp-1 expression. 3 × 104 cells were seeded in triplicate in 96-well plates with or without IL-4 (10 ng/ml) and anti-CD40 (5 μg/ml) for 48 h. Cell proliferation was measured using an MTT assay (Roche Diagnostic). Cell surface markers (anti-CD23 and anti-CD138) were analyzed using a FACScalibur flow cytometer (Becton Dickinson) with CELLQuest software. IgM secretion in the supernatant of cultured B cells was detected using an ELISA kit according to the manufacturer's instruction (BETHYL Laboratories, Inc.).
Small Interfering RNA Transduction.
Small interfering (si)RNA duplexes were synthesized and purified by Dharmacon Inc. The HACS1 target sequence is GGACAGAGCTCATCAAGTGTT. 106 BJAB cells were transfected with 5 μl of 20 μM HACS1-specific siRNA or control scrambled siRNA by electroporation (Amaxa). 48 h after transfection, an aliquot of cells were harvested, treated, and processed for immunoblotting studies. Cells were seeded at 104 cells/well in triplicate in 96-well plates with or without anti-IgM (5 μg/ml) and IL-4 (25 ng/ml) for 72 h. Cell proliferation was measured using an MTT assay.
Results
HACS1 Is Expressed in Activated Human B Cells.
HACS1 was sequenced from malignant plasma cells and was subsequently found to be highly expressed in cell lines from other human B cell malignancies. Our immunohistochemistry analysis of murine lymphoid tissues demonstrated expression predominantly in the lymph node sinusoids and splenic red pulp rather than in lymph nodules and splenic white pulp that contains a more dense mass of lymphocytes (Fig. 1). Using in silico analysis of microarray data from B lymphocytes (4), we found that HACS1 gene expression was low in memory and naïve B cells but had elevated expression in activated human B cells treated with IL-4, CD40L, and anti-IgM (Fig. 2, A and B).
Figure 1.

Immunohistochemistry of mouse spleen and lymph node. Hacs1 is expressed in mouse splenic red pulp and lymph node sinusoids. Low power views of spleen (A) and lymph node (B) stained with anti-HACS1. The white pulp (W) and red pulp (R) in spleen are shown in both magnifications. High power views of spleen and lymph node stained with anti-HACS1 are shown in the bottom panel. Hacs1 is expressed in the cytoplasm of most of the positive cells but is also present in nuclei of some cells (inset in spleen bottom panel).
Figure 2.
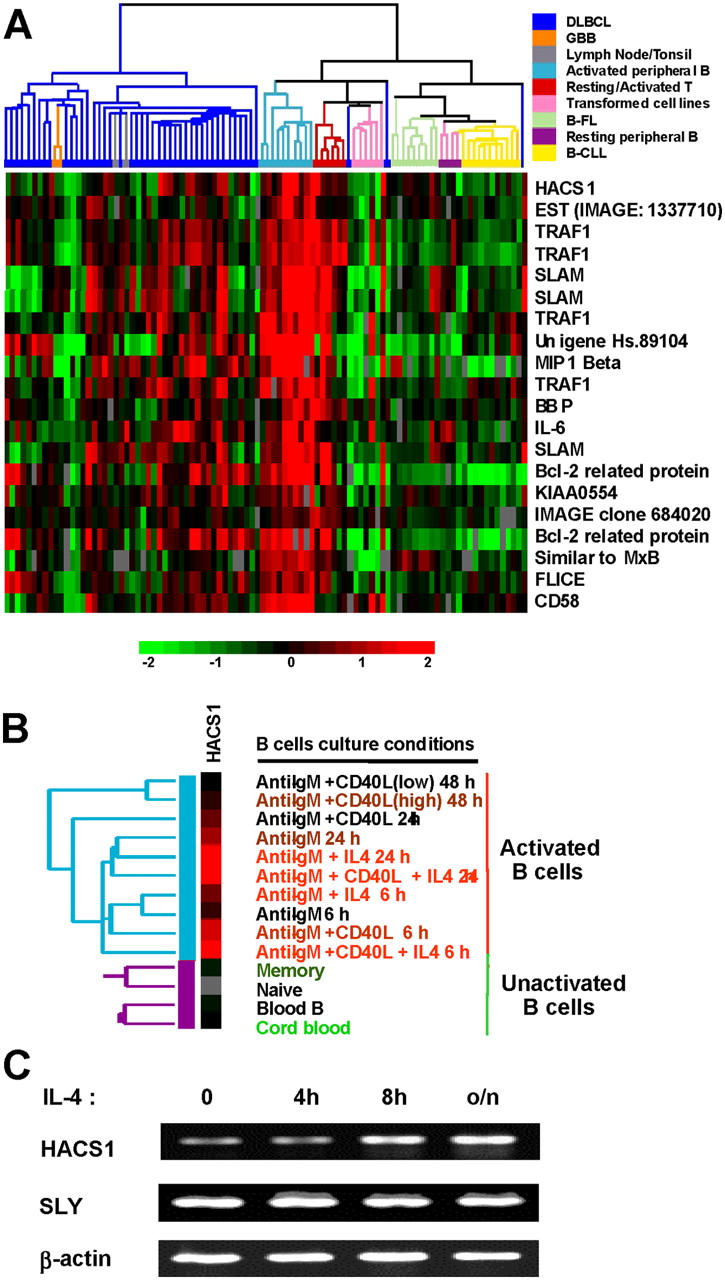
In silico analysis of HACS1 expression from microarray hybridization data of lymphocyte samples. (A) HACS1 clustered together with known signaling proteins and uncharacterized genes and ESTs. (B) HACS1 can be up-regulated in B cells under different culture conditions such as addition of IL-4, anti-IgM, and CD40L. Data was mined from Alizadeh et al. (4) through SOURCE (5). (C) Gene-specific RT-PCR analysis of HACS1 and related gene HACS2/Sly shows only HACS1 can be up-regulated by IL-4, confirming the microarray data.
Hierarchical cluster analysis (7) of HACS1 demonstrated it clustering with other genes involved in signaling such as the TNF receptor–associated protein 1 (TRAF1), signaling lymphocytic activation molecule (SLAM) precursor, the chemokine MIP1β, IL-6, DEC205 (IMAGE clone1337710), and novel uncharacterized genes such as UniGene cluster Hs.89104, KIAA0554, and IMAGE clone 684040 (Fig. 2 A). Interestingly, the expression pattern of both HACS1 and DEC205 overlaps suggesting that the function of HACS1 may be related with that of DEC205. In particular, we have found HACS1 to be strongly expressed in cultured DCs (not depicted).
To confirm our microarray observations, we performed gene-specific RT-PCR. Since HACS1 belongs to a novel gene family of adaptor proteins that includes a highly homologous gene, HACS2/SLY (1, 2), we looked at the expression of both genes in splenic B220+ B cells. Although transcripts of both Hacs1 and Hacs2/Sly are present in mouse B cells, our results indicated that only Hacs1 was induced by IL-4, whereas Hacs2/Sly basal RNA level remained unchanged in the presence of IL-4 (Fig. 2 C).
HACS1 Protein Is Up-Regulated by IL-4 and Other B Cell Activators in Both Human and Murine Spleen B Cells.
To verify whether our in silico results are consistent at the protein level, Western analysis was conducted on purified human peripheral blood CD19+ B cells treated with IL-4, anti-IgM, and anti-CD40. As shown in Fig. 3 A, human peripheral B cells have a low, basal level expression of HACS1 protein, but its expression was greatly induced by IL-4, anti-IgM, or anti-CD40 after overnight treatment. Since IL-13 receptor (IL-13R) shares the IL-4 receptor (IL-4R) α chain (8, 9), we next examined the effects of IL-13 on HACS1 expression. Not surprisingly, HACS1 expression was also induced by IL-13 in human peripheral B cells (Fig. 3 B). Accordingly, when we looked at HACS1 expression in four Hodgkin lymphoma (HL) lines, HACS1 expression was detected in three cell lines that express IL-13 (10, 11) but not in one HL cell line (L540) not expressing IL-13 (Fig. 3 B).
Figure 3.

HACS1 was up-regulated by IL-4, IL-13, anti-IgM, and anti-CD40 in human peripheral B cells. (A) Purified human peripheral CD19+ cells were incubated with 10 ng/ml IL-4 or 1 μg/ml anti-IgM or 1 μg/ml anti-CD40 overnight, and then cells were harvested and lysed and HACS1 expression was analyzed by Western blot. (B) Western blot analysis of HACS1 expression in CD19+ cells induced by IL-13 (10 ng/ml overnight) and in human HL cell lines L1236, HDLM2, and L428 and L540. K562 (erythroleukemia) served as a positive control (1).
The effects of B cell activators on HACS1 expression were next tested on primary murine splenic B220+ B cells. Similar to human CD19+B cells, a low expression level of the Hacs1 protein was detected in resting B cells from mouse spleen but was induced by IL-4 in a dose-dependent manner (Fig. 4 A). Interestingly, the induced Hacs1 protein in B220+ cells usually appeared as triplet bands, which might represent protein modifications or alternatively spliced isoforms. Up-regulation of the Hacs1 protein became apparent after 6 h of treatment as seen on a Western blot of IL-4–stimulated B220+ B cells (Fig. 4 B). These findings were consistent with the results using confocal immunofluorescence microscopy examining Hacs1 protein in B220+ B cells 6 h after IL-4 stimulation (Fig. 4 C).
Figure 4.
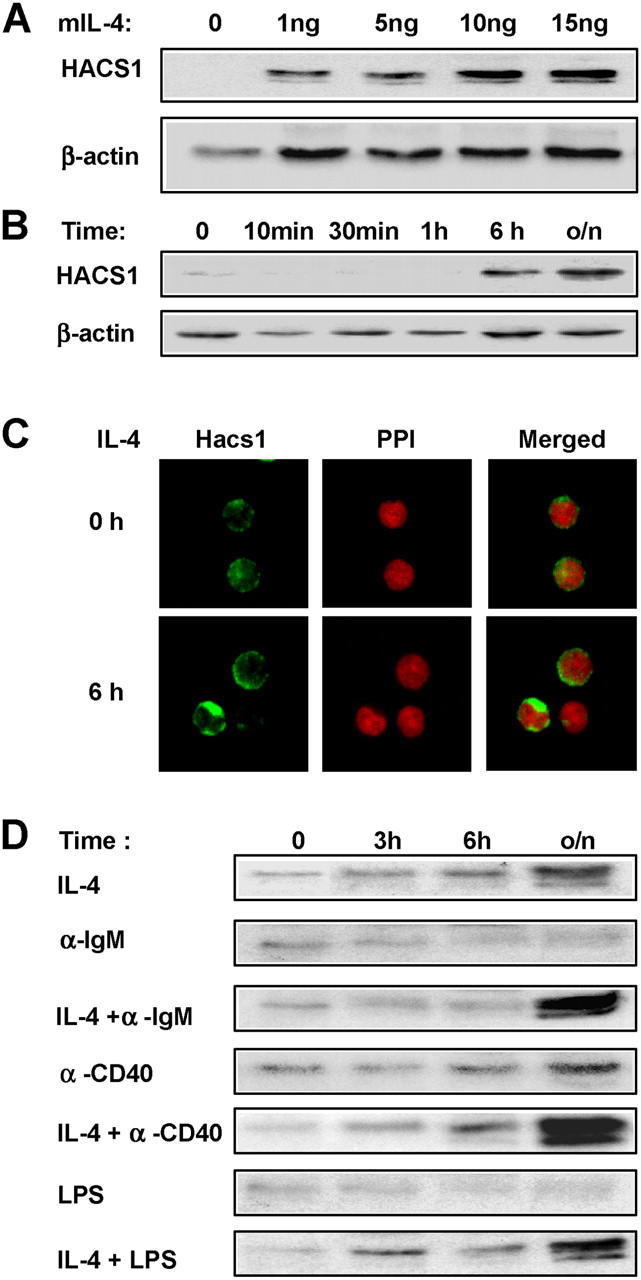
Hacs1 was up-regulated by IL-4 and other B cell activators in B220+ murine splenocytes. (A) Purified murine B220+ splenocytes were incubated with an increasing amount of IL-4 overnight or (B) incubated with 10 ng/ml of IL-4 for the indicated time, and then cells were harvested, lysed, and analyzed by Western blot. (C) Immunofluorescence staining of Hacs1 protein in B220+ mouse spleen B cells in vitro stimulated with IL-4. The up-regulation of Hacs1 expression (green) was evident by 6 h poststimulation as shown on the left (green). The nuclei were stained red with propidium iodide (PPI) as shown on the middle panel. The right panel shows the two stained images merged together. (D) B220+ cells were incubated with various B cell activators alone or with IL-4 plus different stimuli for the indicated time, and then expression of Hacs1 was analyzed by Western blot.
Similar to human CD19+ B cells, when murine B220+ B cells were treated with other known B cell activators including anti-IgM, anti-CD40, and LPS, Hacs1 gene expression increased, albeit at a lower level for anti-IgM and LPS stimulation. However, when combined with IL-4, synergistic effects on Hacs1 expression were observed, especially between IL-4 and anti-CD40 (Fig. 4 D). Other cytokines including IL-2, IL-3, IL-6, IL-7, IL-10, and IL-13, had no effect on Hacs1 expression in murine B cells (not depicted), suggesting that the IL-4 signaling pathway is the dominant signaling mechanism for Hacs1 expression.
Up-Regulation of HACS1 by IL-4 Is STAT6 Dependent and Diminished by Inhibition of PI 3-Kinase and PKC.
To dissect the IL-4 signaling pathway which up-regulates HACS1, three known IL-4 receptor signaling mechanisms, the Stat6, PI 3-kinase, and Ras/mitogen-activated protein kinase (MAPK) pathways, were independently investigated. After IL-4 stimulation of murine splenic B cells, activation of Stat6 became evident (Fig. 5 A). Similarly, activation of PI 3-kinase by IL-4 was observed by increased phosphorylation of Akt, a downstream target of PI 3-kinase (Fig. 5 A). To determine whether IL-4 regulates Hacs1 levels via Stat6, we demonstrated the absence of Hacs1 induction in IL-4–stimulated B220+ spleen B cells from a Stat6-deficient mouse (Fig. 5 B). Thus, Stat6 is absolutely required for Hacs1 induction by IL-4. By searching for putative transcription factor binding elements in the 5′ putative promoter region of HACS1, we identified two Stat6 binding elements (TTCAACAGAA and TTCAATGGAA) at 1.4 and 1.5 kb upstream of the putative start codon, suggesting that Stat6 can bind to the HACS1 promoter to exert regulatory function.
Figure 5.

Up-regulation of Hacs1 by IL-4 via Stat6, PI 3-kinase, and PKC pathways. (A) B220+ cells were incubated with 10 ng/ml IL-4 for the indicated time, and the tyrosine phosphorylation of Stat6 and Akt were analyzed by Western blot. (B) Stat6 is necessary for up-regulation of Hacs1 by IL-4. B220+ cells were purified from Stat6-deficient mice (Stat6−/−) and control mice (Balb/c and J129) and then incubated with IL-4 (10 ng/ml) for 18 h. Expression of Hacs1 was analyzed by Western blot. (C) Inhibition of PI 3-kinase and PKC impaired up-regulation of Hacs1 by IL-4 in B220+ murine splenocytes. B220+ cells were pretreated without or with different inhibitors (100 nM wortmannin [wort], 10 μM PD98059 [PD], 20 nM rapamycin [Rapa], and 5 μm Bis I) for 15 min, and then 10 ng/ml IL-4 were added and incubated for 8 h. The cells were lysed and analyzed by Western blot.
To further investigate whether other signaling pathways activated by IL-4 are also engaged in the up-regulation of Hacs1, we employed a variety of chemical inhibitors. IL-4 alone was reported not to induce activation of the MAPK pathway in murine splenic B cells (12, 13). In agreement with this notion, addition of PD98059, a MAPK inhibitor, to IL-4–treated spleen B cells did not affect the expression of Hacs1 (Fig. 5 C), suggesting that Hacs1 regulation does not go through the Ras/MAPK pathways. In contrast, when cells were pretreated with a PI 3-kinase inhibitor, wortmannin, Hacs1 up-regulation was diminished (Fig. 5 C), but addition of rapamycin, an inhibitor of FRAP/mTOR and p70S6, downstream effectors of PI 3-kinase, did not affect the up-regulation of Hacs1 by IL-4. These results suggest that the PI 3-kinase pathway is involved in IL-4–mediated Hacs1 expression but not via activation of p70S6 kinase. When cells were pretreated with a pan-PKC inhibitor Bis1, the up-regulation of Hacs1 by IL-4 was diminished but not abolished (Fig. 5 C), implying that up-regulation of Hacs1 by IL-4 might go through the PI 3-kinase/PKC pathway. Since tyrosine phosphorylation of Stat6 stimulated by IL-4 was not decreased by either wortmannin or Bis I treatment, whereas expression of Hacs1 was significantly reduced, the Stat6 pathway alone cannot be responsible for IL-4–mediated up-regulation of Hacs1. Thus, signaling via the PI 3-kinase/PKC pathway and Stat6 pathway appears to play important roles in promoting Hacs1 expression.
Up-Regulation of HACS1 Involves the NF-κB Pathway.
Since activation of PI 3-kinase, in particular the PKC pathway, is involved in induction of Hacs1 by IL-4, we further assessed which downstream transcription factors might be activated and function in this pathway. Activation of PI 3-kinase by IL-4 has been demonstrated to induce different PKC isoforms including δ, ɛ, η, and ζ (14–16). IL-4–activated PKCζ has been shown to enhance germline Cɛ transcription through activation of NF-κB in a human B lymphoma cell line (15, 17). Recent evidence from PKCζ knockout mice demonstrated that this kinase is important for NF-κB transcriptional activity in both embryonic fibroblast and B cells (18). In addition, disruption of the PKCζ locus not only resulted in inhibition of cell proliferation and survival but also impaired the transcription of NF-κB–dependent genes in B cells (18). To determine whether IL-4–activated PKCζ and up-regulation of Hacs1 by IL-4 is associated with nuclear translocation of NF-κB in murine splenic B cells, Western blotting was performed on B220+ cells treated with IL-4 and Bay11-7082, a cell-permeable chemical inhibitor of NF-kB. As shown in Fig. 6, A and B, upon IL-4 stimulation PKCζ was highly phosphorylated and the nuclear translocation of NF-κB protein p50 was induced in B220+ cells. When B220+ cells were pretreated with Bay11-7082, the IL-4–induced nuclear expression of NF-κB but not Stat6 was abrogated. Treatment of B220+ cells with Bay11-7082 and another NF-κB inhibitor PDTC also resulted in a dose-dependent inhibition of IL-4 and anti-CD40–induced Hacs1 expression (Fig. 6 C), suggesting that Hacs1 expression requires NF-κB activity. Subsequent database search and analysis of the HACS1 putative promoter region revealed a consensus binding sequence for NF-κB at ∼1.8 kb upstream of the HACS1 start codon.
Figure 6.

IL-4–activated PKCζ and up-regulation of Hacs1 was blocked by inhibition of NF-κB. (A) IL-4 induced phosphorylation of PKCζ in murine splenic B cells. B220+ cells were incubated with 10 ng/ml IL-4 for the indicated time, and expression of phosphorylated PKCζ was analyzed by Western blot. (B and C) Inhibition of NF-κB blocked nuclear expression of NF-κB and the subsequent up-regulation of Hacs1 by IL-4. B220+ cells were pretreated with or without Bay11-7082 or PDTC for 15 min and then incubated with 10 ng/ml IL-4 for the indicated time. The nuclear extracts or whole cell lysates were prepared and analyzed for expression of NF-κB p50 and Stat6 in nuclei (B) or for expression of Hacs1 in whole cells (C) by Western blot.
Association of HACS1 with Tyrosine-phosphorylated Proteins in Stimulated B Cells.
To further investigate the role of HACS1 in B cell signaling, a human B cell line (BJAB) was stimulated with or without anti-IgM, and the HACS1-interacting proteins were examined. Upon stimulation with anti-IgM, endogenous HACS1 inducibly associated with multiple tyrosine-phosphorylated proteins, indicating HACS1 may function as an adaptor protein downstream of the B cell receptor (BCR) (Fig. 7 A).
Figure 7.

HACS1 associates with phosphotyrosine-containing proteins in stimulated B cells. (A) Lysates from human BJAB cells were immunoprecipitated with anti-HACS1 antibody and preimmune serum control after stimulation with or without goat anti–human IgM for 5 min. The presence of tyrosine-phosphorylated proteins associated with HACS1 were assessed by immunoblotting with an antiphosphotyrosine antibody (4G10). Reblotting shows the level of HACS1 in the BJAB cell line. (B) Human BJAB cells were electroporated with the cytoplasmic tail of PIR-B using a pEF(HA)2PIR-B construct or the pEF(HA)2 vector alone. After stimulation with or without goat anti–human IgM for 5 min, immunoprecipitation was performed with anti-HACS1 and control IgG antibodies. Western blotting was performed with anti-HA antibody (top) and anti-HACS1 antibody (bottom), showing that the cytoplasmic tail of PIR-B binds to HACS1 in vitro.
To identify HACS1 binding proteins important in the IL-4 signaling pathway, a yeast two-hybrid screen of a mouse spleen library was conducted. From this screen, 18 clones remained after elimination of false positives. Each clone contained insert after restriction digest, and every clone was sequenced. 1 of the 18 clones was identified as PIR-B, a protein involved in B cell suppression. The PIR-B fragment obtained from the yeast-two hybrid screen was 386 residues in length and included the transmembrane domain and the entire cytoplasmic tail of the receptor (amino acids 456–841). Therefore, we deemed PIR-B a potential binding partner of HACS1 in our IL-4 B cell model. PIR-B is known to be constitutively tyrosine-phosphorylated in primary B lymphocytes and negatively regulates the B cell response (19, 20). Furthermore, IL-4 has been shown to effect inhibitory receptor expression levels and contribute to cellular activation. To initially test the HACS1–PIR-B interaction, we conducted in vitro experiments. BJAB cells were electroporated with a construct containing the cytoplasmic tail of PIR-B which was then shown to bind preferentially to endogenous HACS1, suggesting that HACS1 and PIR-B can associate in human B cells under these experimental conditions (Fig. 7 B). However, association studies of HACS1 with endogenous PIR-B in primary murine B cells proved unsuccessful, although HACS1 was found to constitutively associate with a phosphotyrosine protein of 110 kD (not depicted).
HACS1 Is Involved in B Cell Activation and Differentiation.
Since HACS1 is up-regulated during B cell activation and is associated with phosphotyrosyl proteins in stimulated B cells, its function could be associated with regulating the cellular response of activated B cells. We investigated whether HACS1 affects B cell activation and differentiation. Activation of B cells by IL-4 and other B cell activators usually result in B cell proliferation, cell surface antigen modification, and differentiation (21). Both IL-4 and anti-CD40 stimulate the proliferation of B cells and enhance the expression of cell surface molecules such as the low affinity Fc receptor for IgE (CD23). When a HACS1 retroviral expression construct was introduced into murine spleen B cells, we found that compared with control cells (vector alone), cell proliferation stimulated by IL-4 and anti-CD40 was inhibited in HACS1-transduced B cells (Fig. 8 A). Similarly, the expression of CD23 (Fig. 8 B) was impaired in those cells. In contrast, expression of this exogenous HACS1 resulted in an enhancement of differentiation of B220+ cells to plasma cells indicated as increased surface CD138 (syndecan-1) expression, IgM secretion, and up-regulation of XBP-1 (Fig. 8, C–F).
Figure 8.

The role of HACS1 is likely involved in B cell activation and differentiation. (A) Effects of HACS1 on IL-4 and CD-40–driven B220+ cell proliferation. Murine splenic B220+ cells were infected as described in Materials and Methods. GFP+ cells were sorted, and 3 × 104 cells were seeded in triplicate in 96-well plates with or without IL-4 (10 ng/ml) or anti-CD40 (5 μg/ml) for 48 h, followed by an MTT assay. The data is representative of three independent experiments. (B and C) Effects of HACS1 on the surface phenotype of B220+ cells. The cells treated as above in triplicates were harvested and combined and stained with PE-conjugated antibodies against CD23 and CD138. Flow cytometry data is converted to mean fluorescence intensity of the surface expression of living cells. The data is representative of three independent experiments. (D) HACS1 enhanced IgM secretion in vitro. Cell culture supernatant was collected from triplicates of each group above, and IgM levels were measured by ELISA assay. The mean of triplicates is shown, which is representative of two independent experiments. (E) Expression of HACS1 in transduced B220+ cells (48 h postinfection) was confirmed by Western blot analysis. (F) Expression of Xbp-1 was enhanced in HACS1-transduced B220+ cells. Briefly, HACS1 and control virus-infected B220+ cells were harvested after 48 h of infection, and total RNA was extracted, quantitated, and analyzed by RT-PCR. (G) HACS1-specific siRNA knocked down endogenous HACS1 in BJAB cells. BJAB cells were transfected with either control scrambled oligos (C) or HACS1-specific siRNA oligos (H) by electroporation. After 48 h of transfection, cells were treated with indicated reagents for 5 min, and the expression of HACS1 was analyzed by Western blot. (H) Knock down of HACS1 in BJAB cells did not significantly affect cell proliferation stimulated by IL-4. 48 h after transduction of control (C) and HACS1 (H) siRNA oligos, 1 × 104 cells were seeded in triplicate in 96-well plates with or without IL-4 (25 ng/ml) or anti-IgM (5 μg/ml) for 72 h, followed by an MTT assay. The data is representative of two independent experiments.
To further investigate the role of HACS1 in B cells, HACS1-specific siRNA was electroporated into BJAB cells, which constitutively express endogenous HACS1. We found that 48 h after transfection, ∼90% of endogenous HACS1 had been knocked down in BJAB cells (Fig. 8 G). Compared with control, knock down of HACS1 only marginally affected cell proliferation in both IL-4 and IgM-treated human B cells (Fig. 8 H).
Discussion
Although our immunohistochemistry analysis of normal mouse lymphoid tissues did not show predominant expression of Hacs1 in regions that contain mostly lymphocytes, in silico mining of published microarray data indicated that the novel adaptor HACS1 can be up-regulated by IL-4, anti-IgM, and ligation of CD40 in human B cells. By RT-PCR and Western blot, we further confirmed induction of HACS1 by IL-4 and other B cell activators 6 h after treatment, suggesting that HACS1 is a secondary response gene induced during B cell activation. IL-13, like IL-4, also up-regulated HACS1 gene expression in human peripheral B cells. Both IL-4 and IL-13 are Th2-derived pleiotropic cytokines that share common functions in human B cells, including modulating surface antigen expression and Ig class switching (22, 23). The IL-4Rα chain is a component of IL-13 receptors, which includes two surface proteins, IL-13Rα1 and IL-13Rα2. Although mouse IL-4 was able to stimulate Hacs1 gene expression in murine splenic B cells, mouse IL-13 failed to up-regulate Hacs1 expression. This is in keeping with the notion that B220+ mouse splenocytes are unresponsive to IL-13 (24) due to the lack of IL-13Rα1 expression (25). IL-2R and IL-7R share a common subunit (IL-2γ chain), which is also shared by IL-4R (14), but IL-2 and IL-7 did not affect HACS1 gene expression, indicating that IL-4Rα chain–mediated signaling is critical to HACS1 induction. Other B cell activators including anti-IgM, anti-CD40, and LPS also stimulated HACS1 gene expression especially when combined with IL-4, implying that signals mediated by these stimuli must have some redundancy or interaction with the IL-4 signaling pathway for the regulation of HACS1.
By dissecting the signaling pathways for the up-regulation of HACS1 by IL-4, we demonstrate that Stat6 is an essential upstream signaling molecule for IL-4–mediated HACS1 expression. This observation is consistent with the known role of Stat 6 in IL-4–mediated gene transcription. Two putative Stat6 binding motif are present within 2 kb upstream of the HACS1 initiation codon implying that Stat6 may directly bind to the HACS1 promoter to regulate its gene expression. However, studies in Stat6 knockout mice demonstrated that Stat6 is necessary for expression of IL-4–responsive genes (26–28), but Stat6 DNA binding is not sufficient to activate transcription. Stat6 usually has been found to cooperate with other transcription factors, such as NF-κB and C-EBP to activate transcription (29, 30). Recently, Stat6 has been shown physically to interact with NF-κB and coactivator p300/CBP. Both of these transcription factors have been shown to enhance Stat6-dependent transcriptional activation of IL-4–responsive promoters (31, 32).
Using specific inhibitors, we further demonstrated that activation of PI 3-kinase is also involved in IL-4–mediated HACS1 induction in murine splenic B cells. The activation of PI 3-kinase has been demonstrated to function during B cell development, growth, and responses (33, 34). In addition to IL-4, PI 3-kinase can also be activated in BCR, CD40, and LPS-mediated signal transduction pathways (35–37). For example, PI 3-kinase can be activated downstream of the BCR by antigen or by the ligation of CD40 on B cells. Therefore, the induction of the HACS1 gene by IL-4, anti-IgM, and anti-CD40 might be explained, at least partially, by their signaling redundancy upon activation of PI 3-kinase.
Upon activation by IL-4, PI 3-kinase converts PI 4,5-bisphosphate into PI 3,4,5-triphosphate that subsequently activates downstream signaling including serine/threonine kinase Akt, PKC family or MAPK family, and p70S6 kinase (14). In our study, the activation of PI 3-kinase by IL-4 in murine splenic B cells resulted in the phosphorylation of Akt, which like HACS1 was diminished when cells were pretreated with 0.1 μm of wortmannin (not depicted). However, treatment of cells with either p70S6 kinase inhibitor or MAPK inhibitor had no effect on HACS1 protein expression, suggesting that the regulation of HACS1 downstream of PI 3-kinase are not associated with these pathways. Instead, induction of HACS1 by IL-4 was impaired by a PKC inhibitor consistent with the known activation of PKC as a downstream effector of PI 3-kinase (38). In our study, we found that PKCζ, an atypical isoform of PKC (aPKC) that is activated by IgM and implicated as a downstream target of PI 3-kinase (18), was activated by IL-4 stimulation in B220+ splenocytes, but antibodies against phosphorylated conventional PKCs, including PKCα, βI, βII, and δ, failed to detect increasing phosphorylation of PKC by IL-4 treatment. These observations suggest that PKCζ might be the major isoform activated by IL-4. However, the involvement of other isoforms of PKC in Hacs1 gene induction cannot be excluded, since IL-4 also has been found to activate η and PKCɛ (14, 39). Furthermore, LPS, which is known for activating various isoforms of PKC such as α, β, ɛ, but not ζ, also enhanced HACS1 expression.
Both PI 3-kinase and PKC have been demonstrated to activate NF-κB pathways (17, 18, 40, 41). NF-κB is associated with the expression of genes involved in B cell development, differentiation, and function (42). Although constitutively active in mature B cells, NF-κB is further induced after stimulation through CD40 or the BCR. In our study, treatment of B cells with Bay11-7082 and PDTC greatly impaired synergistic effects of IL-4 and CD40 on Hacs1 expression, indicating that nuclear expression of NF-κB is critical to Hacs1 induction. In agreement with this observation, our data mining study indicated that the expression pattern of IL-4 induced HACS1 clustered together with characterized NF-κB targets such as TRAF1 (43) and IL-6 (44). Consistently, the HACS1 promoter region contains a putative NF-κB binding site (tggcctTTCC) at −1.8kb. Further study of the role of NF-κB using spleen B cells from an NF-κB p50-deficient mouse, however, displayed normal induction of Hacs1 by IL-4 compared with control wild-type mouse, thus suggesting that other members of the NF-κB family are responsible for Hacs1 expression. For example, a recent study showed that c-Rel is a selective activator of a novel IL-4/CD40-responsive element that contains overlapping sites for c-Rel and Stat6. This suggests that IL-4 and CD40 cross-linking regulate the human Ig γ4 germline promoter through Stat6/NF-κB cooperation (45). The cooperation between the Stat6 pathway and the NF-κB may also explain why the combination of IL-4 and anti-CD40 provides an additive effect on Hacs1 up-regulation.
Our data mining study found that HACS1 clustered together with SLAM precursor and DEC205. SLAM belongs to the Ig superfamily of receptors and has been shown to enhance cellular proliferation, production of inflammatory cytokines, and Ig secretion (46). DEC205 is an endocytic receptor with homology to macrophage mannose receptor that is involved in antigen processing in DCs (47). DEC205 also interacts with the IL-4R to modulate IL-4R signal transduction in B cells (48). Interestingly, in addition to the expression of HACS1 in B cells, we found, like DEC205 and SLAM, that HACS1 is also highly expressed in DCs, suggesting HACS1 may function as those two molecules involved in regulating the cellular response of immune cells.
Peripheral B cell activation is inhibited by a variety of cell surface receptors including CD22, FcγRII, CD72, and PIR-B (49). Each of these inhibitory molecules contains immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in their cytoplasmic domain. PIR-B has four ITIMs with the tyrosine in the third ITIM playing a crucial role in mediating the inhibitory signal in BCR activation (50). PIR-B is expressed on the surface of B cells, macrophages, DCs, and myeloid lineage cells (51). PIR-B associates with the protein tyrosine kinase Lyn and depends on the protein tyrosine phosphatase SHP-1, and possibly SHP-2, to mediate its inhibitory effect since SHP-1/SHP-2 double-deficient B cells show a reduction in PIR-B–mediated inhibition (50). Interestingly, previous work has shown that IL-4 reduces the expression of PIR-B at the mRNA and protein level and this effect is mediated by Stat6 (52). Therefore, IL-4 abolishes the inhibitory effect of PIR-B and contributes to B cell activation. A similar effect has been demonstrated recently in DCs for the inhibitory receptor ILT-2, the closest human family member to PIR-B, which was shown to be down-regulated after DC activation (53). Our yeast two-hybrid and in vitro studies suggested that HACS1 may associate with PIR-B. Although we were unable to demonstrate that the inhibitory receptor PIR-B and HACS1 associate in a primary B cell model, we cannot exclude the association of these proteins. Probably the binding of these two proteins is weak and requires a specific assay condition or a specific stimulation.
We have also shown that HACS1 interacts with several inducibly tyrosine-phosphorylated proteins in BJAB cells (155, 135, 120, 95, 85, 70, 65, and 40 kD) after surface cross-linking of the BCR. These proteins are additional potential HACS1 binding partners, and ongoing studies to identify these proteins will assist in delineating the function of HACS1 downstream of the BCR.
By introducing a HACS1 expression construct into LPS-stimulated murine splenic B cells, we found that expression of this exogenous HACS1 resulted in an enhancement of differentiation of B220+ cells to plasma cells which are characterized by up-regulation of CD138, IgM secretion, and expression of Xbp-1. Xbp-1 is the first transcription factor shown to be selectively and specifically required for the terminal differentiation of B lymphocytes to plasma cells (54). Xbp-1–deficient mice possess normal numbers of activated B lymphocytes and form normal germinal centers, but they have no plasma cells and secrete very little Ig (54). IL-4 was discovered recently as the only cytokine to control Xbp-1 expression in mature B cells through the Stat6 pathway (55). In addition to IL-4, LPS also stimulates Xbp-1 expression and B cell differentiation. Overexpression of HACS1 appears to enhance the B cell differentiation induced by LPS. We noticed that enhancement of B cell differentiation by HACS1 is more remarkable when a lower dose of LPS (5 μg/ml) was used to pretreat B cells. HACS1 alone seems unlikely to be sufficient to induce B cell terminal differentiation since overexpression of HACS1 in BCL-1 cells (which can be differentiated to plasma cells by IL-2 and IL-5 stimulation) failed to up-regulate Xbp-1 and induce IgM secretion (not depicted). These results suggest that HACS1 may function in a network of pathways that is required for B cell differentiation. Using siRNA technology, we successfully knocked down HACS1 expression in the K562 (not depicted) and BJAB cell lines. We found that knock down of HACS1 in the K562 (not depicted) and BJAB cells did not significantly affect cell proliferation.
In summary, our study demonstrated that HACS1 is up-regulated upon B cell activation mainly through the IL-4–mediated signal transduction pathway. Induction of HACS1 by IL-4 involves multiple signaling cascades including activation of Stat6, PI 3-kinase, PKC kinases, and NF-κB, indicating that the function of HACS1 is likely a component of the signaling cascades leading to B cell activation and differentiation.
Acknowledgments
We thank J. Zhang, C. Siminovitch, P. Ohashi, and B. Neel for providing useful reagents and for their helpful suggestions.
Y.X. Zhu is a recipient of Multiple Myeloma Research Foundation (MMRF) Research award. S. Benn is a recipient of the Hospital for Sick Children RESTRACOMP award. J.O. Claudio is a recipient of International Myeloma Foundation Junior Grant award. A.K. Stewart is funded by grants from the National Cancer Institute of Canada, MMRF, Canadian Institutes of Health Research (CIHR), and ABC Foundation, and C.J. McGlade by CIHR.
The authors have no conflicting financial interests.
Y.X. Zhu and S. Benn contributed equally to this work.
Abbreviations used in this paper: BCR, B cell receptor; Bis I, bisindolylmaleimide I; EST, expressed sequence tag; HL, Hodgkin lymphoma; ITIM, immunoreceptor tyrosine-based motif; MAPK, mitogen-activated protein kinase; PDTC, pyrrolidinedithiocarbamate; PI, phosphatidylinositol; PKC, protein kinase C; SAM, sterile alpha motif; siRNA, small interfering RNA; SH3, Src homology 3.
References
- 1.Claudio, J.O., Y.X. Zhu, S.J. Benn, A.H. Shukla, C.J. McGlade, N. Falcioni, and A.K. Stewart. 2001. HACS1 encodes a novel SH3-SAM adaptor protein differentially expressed in normal and malignant hematopoietic cells. Oncogene. 20:5373–5377. [DOI] [PubMed] [Google Scholar]
- 2.Beer, S., A.B. Simins, A. Schuster, and B. Holzmann. 2001. Molecular cloning and characterization of a novel SH3 protein (SLY) preferentially expressed in lymphoid cells. Biochim. Biophys. Acta. 1520:89–93. [DOI] [PubMed] [Google Scholar]
- 3.Gitton, Y., N. Dahmane, S. Baik, A. Ruiz i Altaba, L. Neidhardt, M. Scholze, B.G. Herrmann, P. Kahlem, A. Benkahla, S. Schrinner, et al. 2002. A gene expression map of human chromosome 21 orthologues in the mouse. Nature 420:586–590. [DOI] [PubMed] [Google Scholar]
- 4.Alizadeh, A.A., M.B. Eisen, R.E. Davis, C. Ma, I.S. Lossos, A. Rosenwald, J.C. Boldrick, H. Sabet, T. Tran, X. Yu, et al. 2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 403:503–511. [DOI] [PubMed] [Google Scholar]
- 5.Diehn, M., G. Sherlock, G. Binkley, H. Jin, J.C. Matese, T. Hernandez-Boussard, C.A. Rees, J.M. Cherry, D. Botstein, P.O. Brown, et al. 2003. SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res. 31:219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plowright, E.E., Z. Li, P.L. Bergsagel, M. Chesi, D.L. Barber, D.R. Branch, R.G. Hawley, and A.K. Stewart. 2000. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood. 95:992–998. [PubMed] [Google Scholar]
- 7.Eisen, M.B., P.T. Spellman, P.O. Brown, and D. Botstein. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA. 95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murata, T., P.D. Noguchi, and R.K. Puri. 1996. IL-13 induces phosphorylation and activation of JAK2 Janus kinase in human colon carcinoma cell lines: similarities between IL-4 and IL-13 signaling. J. Immunol. 156:2972–2978. [PubMed] [Google Scholar]
- 9.Matthews, D.J., L. Hibbert, K. Friedrich, A. Minty, and R.E. Callard. 1997. X-SCID B cell responses to interleukin-4 and interleukin-13 are mediated by a receptor complex that includes the interleukin-4 receptor alpha chain (p140) but not the gamma c chain. Eur. J. Immunol. 27:116–121. [DOI] [PubMed] [Google Scholar]
- 10.Kapp, U., W.C. Yeh, B. Patterson, A.J. Elia, D. Kagi, A. Ho, A. Hessel, M. Tipsword, A. Williams, C. Mirtsos, et al. 1999. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J. Exp. Med. 189:1939–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skinnider, B.F., A.J. Elia, R.D. Gascoyne, B. Patterson, L. Trumper, U. Kapp, and T.W. Mak. 2002. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood. 99:618–626. [DOI] [PubMed] [Google Scholar]
- 12.Richards, J.D., S.H. Dave, C.H. Chou, A.A. Mamchak, and A.L. DeFranco. 2001. Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J. Immunol. 166:3855–3864. [DOI] [PubMed] [Google Scholar]
- 13.Zamorano, J., A.E. Kelly, J. Austrian, H.Y. Wang, and A.D. Keegan. 2001. Costimulation of resting B lymphocytes alters the IL-4-activated IRS2 signaling pathway in a STAT6 independent manner: implications for cell survival and proliferation. Cell Res. 11:44–54. [DOI] [PubMed] [Google Scholar]
- 14.Nelms, K., A.D. Keegan, J. Zamorano, J.J. Ryan, and W.E. Paul. 1999. The IL-4 receptor: signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17:701–738. [DOI] [PubMed] [Google Scholar]
- 15.Ikizawa, K., K. Kajiwara, Y. Basaki, T. Koshio, and Y. Yanagihara. 1996. Evidence for a role of phosphatidylinositol 3-kinase in IL-4-induced germline C epsilon transcription. Cell. Immunol. 170:134–140. [DOI] [PubMed] [Google Scholar]
- 16.Gomez, J., A. Garcia, L.R. Borlado, P. Bonay, A.C. Martinez, A. Silva, M. Fresno, A.C. Carrera, C. Eicher-Streiber, and A. Rebollo. 1997. IL-2 signaling controls actin organization through Rho-like protein family, phosphatidylinositol 3-kinase, and protein kinase C-zeta. J. Immunol 158:1516–1522. [PubMed] [Google Scholar]
- 17.Yanagihara, Y., Y. Basaki, K. Ikizawa, and K. Kajiwara. 1997. Possible role of nuclear factor-kappa B activity in germline C epsilon transcription in a human Burkitt lymphoma B cell line. Cell. Immunol. 176:66–74. [DOI] [PubMed] [Google Scholar]
- 18.Martin, P., A. Duran, S. Minguet, M.L. Gaspar, M.T. Diaz-Meco, P. Rennert, M. Leitges, and J. Moscat. 2002. Role of zeta PKC in B-cell signaling and function. EMBO J. 21:4049–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho, L.H., T. Uehara, C.C. Chen, H. Kubagawa, and M.D. Cooper. 1999. Constitutive tyrosine phosphorylation of the inhibitory paired Ig-like receptor PIR-B. Proc. Natl. Acad. Sci. USA. 96:15086–15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ujike, A., K. Takeda, A. Nakamura, S. Ebihara, K. Akiyama, and T. Takai. 2002. Impaired dendritic cell maturation and increased T(H)2 responses in PIR-B(−/−) mice. Nat. Immunol. 3:542–548. [DOI] [PubMed] [Google Scholar]
- 21.Maliszewski, C.R., K. Grabstein, W.C. Fanslow, R. Armitage, M.K. Spriggs, and T.A. Sato. 1993. Recombinant CD40 ligand stimulation of murine B cell growth and differentiation: cooperative effects of cytokines. Eur. J. Immunol. 23:1044–1049. [DOI] [PubMed] [Google Scholar]
- 22.Defrance, T., P. Carayon, G. Billian, J.C. Guillemot, A. Minty, D. Caput, and P. Ferrara. 1994. Interleukin 13 is a B cell stimulating factor. J. Exp. Med. 179:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Punnonen, J., G. Aversa, B.G. Cocks, A.N. McKenzie, S. Menon, G. Zurawski, R. de Waal Malefyt, and J.E. de Vries. 1993. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc. Natl. Acad. Sci. USA. 90:3730–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zurawski, G., and J.E. de Vries. 1994. Interleukin 13, an interleukin 4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol. Today. 15:19–26. [DOI] [PubMed] [Google Scholar]
- 25.Andrews, R., L. Rosa, M. Daines, and G. Khurana Hershey. 2001. Reconstitution of a functional human type II IL-4/IL-13 receptor in mouse B cells: demonstration of species specificity. J. Immunol. 166:1716–1722. [DOI] [PubMed] [Google Scholar]
- 26.Shimoda, K., J. van Deursen, M.Y. Sangster, S.R. Sarawar, R.T. Carson, R.A. Tripp, C. Chu, F.W. Quelle, T. Nosaka, D.A. Vignali, et al. 1996. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 380:630–633. [DOI] [PubMed] [Google Scholar]
- 27.Linehan, L.A., W.D. Warren, P.A. Thompson, M.J. Grusby, and M.T. Berton. 1998. STAT6 is required for IL-4-induced germline Ig gene transcription and switch recombination. J. Immunol. 161:302–310. [PubMed] [Google Scholar]
- 28.Takeda, K., T. Tanaka, W. Shi, M. Matsumoto, M. Minami, S. Kashiwamura, K. Nakanishi, N. Yoshida, T. Kishimoto, and S. Akira. 1996. Essential role of Stat6 in IL-4 signalling. Nature. 380:627–630. [DOI] [PubMed] [Google Scholar]
- 29.Delphin, S., and J. Stavnezer. 1995. Characterization of an interleukin 4 (IL-4) responsive region in the immunoglobulin heavy chain germline epsilon promoter: regulation by NF-IL-4, a C/EBP family member and NF-kappa B/p50. J. Exp. Med. 181:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warren, W.D., K.L. Roberts, L.A. Linehan, and M.T. Berton. 1999. Regulation of the germline immunoglobulin Cgamma1 promoter by CD40 ligand and IL-4: dual role for tandem NF-kappaB binding sites. Mol. Immunol. 36:31–44. [DOI] [PubMed] [Google Scholar]
- 31.Shen, C.H., and J. Stavnezer. 1998. Interaction of stat6 and NF-kappaB: direct association and synergistic activation of interleukin-4-induced transcription. Mol. Cell. Biol. 18:3395–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gingras, S., J. Simard, B. Groner, and E. Pfitzner. 1999. p300/CBP is required for transcriptional induction by interleukin-4 and interacts with Stat6. Nucleic Acids Res. 27:2722–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki, T., A. Suzuki, J. Sasaki, and J.M. Penninger. 2002. Phosphoinositide 3-kinases in immunity: lessons from knockout mice. J. Biochem. (Tokyo). 131:495–501. [DOI] [PubMed] [Google Scholar]
- 34.Clayton, E., G. Bardi, S.E. Bell, D. Chantry, C.P. Downes, A. Gray, L.A. Humphries, D. Rawlings, H. Reynolds, E. Vigorito, and M. Turner. 2002. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 196:753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gold, M.R., V.W. Chan, C.W. Turck, and A.L. DeFranco. 1992. Membrane Ig cross-linking regulates phosphatidylinositol 3-kinase in B lymphocytes. J. Immunol. 148:2012–2022. [PubMed] [Google Scholar]
- 36.Ren, C.L., T. Morio, S.M. Fu, and R.S. Geha. 1994. Signal transduction via CD40 involves activation of lyn kinase and phosphatidylinositol-3-kinase, and phosphorylation of phospholipase C gamma 2. J. Exp. Med. 179:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bone, H., and N.A. Williams. 2001. Antigen-receptor cross-linking and lipopolysaccharide trigger distinct phosphoinositide 3-kinase-dependent pathways to NF-kappa B activation in primary B cells. Int. Immunol. 13:807–816. [DOI] [PubMed] [Google Scholar]
- 38.Le Good, J.A., W.H. Ziegler, D.B. Parekh, D.R. Alessi, P. Cohen, and P.J. Parker. 1998. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 281:2042–2045. [DOI] [PubMed] [Google Scholar]
- 39.Ikizawa, K., K. Kajiwara, K. Izuhara, and Y. Yanagihara. 2001. PKCdelta and zeta mediate IL-4/IL-13-induced germline epsilon transcription in human B cells: a putative regulation via PU.1 phosphorylation. Biochem. Biophys. Res. Commun. 288:34–41. [DOI] [PubMed] [Google Scholar]
- 40.Ozes, O.N., L.D. Mayo, J.A. Gustin, S.R. Pfeffer, L.M. Pfeffer, and D.B. Donner. 1999. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 401:82–85. [DOI] [PubMed] [Google Scholar]
- 41.Lallena, M.J., M.T. Diaz-Meco, G. Bren, C.V. Paya, and J. Moscat. 1999. Activation of IkappaB kinase beta by protein kinase C isoforms. Mol. Cell. Biol. 19:2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gugasyan, R., R. Grumont, M. Grossmann, Y. Nakamura, T. Pohl, D. Nesic, and S. Gerondakis. 2000. Rel/NF-kappaB transcription factors: key mediators of B-cell activation. Immunol. Rev. 176:134–140. [DOI] [PubMed] [Google Scholar]
- 43.Hinz, M., P. Loser, S. Mathas, D. Krappmann, B. Dorken, and C. Scheidereit. 2001. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood. 97:2798–2807. [DOI] [PubMed] [Google Scholar]
- 44.Libermann, T.A., and D. Baltimore. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 10:2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agresti, A., and D. Vercelli. 2002. c-Rel is a selective activator of a novel IL-4/CD40 responsive element in the human Ig gamma4 germline promoter. Mol. Immunol. 38:849–859. [DOI] [PubMed] [Google Scholar]
- 46.Veillette, A., and S. Latour. 2003. The SLAM family of immune-cell receptors. Curr. Opin. Immunol. 15:277–285. [DOI] [PubMed] [Google Scholar]
- 47.Jiang, W., W.J. Swiggard, C. Heufler, M. Peng, A. Mirza, R.M. Steinman, and M.C. Nussenzweig. 1995. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 375:151–155. [DOI] [PubMed] [Google Scholar]
- 48.McKay, P.F., N. Imami, M. Johns, D.A. Taylor-Fishwick, L.M. Sedibane, N.F. Totty, J.J. Hsuan, D.B. Palmer, A.J. George, B.M. Foxwell, et al. 1998. The gp200-MR6 molecule which is functionally associated with the IL-4 receptor modulates B cell phenotype and is a novel member of the human macrophage mannose receptor family. Eur. J. Immunol. 28:4071–4083. [DOI] [PubMed] [Google Scholar]
- 49.Takai, T., and M. Ono. 2001. Activating and inhibitory nature of the murine paired immunoglobulin-like receptor family. Immunol. Rev. 181:215–222. [DOI] [PubMed] [Google Scholar]
- 50.Maeda, A., M. Kurosaki, M. Ono, T. Takai, and T. Kurosaki. 1998. Requirement of SH2-containing protein tyrosine phosphatases SHP-1 and SHP-2 for paired immunoglobulin-like receptor B (PIR-B)–mediated inhibitory signal. J. Exp. Med. 187:1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Timms, J.F., K. Carlberg, H. Gu, H. Chen, S. Kamatkar, M.J. Nadler, L.R. Rohrschneider, and B.G. Neel. 1998. Identification of major binding proteins and substrates for the SH2-containing protein tyrosine phosphatase SHP-1 in macrophages. Mol. Cell. Biol. 18:3838–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rudge, E.U., A.J. Cutler, N.R. Pritchard, and K.G. Smith. 2002. Interleukin 4 reduces expression of inhibitory receptors on B cells and abolishes CD22 and Fc gamma RII-mediated B cell suppression. J. Exp. Med. 195:1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ju, X.S., C. Hacker, B. Scherer, V. Redecke, T. Berger, G. Schuler, H. Wagner, G.B. Lipford, and M. Zenke. 2004. Immunoglobulin-like transcripts ILT2, ILT3 and ILT7 are expressed by human dendritic cells and down-regulated following activation. Gene. 331:159–164. [DOI] [PubMed] [Google Scholar]
- 54.Reimold, A.M., N.N. Iwakoshi, J. Manis, P. Vallabhajosyula, E. Szomolanyi-Tsuda, E.M. Gravallese, D. Friend, M.J. Grusby, F. Alt, and L.H. Glimcher. 2001. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 412:300–307. [DOI] [PubMed] [Google Scholar]
- 55.Iwakoshi, N.N., A.H. Lee, P. Vallabhajosyula, K.L. Otipoby, K. Rajewsky, and L.H. Glimcher. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4:321–329. [DOI] [PubMed] [Google Scholar]