

Abstract
About 30% of cases of the autosomal recessive immunodeficiency disorder hemophagocytic lymphohistiocytosis are believed to be caused by inactivating mutations of the perforin gene. We expressed perforin in rat basophil leukemia cells to define the basis of perforin dysfunction associated with two mutations, R225W and G429E, inherited by a compound heterozygote patient. Whereas RBL cells expressing wild-type perforin (67 kD) efficiently killed Jurkat target cells to which they were conjugated, the substitution to tryptophan at position 225 resulted in expression of a truncated (∼45 kD) form of the protein, complete loss of cytotoxicity, and failure to traffic to rat basophil leukemia secretory granules. By contrast, G429E perforin was correctly processed, stored, and released, but the rat basophil leukemia cells possessed reduced cytotoxicity. The defective function of G429E perforin mapped downstream of exocytosis and was due to its reduced ability to bind lipid membranes in a calcium-dependent manner. This study elucidates the cellular basis for perforin dysfunctions in hemophagocytic lymphohistiocytosis and provides the means for studying structure–function relationships for lymphocyte perforin.
Keywords: HLH, cytotoxic granule, NK cell, CTL, immunodeficiency
Introduction
Perforin, a membrane-disruptive protein secreted by cytotoxic T lymphocytes and natural killer cells (collectively cytotoxic lymphocytes [CLs]), is essential for the death of virus-infected or transformed cells targeted for destruction through the granule exocytosis pathway (1–6). Perforin-deficient mice and humans are severely immunosuppressed. Perforin-null mice lack resistance to many viruses and intracellular pathogens such as Listeria monocytogenes (1). Their suppression of many experimental metastases (7) and carcinogen-induced tumors (8) is also deficient, and more than half develop spontaneous, highly aggressive B cell lymphomas as they age (9), indicating a fatal lapse of tumor immune surveillance (10).
A syndrome of perforin deficiency was recently described in humans, in that ∼30% of immunodeficient children presenting with the autosomal recessive disorder hemophagocytic lymphohistiocytosis (HLH) carry mutations in both their perforin alleles (11–13). The CLs of these children are unable to impart a lethal hit to target cells and thus fail to clear APCs, resulting in uncontrolled activation and accumulation of macrophages and overproduction of proinflammatory cytokines. Clinically, this is manifested as fever, liver and spleen enlargement, and hemophagocytosis in the spleen, liver, and bone marrow (14). The CLs of these patients show a marked reduction of perforin content, which is thought to indicate failed expression, inaccurate trafficking and/or instability of mutated perforin. Overall, the clinical and pathological findings in HLH are strikingly reminiscent of perforin-deficient mice infected with lymphocytic choriomeningitis virus, as these animals also show expansion of T- and antigen-presenting cell numbers and are unable to down-regulate the immune response (10).
Perforin is released from the secretory granules of CLs with granzymes, a family of proapoptotic serine proteases, and is thought to mediate their trafficking to the target cell cytosol where the proteolytic activity of granzymes imposes apoptotic cell death (15). Purified perforin applied alone at high concentration can also induce osmotic lysis, which may also occur under some physiologically relevant conditions (15). However, at the molecular and cellular levels very little is known of how perforin achieves its functions. This sizeable gap in our knowledge exists largely because it has not been possible to develop robust prokaryotic or eukaryotic models for expressing perforin or its putative domains. Some years ago, it was shown that rat basophil leukemia (RBL) cells transfected to express perforin acquired the capacity to lyse erythrocytes (16), whereas cells that coexpressed perforin and granzyme B were able to kill some nucleated target cells (17). However, these RBL transfectants failed to express perforin in sufficient quantity, or expression was too unstable to enable ongoing studies. A further study reported expression and partial purification of perforin expressed in baculovirus-infected insect cells (18); however, the yield was low and the recovered perforin was denatured and had poor activity.
In the current study, we revisited perforin expression in RBL cells and developed robust expression systems to investigate, for the first time, the molecular and cellular basis for perforin dysfunction associated with two missense mutations reported in an HLH patient. Our results indicate the feasibility of using RBL-based and other perforin expression modalities to study both the basis of the HLH immune deficiency and, more broadly, structure–function relationships for perforin.
Materials and Methods
Cell Culture.
The cell lines RBL-2H3 cells (rat basophil leukemia; American Type Culture Collection), which will be referred to in the text as RBL, and 293T (human embryonic kidney) were maintained in DMEM medium supplemented with 10% FCS, 2 mM glutamine, and 100 μg/ml each of streptomycin and penicillin in a humidified incubator at 37°C. Jurkat T cells were maintained in RPMI-1640 medium supplemented as above. RBL and 293T cells were detached from culture flasks using trypsin-EDTA solution (CSL Ltd.) at 37°C.
Transient Transfection of RBL Cells.
Mature human and mouse perforin each have 534 amino acids. However, the leader sequence of human perforin is one amino acid longer than that of the mouse. This results in a difference in conventional amino acid numbering such that amino acids at positions 225 and 429 mutated in HLH Patient #5 (11) correspond to residues 224 and 428 in the mouse protein, as noted throughout Results and Discussion. Importantly, arginine 225 is a nonconserved residue with threonine being present in mouse perforin. To demonstrate the equivalence of arginine and threonine at this position, we generated the T224R variant and, subsequently, the T224W mutant, which corresponds to R225W in Patient #5 (11). The mutations were introduced using the Transformer (Stratagene) site-directed mutagenesis system according to the manufacturer's instructions. The resultant and the WT cDNA were cloned into the pIRES2-EGFP expression vector (CLONTECH Laboratories, Inc.). Fcɛ receptor–expressing RBL cells were grown to near confluence in 175-cm2 flasks, harvested, washed twice, and resuspended at 108 cells/ml in serum-free DMEM. 200 μL of the cell suspension was mixed with 20 μg pIRES2-EGFP containing the WT or mutated perforin cDNA or vector DNA alone, incubated at room temperature for 10 min, and electroporated in 4-mm electroporation cuvettes and Bio-Rad Laboratories pulser at 500 μF and 0.25 V. After 10 min at RT, the cells were transferred into complete DMEM. Cells were harvested 18–20 h later, and GFP-expressing cells were sorted by flow cytometry (FACStar; Beckton Dickinson).
Generation of Recombinant Retroviruses and Stable Expression of Perforin in RBL Cells.
The missense perforin mutation, G428E, corresponding to the human G429E (identified in another perforin allele in Patient #5; [reference 11]), was generated using the Quick-Change site–directed mutagenesis system (Stratagene) according to the manufacturer's instructions. The cDNAs encoding mouse WT and G428E perforin were subcloned into the retroviral expression vector MSCV, which contains an internal ribosome entry site for GFP expression (19). For retroviral transduction of RBL cells, viral supernatant was generated by cotransfecting the MSCV plasmids with an amphotropic packaging plasmid into 293T cells by calcium phosphate precipitation. After 48 h, the viral supernatant was harvested and added to RBL cells every 12 h for 3 d. The population of cells with the greatest GFP expression (up to 5% of total cells) was subsequently purified by flow cytometry and analyzed for perforin expression.
Assessing the Cytotoxicity of Transfected RBL Cells.
The cytotoxic capacity of RBL cells was analyzed using Jurkat T cell targets in a 4-h 51Cr release assay as described previously (16). Briefly, the surface of 51Cr-labeled Jurkat cells was derivatized with a 1 mM solution of trinitrobenzosulfonic acid in PBS, (pH 7.4) for 15 min at 37°C and washed with unsupplemented DMEM three times. The transfected RBL cells were harvested and incubated with antitrinitrophenol IgE mAb (2 μg/ml) at 37°C for 15 min and washed with unsupplemented DMEM three times. RBL and Jurkat cells were coincubated at various effector to target (E:T) ratios at 37°C in 200 μl serum-free DMEM supplemented with 1% BSA for 4 h in 96-well plates. The supernatant was then harvested and the released 51Cr measured in a gamma counter. The total 51Cr content of Jurkat cells was estimated using 5% Triton X-100–lysed cells. The percentage-specific chromium release was calculated as 100× ([experimental release − spontaneous release]/[total release – spontaneous release]) and is shown as mean ± SD.
Isolation of Lysosomal Granules from RBL Cells.
Perforin was isolated from 109 stably expressing RBL cells by nitrogen cavitation and Percoll density fractionation as described (20). To distinguish granule-enriched fractions from other subcellular fractions, the activity of the RBL granule marker enzyme, β-hexosaminidase, was measured as follows. 50 μL of each fraction was mixed with 30 μl 8 mM p-nitrophenyl N-acetyl-β-d-glucosaminide (Sigma-Aldrich) and 10 μl 0.5 M sodium acetate, pH 5.0, at RT for 30 min. The reaction was stopped by adding 150 μl 50 mM NaOH, and the absorbance was measured at 405 nm.
Expression of Recombinant Perforin and Membrane-binding Assay.
Perforin cDNA was cloned into the pFastBac vector and overexpressed in Sf-21 cells using a Back-to-Back kit (Invitrogen) and perforin was purified, all according to the manufacturer's instructions. Small amounts of recombinant WT and the G428E perforin mutant protein were obtained. To study the calcium-dependent membrane binding of perforin, 2 × 107 sheep RBCs were resuspended in 200 μl 20 mM Hepes-150 mM NaCl buffer (pH 7.4) supplemented with 1 mM CaCl2. An aliquot of the purified perforin was added to the cell suspension for 5 min on ice. The cells were pelleted at 16,000 g for 10 s, the supernatant promptly removed, and the cells lysed in ice cold water. The lysate was centrifuged for 20 min at 16,000 g at 4°C. The pellet was washed once, dissolved in SDS-PAGE loading buffer, and analyzed by Western blotting.
Immunoperoxidase Staining.
Approximately 1,000 RBL cells were seeded in each well of an 8-well chamber slide 1 d before staining and cultured overnight. In some experiments, cells were induced to undergo degranulation by transient incubation with TNP-labeled tumor target cells. The RBL cells were fixed for 10 min at RT in 3.7% paraformaldehyde, washed three times in PBS, permeabilized in 0.1% Triton X-100, 0.5% BSA for 5 min, and then washed as before. The cells were treated with periodic acid (0.5%) for 10 min, and endogenous peroxidase activity was quenched with 0.3% H2O2 for 15 min. Blocking buffer (1% BSA/1% skim milk powder in PBS) was added for 30 min before the rat antiperforin mAb P1-8 (21). Bound Ig was detected with biotinylated donkey anti–rat IgG (Jackson ImmunoResearch Laboratories), streptavidin-HRP (Dako) for 10 min, and the chromogen diaminobenzidine (Dako). Finally, cells were counterstained with eosin and viewed by light microscopy.
Western Blotting.
Cell lysates from stable or transiently transfected RBL cells or granule extracts were resolved on 10% SDS-PAGE (Tris-Glycine) gels, transferred to PVDF membranes, and assayed for perforin content using rat antiperforin mAb PI-8 (provided by Dr. Hideo Yagita, Juntendo Univeristy, Tokyo, Japan) (21) and anti–rat HRP-conjugated Ig. The signal was detected using chemiluminescence (Amersham Biosciences).
Results and Discussion
In ∼30% of cases, the severe immunodeficiency of HLH has been attributed to mutations in the PFN1 gene (11–13). Although mutations that cause a frame-shift or premature termination would be expected to abrogate perforin function, others result in single amino acid substitutions whose effect on perforin function is unknown. The appreciation that perforin mutations can result in immune dysfunction offers a unique opportunity to characterize perforin structure–function relationships. To date, however, such studies have been greatly hindered by a lack of appropriate methodologies for expressing perforin in cells capable of granule-mediated cytotoxicity. Perforin's capacity to disrupt lipid membranes means that few cell lines can accurately process and store perforin in secretory vesicles and then release it in a regulated fashion after conjugate formation. No appropriate human cell lines exist to attempt such an analysis. However, standard transfection methodologies in RBL cells have shown previously that perforin expression confers the capacity to lyse erythrocytes (16), whereas coexpression of perforin and either granzyme A or B was necessary to effectively kill nucleated cells (17). Despite this, perforin expression was not sufficiently stable in RBL cells to permit ongoing studies. To address these issues, we revisited perforin expression in RBL cells but applied efficient expression and cell sorting technologies to compare the function of WT and mutated perforin. For our analysis, we chose to study HLH Patient #5, a compound heterozygote with perforin missense mutations R225W and G429E (11,12). The R225W mutation has also been identified in several unrelated individuals (13). We had shown previously that overexpressing human perforin in rodent CTLs could disrupt the processing of endogenous perforin, suggesting that human and rodent perforin may be processed in subtly different ways (22). Therefore, we introduced mutations equivalent to those of P5 into mouse perforin and compared the function of these molecules with WT mouse perforin.
We initially devised a rapid method for transient transfection of RBL cells, using the vector pIRES2-EGFP. 1 d after electroporation, fluorescent cells were sorted and immediately used in a 51Cr release assay with Jurkat target cells to which they were conjugated. The efficiency of electroporation was as high as 40%, and up to 106 GFP-expressing cells were obtained per electroporation. Although G429 is conserved in human, mouse, and rat perforin, R225 is not invariant and corresponds to T224 in mouse perforin. To confirm the functional equivalence of arginine and threonine at this position, we generated RBL cells expressing T224R mouse perforin and found they were as efficient in the 51Cr release assay as WT perforin-transfected cells. However, expressing perforin with tryptophan at the same position (T224W) resulted in complete loss of cytolytic function (Fig. 1). As expected, the WT protein had an apparent molecular mass of ∼67 kD; however, the introduction of tryptophan resulted in the appearance of truncated (∼45 kD) perforin (Fig. 1), suggesting the mutation facilitated proteolytic cleavage/ processing of perforin. Furthermore, immunohistochemistry analysis of transfected cells indicated mislocalisation of T224W, possibly due to a loss of putative signaling motif(s). Whereas WT perforin produced a punctate appearance consistent with packaging in secretory granules, T224W perforin produced diffuse staining throughout the RBL cell cytoplasm (Fig. 2 A).
Figure 1.

Reduced cytotoxic activity and truncation of T224W mouse perforin expressed in RBL cells. Perforin-dependent 51Cr release from TNP-labeled Jurkat cells coincubated with transiently transfected, sorted RBL cells for 4 h in the presence of anti-TNP IgE. The data points are shown as the mean ± SD of triplicate samples and are representative of three similar assays. The Western blot (right) shows truncation of T224W perforin expressed in two independent transfection experiments (T224W-1 and T224W-2) compared with WT and T224R perforin.
Figure 2.
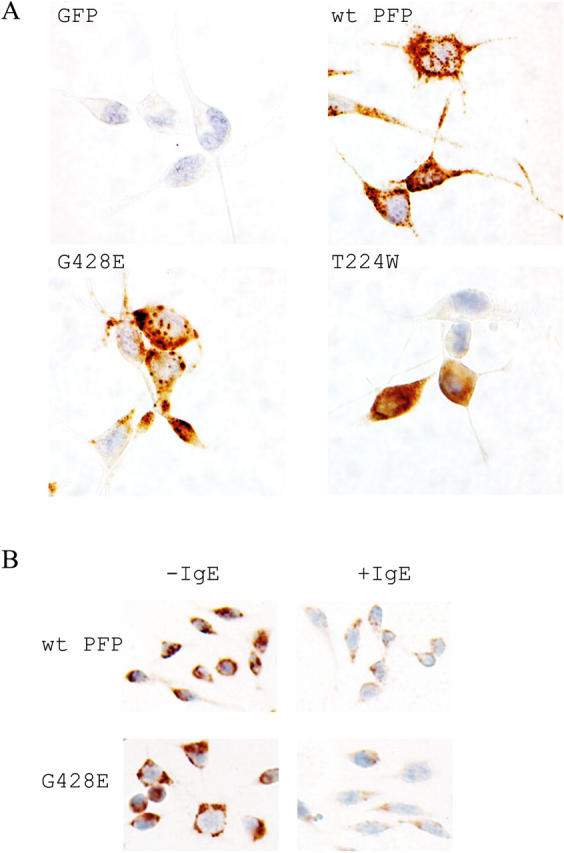
T224W and G428E perforin localize differently in RBL cells. (A) Immunohistochemistry of perforin-expressing RBL cells demonstrated with antiperforin antibody PI-8 and counterstained with eosin. (B) RBL cells either unlabeled or labeled with α-TNP–IgE were stained as in A, after degranulation was induced by transient incubation with TNP-labeled target cells. Magnification, 400×.
When we similarly analyzed the effect of the G428E (G429E in humans) mutation co-inherited by Patient #5 (11), we observed a reduced level of 51Cr release compared with RBL cells expressing WT perforin (unpublished data). To accurately quantify this reduced activity, we produced cell lines that stably expressed WT and G428E perforin. Retrovirus-transduced RBL cells were analyzed on a flow cytometer, and the most highly fluorescent cells (0.2–5% of the total population) were sorted and expanded in culture resulting in >93% GFP-positive cells some days later. These cells expressed perforin at levels equivalent to IL-18/IL-21–activated mouse primary NK cells (Fig. 3 A). Perforin expression and cytotoxic function remained stable over many weeks of continuous culture (unpublished data). Consistent with our transient transfection experiments, RBL cells expressing WT perforin were efficient in lysing Jurkat target cells across a broad range of E:T ratios (Fig. 3 B). To determine the difference in cytolytic activity between WT and G428E perforin, the E:T ratios required to produce equivalent levels of 51Cr release were compared. We found that RBL cells expressing similar levels of G428E were three to four times less efficient at inducing chromium release (Fig. 3 B).
Figure 3.

Reduced cytotoxic activity but normal apparent molecular mass of G428E mouse perforin expressed in RBL cells. (A) Western blot showing perforin expression in stably transduced RBL cells compared with IL18/IL-21–activated mouse NK cells and empty vector-expressing cells (GFP). (B) Perforin-dependent 51Cr release from TNP-labeled Jurkat cells coincubated for 4 h in the presence of anti-TNP IgE with RBL cells stably expressing WT or G428E perforin. The data points are shown as the mean ± SD of three independent experiments. The Western blot (right) shows that G428E comigrates with WT perforin. GFP is the empty vector control. (C) RBL cells stably overexpressing WT or G428E perforin or the empty vector (GFP) were lysed and fractionated on a Percoll density gradient. Fractions were then analyzed for their perforin content by Western blotting and their β-hexosaminidase activity (as described in Materials and Methods).
We went on to investigate the reason for the reduced cytotoxicity of G428E perforin. As demonstrated by immunoblotting (Fig. 3 B), this was not due to protein cleavage or degradation. To rule out incorrect trafficking to secretory granules, we examined the intracellular localization of WT and G482E perforin in stably transduced RBL cells. Finding normal quantities of mutated perforin in the granules would further exclude a significant defect in gene transcription, mRNA stability or translation, or protein folding. When lysates of RBL cells expressing WT or G428E perforin were fractionated on a Percoll gradient and analyzed by Western blot, perforin was consistently localized in the fractions containing maximal β-hexosamidase activity, a marker of the lysosome-like secretory granules (21) (Fig. 3 C). The correct targeting of perforin was also confirmed through immunohistochemical staining, as both WT and G428E perforin demonstrated indistinguishable punctate cytoplasmic staining (Fig. 2 A). G428E perforin was also released by exocytosis as efficiently as WT perforin upon RBL Fcɛ receptor cross-linking (Fig. 2 B).
Since G428E perforin was expressed at equivalent levels to WT perforin and correctly targeted to, and released from, granules (Fig. 2 and Fig. 3, B and C), the mutation was likely to affect a postsynaptic function of perforin. To test this possibility, we generated and purified recombinant WT and G428E perforin using a baculovirus expression system and tested their ability to bind to sheep RBC membranes in a calcium-dependent manner. Whereas WT perforin displayed strong calcium-dependent plasma membrane binding with essentially all the added perforin bound, the binding of G428E perforin was markedly reduced (Fig. 4). Consistent with this observation, the cytolytic activity of the recombinant G428E mutant was ∼5% of that of WT perforin (unpublished data). Although RBL cells have been used as a read-out of perforin function for many years, a perceived weakness of the model is that perforin exerts its cytolytic effects in the absence of granzyme B. Exposure of target cells to recombinant G428E-perforin with granzyme B did not rescue the perforin phenotype (unpublished data). Therefore, our findings strongly suggested that the diminished activity of G428E-perforin was due to diminished target cell membrane binding, rather than the absence of granzymes. Based on modeling studies and biochemical analysis of CTL clones, Uellner and colleagues found that perforin's COOH terminus strongly resembles that of calcium-dependent membrane-binding C2 family of protein domains, some of which are involved in vesicular trafficking at neuronal synapses (23). Importantly, G428 is located in the C2 domain of perforin, immediately adjacent to one of five highly conserved aspartate residues believed to be essential for calcium binding (23). We postulate that the G428E substitution may interfere with calcium binding to the C2 domain, leading to decreased affinity of perforin for lipids in the target cell membrane (23). The apparent quantitative differences in G428E-perforin–induced cytolysis in the RBL-based and recombinant models probably reflects differences in the kinetics of perforin–target cell membrane interaction in the context of a cell conjugate (RBL model) and in solution (purified recombinant perforin).
Figure 4.

The G428E mutation significantly reduces calcium-dependent membrane binding of soluble perforin. Equal quantities of recombinant WT and mutant perforin were tested for their capacity to bind to sheep erythrocytes in the absence (−) or presence (+) of 1 mM CaCl2. The total input of perforin in each case is shown as C.
This is the first study to successfully define the functional basis of naturally occurring perforin mutations that when co-inherited, lead to the catastrophic immunosuppression seen in HLH. Surprisingly, we demonstrated that partial loss of perforin function may be sufficient to bring about fatal disease. Whereas the T224W mutation (corresponding to R225W in humans) resulted in protein instability and complete loss of RBL cytotoxic function, G428E (G429E in humans) was only partially inactivating as RBL cells retained ∼25–30% of WT lytic activity. It is not clear whether this substitution would lead to disease when inherited in the homozygous state; however, when combined with the completely inactive R225W allele, the overall cytolytic activity of perforin appears to be insufficient to rescue the patient. Based on the result of our RBL assays, one could predict that CTL expressing equal quantities of T224W- and G428E-perforin would have some residual but markedly reduced cytotoxic activity. In fact, the NK cells of Patient #5 did exhibit ∼15% lytic activity of control samples (11,12). The concordance of our data with the clinical findings in this case provides evidence that our experimental approaches should provide a robust basis for understanding other perforin mutations identified in HLH.
Acknowledgments
We thank Dr. Morgan Wallace (Peter MacCallum Cancer Center) for purified mouse NK cells.
This work was supported by program grants from the National Health and Medical Research Council (NHMRC), Australia (to M.J. Smyth and J.A. Trapani) and the Juvenile Diabetes Research Foundation (to J.A.T). M.J. Smyth, P.K. Darcy, and J.A. Trapani also receive fellowships from NHMRC.
The authors have no conflicting financial interests.
I. Voskoboinik and M.-C. Thia contributed equally to this work.
References
- 1.Trapani, J.A. 1998. Dual mechanisms of apoptosis induction by cytotoxic lymphocytes. Int. Rev. Cytol. 182:111–192. [DOI] [PubMed] [Google Scholar]
- 2.Masson, D., and J. Tschopp. 1985. Isolation of a lytic, pore-forming protein (perforin) from cytolytic T-lymphocytes. J. Biol. Chem. 260:9069–9072. [PubMed] [Google Scholar]
- 3.Young, J.D., Z.A. Cohn, and E.R. Podack. 1986. The ninth component of complement and the pore-forming protein (perforin 1) from cytotoxic T cells: structural, immunological, and functional similarities. Science. 233:184–190. [DOI] [PubMed] [Google Scholar]
- 4.Podack, E.R., and G. Dennert. 1983. Assembly of two types of tubules with putative cytolytic function by cloned natural killer cells. Nature. 302:442–445. [DOI] [PubMed] [Google Scholar]
- 5.Dennert, G., and E.R. Podack. 1983. Cytolysis by H-2-specific T killer cells. Assembly of tubular complexes on target membranes. J. Exp. Med. 157:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millard, P.J., M.P. Henkart, C.W. Reynolds, and P.A. Henkart. 1984. Purification and properties of cytoplasmic granules from cytotoxic rat LGL tumors. J. Immunol. 132:3197–3204. [PubMed] [Google Scholar]
- 7.Smyth, M.J., K.Y. Thia, E. Cretney, J.M. Kelly, M.B. Snook, C.A. Forbes, and A.A. Scalzo. 1999. Perforin is a major contributor to NK cell control of tumor metastasis. J. Immunol. 162:6658–6662. [PubMed] [Google Scholar]
- 8.Street, S.E., E. Cretney, and M.J. Smyth. 2001. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood. 97:192–197. [DOI] [PubMed] [Google Scholar]
- 9.Smyth, M.J., K.Y. Thia, S.E. Street, D. MacGregor, D.I. Godfrey, and J.A. Trapani. 2000. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 192:755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapani, J.A., and M.J. Smyth. 2002. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2:735–747. [DOI] [PubMed] [Google Scholar]
- 11.Stepp, S.E., R. Dufourcq-Lagelouse, F. Le Deist, S. Bhawan, S. Certain, P.A. Mathew, J.I. Henter, M. Bennett, A. Fischer, G. de Saint Basile, et al. 1999. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 286:1957–1959. [DOI] [PubMed] [Google Scholar]
- 12.Feldmann, J., F. Le Deist, M. Ouachee-Chardin, S. Certain, S. Alexander, P. Quartier, E. Haddad, N. Wulffraat, J.L. Casanova, S. Blanche, et al. 2002. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphohistiocytosis. Br. J. Haematol. 117:965–972. [DOI] [PubMed] [Google Scholar]
- 13.Molleran Lee, S., J. Villanueva, J. Sumegi, K. Zhang, K. Kogawa, J. Davis, and A.H. Filipovich. 2004. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J. Med. Genet. 41:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henter, J.I., M. Arico, G. Elinder, S. Imashuku, and G. Janka. 1998. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol. Oncol. Clin. North Am. 12:417–433. [DOI] [PubMed] [Google Scholar]
- 15.Browne, K.A., E. Blink, V.R. Sutton, C.J. Froelich, D.A. Jans, and J.A. Trapani. 1999. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol. Cell. Biol. 19:8604–8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shiver, J.W., and P.A. Henkart. 1991. A noncytotoxic mast cell tumor line exhibits potent IgE-dependent cytotoxicity after transfection with the cytolysin/perforin gene. Cell. 64:1175–1181. [DOI] [PubMed] [Google Scholar]
- 17.Nakajima, H., H.L. Park, and P.A. Henkart. 1995. Synergistic roles of granzymes A and B in mediating target cell death by rat basophilic leukemia mast cell tumors also expressing cytolysin/perforin. J. Exp. Med. 181:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu, C.C., P.M. Persechini, and J.D. Young. 1994. Characterization of recombinant mouse perforin expressed in insect cells using the baculovirus system. Biochem. Biophys. Res. Commun. 201:318–325. [DOI] [PubMed] [Google Scholar]
- 19.Zhou, W., D.R. Clouston, X. Wang, L. Cerruti, J.M. Cunningham, and S.M. Jane. 2000. Induction of human fetal globin gene expression by a novel erythroid factor, NF-E4. Mol. Cell. Biol. 20:7662–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis, J.E., V.R. Sutton, K.A. Browne, and J.A. Trapani. 2003. Purification of natural killer cell cytotoxic granules for assaying target cell apoptosis. J. Immunol. Methods. 276:59–68. [DOI] [PubMed] [Google Scholar]
- 21.Kawasaki, A., Y. Shinkai, Y. Kuwana, A. Furuya, Y. Iigo, N. Hanai, S. Itoh, H. Yagita, and K. Okumura. 1990. Perforin, a pore-forming protein detectable by monoclonal antibodies, is a functional marker for killer cells. Int. Immunol. 2:677–684. [DOI] [PubMed] [Google Scholar]
- 22.Thia, K.Y., M.J. Smyth, and J.A. Trapani. 1993. Expression of human perforin in a mouse cytotoxic T lymphocyte cell line: evidence for perturbation of granule-mediated cytotoxicity. J. Leukoc. Biol. 54:528–533. [DOI] [PubMed] [Google Scholar]
- 23.Uellner, R., M.J. Zvelebil, J. Hopkins, J. Jones, L.K. MacDougall, B.P. Morgan, E. Podack, M.D. Waterfield, and G.M. Griffiths. 1997. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J. 16:7287–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]