

Abstract
Natural killer (NK) cells express multiple activating receptors that initiate signaling cascades through DAP10- or immunoreceptor tyrosine-based activation motif–containing adapters, including DAP12 and FcRγ. Among downstream signaling mediators, the guanine nucleotide exchange factor Vav1 carries out a key role in activation. However, whether Vav1 regulates only some or all NK cell–activating pathways is matter of debate. It is also possible that two other Vav family molecules, Vav2 and Vav3, are involved in NK cell activation. Here, we examine the relative contribution of each of these exchange factors to NK cell–mediated cytotoxicity using mice lacking one, two, or all three Vav proteins. We found that Vav1 deficiency is sufficient to disrupt DAP10-mediated cytotoxicity, whereas lack of Vav2 and Vav3 profoundly impairs FcRγ- and DAP12-mediated cytotoxicity. Our results provide evidence that these three Vav proteins function specifically in distinct pathways that trigger NK cell cytotoxicity.
Keywords: DAP12, NKG2D, adapters, GEF, FcRγ
Introduction
NK cells express multiple activating receptors with diverse structures and specificities. In mice, prototypic receptors include NKG2D, Ly49D, Ly49H, and the low affinity receptor for IgG, FcγRIIIa. NKG2D recognizes endogenous MHC class I–related molecules including, the retinoic acid–inducible proteins Rae1α-ɛ, the minor histocompatibility antigen H60, and the UL16-binding proteinlike transcript (MULT; references 1, 2). These molecules are expressed at high levels primarily in virally infected and tumor cells (1, 2). Ly49D triggers killing of allogeneic bone marrow cells and Chinese hamster ovary (CHO) cells by recognizing murine and hamster class I molecules (3, 4). Ly49H mediates selective recognition of m157, a murine cytomegalovirus–encoded class I–like molecule that is expressed on infected cells (5, 6). FcγRIIIa promotes lysis of IgG-coated target cells by antibody-dependent, cell-mediated cytotoxicity (ADCC).
NKG2D, Ly49D, Ly49H, and FcγRIIIa lack cytoplasmic signaling elements and can deliver stimulatory signals only by associating with transmembrane adaptor proteins. Ly49D and Ly49H signal through DAP12 (also called KARAP), whereas FcγRIIIa associates with the γ chain of Fc receptors (FcRγ) and CD3ζ (7, 8). DAP12, FcRγ, and CD3ζ contain immunoreceptor tyrosine-based activation motifs (ITAMs) that are phosphorylated and function as docking sites for Syk and ZAP70 protein tyrosine kinases (7, 8). NKG2D includes two isoforms that differ in their cytoplasmic domains and associated adaptors (9, 10). NKG2D-long (NKG2D-L) signals through DAP10, a unique adaptor containing a YxNM motif that recruits phosphatidylinositol-3-kinase (PI-3K; reference 11) and Grb-2. NKG2D-short (NKG2D-S) associates with both DAP10 and DAP12 (9, 10). Syk, ZAP70, and PI-3K initiate signaling cascades that lead to activation of mitogen-activated protein kinases (MEKs), phospholipase C (PLC)γ, Ca2+ mobilization, secretion of IFN-γ, and release of cytotoxic granules that contain perforin and granzymes (7, 8).
Vav proteins are critical signaling mediators that connect protein tyrosine and inositol kinases with effector responses in NK cells (12, 13). Vav proteins include three closely related guanine nucleotide exchange factors (Vav1, Vav2, and Vav3) that activate small GTP-binding proteins belonging to the Rho-Rac/Cdc42 family, including Rac1, RhoA, and RhoG (14). Rac1 sequentially activates p21-activated kinase 1 (PAK1), MEK, and extracellular signal-regulated kinase (ERK)1/2. In addition, Vav proteins perform adaptor functions by binding the Src homology 2 domain-containing leukocyte phosphoprotein (SLP)76, which is part of a complex including PLCγ and the linker for activation of T cells (LAT; references 14, 15). In vitro studies demonstrated that Vav1, Vav2, and Rac1 are activated in human NK cells after challenge with tumor cells (16, 17) or engagement of FcγRIIIa (17), integrins (18, 19), and NKG2D (20) with cognate ligands or specific antibodies. Moreover, blocking Vav1 or Rac1 function using antisense oligonucleotides for Vav1 or a dominant-negative form of Rac1 inhibits NK cell adhesion to target cells, cytotoxicity and secretion of IFN-γ (16, 17, 20). Consistent with these results, in vivo studies demonstrated that Vav1 deficiency reduces “natural” cytotoxicity, the ability of NK cells to lyse various tumor cells (21, 22). However, the impact of Vav1 deficiency on ADCC was controversial (21, 22) and INF-γ secretion was not affected in Vav1ko mice (21), suggesting that Vav1 may not regulate all NK cell effector functions. The possible involvement of other Vav family members in NK cell activation has not been addressed in vivo.
Here, we investigate the requirement for Vav proteins in cytotoxicity mediated by several different activating NK cell receptors using mice lacking one, two, or three Vav proteins. We show that lack of Vav1 primarily impairs the NKG2D–DAP10 cytolytic pathway, whereas lack of Vav2 and Vav3 reduces cytotoxicity triggered by receptors that signal through ITAM-containing adapters. Our findings demonstrate an unexpected specialization of Vav proteins in regulating different cytotoxic pathways.
Materials and Methods
Mice.
Vav1ko, Vav2ko, Vav3ko, Vav2,3ko, Vav1,2,3ko, DAP10ko, and DAP12ko mice have been described previously (10, 23).
RNase Protection Assay (RPA).
Riboprobes included cDNA fragments of murine Vav1 (nucleotide [nt] 664–862), Vav2 (nt 652–800), and Vav3 (nt 676–852). RPA was performed by standard techniques.
Cytotoxicity Assays.
NK cells were purified from spleens with DX5 microbeads (Miltenyi Biotec), cultured for different periods of time in RPMI 1640 with 10% heat-inactivated FCS (Hyclone) and 1,000 U/ml rIL-2 (Roche) and tested against target cells by standard 51Cr release assay. Target cells used to assess natural cytotoxicity included YAC-1, CHO, RMAS, RMAS-Rae1γ (10), Baf3, and Baf3-m157 (6). ADCC was evaluated using EL-4 cells coated with the anti-Thy1.2 antibody TIB-107 (American Type Culture Collection). Cytotoxicity was blocked with antibodies against Ly49D (4), Ly49H (6), or NKG2D (10). In some experiments, NK cells were pretreated for 20 min at 37°C with the MEK inhibitor PD98059 (Calbiochem) before the addition of antibodies and 51Cr-labeled cells.
Biochemical Analysis.
WT, DAP10ko, DAP12ko, Vav1ko, and Vav1,2,3ko NK cells were cultured for different periods of time in IL-2. After incubation with mAb HB-197 (Fc block; American Type Culture Collection) for 15 min on ice to block Fc receptors, NK cells (4 × 106/sample) were treated with biotinylated anti–mouse NKG2D (1 μg/106 cells) for 15 min on ice followed by streptavidin (1.51 μg/106 cells) at 37°C for 2 and 5 min. Alternatively, NK cells were conjugated at 37°C for 5 min with CHO cells. NK cell lysates were analyzed by SDS-PAGE and immunoblotting using antibodies against pERK1/2 (Thr202/Tyr204) and ERK1/2 (New England Biolabs Inc.).
Online Supplemental Material.
In Fig. S1, Vav1, 2, and 3 are expressed in NK cells, but are not required for NK cell maturation. Fig. S2 shows NKG2D-mediated cytotoxicity in Vav1ko and DAP10ko NK cells. Fig. S3 depicts lysis of RMA-S/Rae1γ and YAC-1 cells by Vav1,2,3ko and Vav2,3ko NK cells ex vivo and early after IL-2 activation. In Fig. S4, Vav deficiency does not affect conjugate formation between WT, Vav-deficient NK cells, and target cells. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20031847/DC1.
Results and Discussion
Vav1, Vav2, and Vav3 Are Expressed in NK Cells, but Are Not Required for NK Cell Maturation and Cell Surface Expression of Activating and Inhibitory Receptors.
To determine which vav genes are expressed in NK cells, we performed a quantitative RPA with riboprobes specific for individual Vav transcripts. All three vav genes were expressed in primary mouse NK cells. Vav1 and Vav3 transcripts were the most abundant, whereas the vav2 gene was expressed at low levels (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20031847/DC1). Consistent with the results of RPA analyses, Vav1 and Vav3 proteins were expressed at very high levels in IL-2–cultured NK cells, whereas the Vav2 protein was less abundant (Fig. S1 B). NK cell development is normal in mice lacking Vav1 (21). Despite severe defects in the production of mature T and B lymphocytes in the periphery (23), healthy Vav1,2,3ko mice (4–5 wk old) harbored normal NK cell numbers in the spleen, as did Vav1ko mice (Fig. S1 C). In addition, Vav1,2,3ko and WT NK cells expressed comparable levels of DX5, IL-2/15Rβ (CD122), NK1.1, NKG2D, Mac-1 (CD11b), and NKG2A on the cell surface and similar percentages of the NK populations expressed Ly49A, Ly49D, and Ly49H (Fig. S1 D and not depicted). Thus, none of the Vav proteins appear to be essential for NK cell maturation or surface expression of activating or inhibitory NK cell receptors.
Vav1 Protein Is Not Necessary for FcRγ- and DAP12-mediated Cytotoxicity.
To determine the function of Vav1 protein in ITAM-dependent signaling, we examined ADCC as well as Ly49H/DAP12- and Ly49D/DAP12-mediated cytotoxicity in Vav1ko and WT mice. Vav1ko and WT NK cells killed EL-4 cells coated with an anti-Thy1.2 antibody equally well (Fig. 1 A). Similarly, Vav1ko NK cells killed Baf3 cells expressing m157 (Baf3-m157; references 5, 6), but not Baf3 cells transfected with a control plasmid (Fig. 1 B). Finally, Vav1-deficient NK cells efficiently lysed CHO cells in a Ly49D-mediated manner (Fig. 1 C). We conclude that FcRIIIA/FcRγ-, Ly49H/DAP12-, and Ly49D/DAP12-mediated cytotoxicity are largely Vav1 independent. Because the DAP12 and FcRγ adapters recruit signaling mediators through cytoplasmic ITAMs, our results indicate that ITAM-mediated cytotoxicity in NK cells does not require Vav1.
Figure 1.
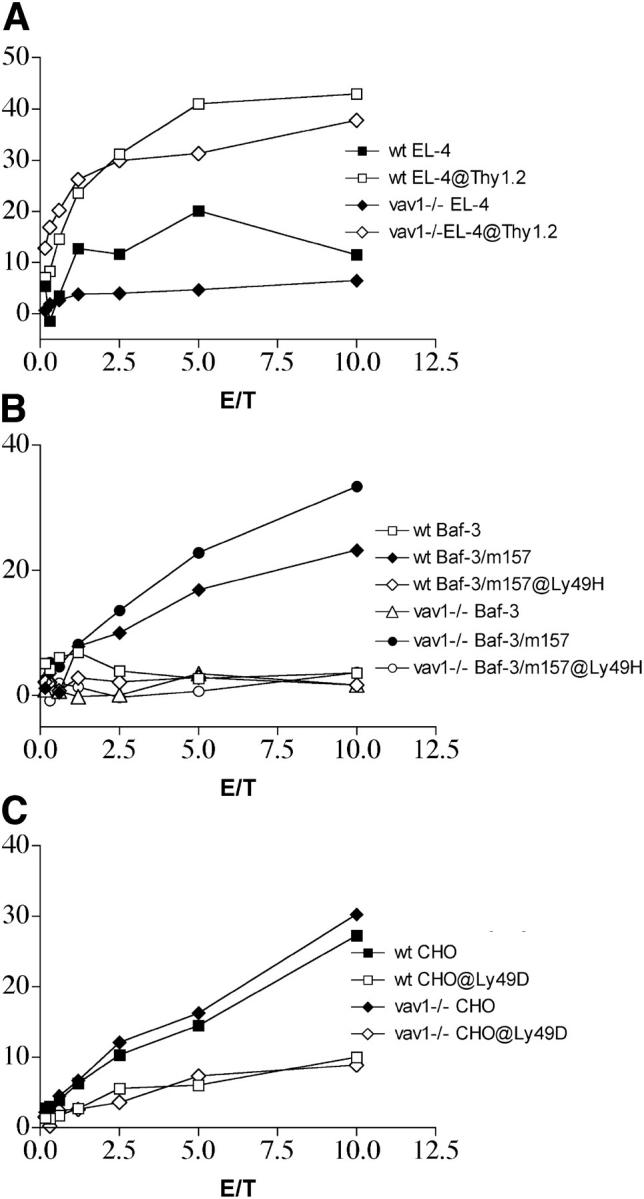
ITAM-dependent cytotoxicity in Vav1ko mice. Cytotoxicity of Vav1ko NK cells was tested against the following: (A) EL-4 cells with or without an anti-Thy1.2 mAb, (B) Baf-3-m157 and mock-transfected Baf-3 with or without a blocking mAb against Ly49H, and (C) CHO cells with or without a blocking anti-Ly49D mAb. NK cells were cultured in IL-2 for 10–15 d, contained >98% NK1.1+/CD3− cells. Vav1ko and WT NK cells exhibited comparable percentages of Ly49H+ and Ly49D+ cells.
Vav1 Protein Is Essential for DAP10-mediated Cytotoxicity.
NK cells express two isoforms of NKG2D: NKG2D-L and NKG2D-S (9). NKG2D-L signals through DAP10, whereas NKG2D-S signals through both DAP12 and DAP10. The relative proportion of NKG2D-L and NKG2D-S in NK cells varies during IL-2 activation. NK cells isolated ex vivo predominantly express NKG2D-L (9), although some NKG2D-S is also present (unpublished data). Early after IL-2 activation (6–10 d), NK cells up-regulate NKG2D-S. After prolonged culture with IL-2 (10–30 d), NKG2D-S expression decreases and NKG2D-L again becomes predominant (9, 10). To determine the function of Vav1 in NKG2D/DAP12- and NKG2D/DAP10-mediated cytotoxicity, we compared cytotoxicity of Vav1ko and WT NK cells at different time points during culture with IL-2. Target cells included RMAS transfectants expressing Rae1γ (RMAS-Rae1γ), control RMAS transfectants, and YAC-1 cells, which express NKG2D ligands and, hence, are lysed to some extent in an NKG2D-dependent fashion (1). All experiments were performed with or without a mAb against NKG2D to precisely define the proportion of target cell lysis mediated by NKG2D. When evaluated for NKG2D-mediated lysis of RMAS-Rae1γ, Vav1ko (day 0) were significantly less efficient than WT NK cells (Fig. 2 A). This difference was less pronounced when Vav1ko were tested during culture with IL-2 (days 6–12), but again became substantial after extended culture (days 12–30; Fig. 2 A). NKG2D-mediated lysis of YAC-1 was also affected by the lack of Vav1 (Fig. 2 B). The severity of this deficit varied in a temporal pattern similar to that observed for lysis of RMAS-Rae1γ, but was less pronounced, especially at day 0. We speculate that the few NKG2D–DAP12 receptor complexes present in fresh NK cells may be sufficient to trigger YAC-1 killing or may synergize with a yet unknown receptor mediating NKG2D-independent lysis of YAC-1. In summary, the IL-2/time-dependent deficit of RMAS-Rae1γ and YAC-1 lysis caused by Vav1 deficiency precisely mirrored the prevalence of NKG2D-L/DAP10 signaling and paralleled that previously observed in DAP10-deficient NK cells (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20031847/DC1, and reference 10). Therefore, we conclude that the NKG2D–DAP10 pathway is largely Vav1 dependent.
Figure 2.

NKG2D-dependent cytotoxicity in Vav1ko mice. Cytolitic activity of Vav1ko NK cells against RMA-S, RMA-S/Rae1γ (A), and YAC-1 (B) in the presence of a blocking anti-NKG2D mAb or control mAb. Effector cells included freshly isolated NK cells (day 0) and NK cells cultured in IL-2 for 6, 15, and 30 d. Reduction of NKG2D-dependent cytotoxicity is particularly evident at day 0 (RMA-S-Rae1γ) and at day 30 (RMA-S-Rae1γ and YAC-1). Note that our RMA-S parental cell line is poorly killed by B6 NK cells although it expresses a low level of MHC class I. Different variants of RMA-S may be more susceptible to NK cell–mediated lysis.
Vav2 and Vav3 Proteins Are Required for FcRγ- and DAP12-mediated Signaling.
Vav2 is expressed in human NK cells and plays a central role in NK cell–mediated cytotoxicity (24). In addition, NK cells express Vav3 (Fig. S1, A and B). Thus, Vav1 may not be required for ITAM signaling due to either preferential use of Vav2 and/or Vav3 in this pathway or simple redundancy of the Vav proteins. To test these hypotheses, we analyzed the impact of individual and combined Vav1, Vav2, and Vav3 deficiencies on NK cell–mediated cytotoxicity. Lack of Vav2 or Vav3 alone did not significantly affect FcRγ-, DAP12-, or DAP10-mediated killing at any time point (unpublished data). In contrast, lack of all Vav proteins completely abrogated ADCC (Fig. 3 A) and severely reduced lysis of Baf3-m157 (Fig. 3 B). However, Vav1,2,3ko NK cells were able to partially lyse Baf3-m157 in a Ly49H-mediated manner at high E:T ratios (15–20%). In addition, Ly49D-mediated lysis of CHO cells by Vav1,2,3ko NK cells remained surprisingly intact (Fig. 3 C). Lack of all three Vav proteins caused a marked reduction in NKG2D-mediated lysis of RMAS-Rae1γ and YAC-1 (Fig. S3, A and B, available at http://www.jem.org/cgi/content/full/jem.20031847/DC1). This defect was more profound than that observed in Vav1ko mice and was evident in resting as well as in IL-2–activated NK cells at any time during culture. We conclude that lack of all three Vav isoforms results in a more substantial defect in NK cell–mediated cytotoxicity than does Vav1 deficiency alone and affects both DAP10-mediated and ITAM-dependent cytotoxicity. The unexpected ability of Vav1,2,3ko NK cells to lyse CHO cells in a Ly49D-mediated manner may suggest that Ly49D/DAP12 signaling is Vav independent. Alternatively, NK cell receptors for CHO cells may include one or more as yet unidentified Vav-independent receptors in addition to Ly49D. These receptors may synergize with Ly49D or provide costimulation to Ly49D, allowing Ly49D signals to reach the threshold required for cytotoxicity.
Figure 3.

ADCC, Ly49H-, and Ly49D-dependent cytotoxicity in Vav1,2,3ko and Vav2,3ko NK cells. Cytotoxicity of Vav1,2,3ko (A and B) and Vav2,3ko (D and E) was tested against EL-4 cells with or without an anti-Thy1.2 mAb as well as Baf-3-m157 and Baf-3. Cytotoxicity of Vav1,2,3ko was also tested against CHO cells with and without a blocking anti-Ly49D antibody. ADCC was abrogated in both Vav1,2,3ko and Vav2,3ko NK cells; Ly49H-dependent killing was severely impaired, whereas Ly49D-mediated CHO killing was considerably conserved (C). NK cells were cultured in IL-2 for 10–15 d. In the CHO killing experiment, Ly49D+ cells represented 35% of WT and 25% of Vav1,2,3ko NK cell cultures.
To determine whether Vav2 and Vav3 proteins are preferentially used for ITAM signaling or Vav proteins are redundant, we tested FcRγ- and DAP12-mediated killing in Vav2/3 double deficient mice. Vav2,3ko NK cells completely lacked the capacity to carry out ADCC (Fig. 3 D) and had an obvious defect in Ly49H-mediated killing (Fig. 3 E). Lysis of RMAS-Rae1γ (Fig. S3 C) and YAC-1 cells (Fig. S3 D) by Vav2,3ko NK cells isolated ex vivo or cultured with IL-2 (short or long periods of time) was variably reduced, probably due to conserved NKG2D/DAP10-mediated signaling and dependent on the relative expression of NKG2D-L/NKG2D-S isoforms in the different NK cell populations tested. We conclude that Vav2/Vav3 and Vav1 are not redundant and that Vav2 and Vav3 proteins specialize in ITAM signaling.
Vav Deficiency Disrupts ERK1/2 Activation Triggered by the DAP10 and DAP12 Pathways.
ERK1/2 activation is critical for promoting NK cell–mediated cytotoxicity (12). Because lack of Vav proteins impairs DAP10- and DAP12-mediated cytotoxicity, we asked the following: (a) do the DAP10 and DAP12 pathways trigger ERK1/2 activation; (b) does the lack of Vav proteins impair DAP10- and/or DAP12-mediated ERK1/2 activation; and (c) is blockade of ERK1/2 activation sufficient to inhibit DAP10- and DAP12-mediated cytotoxicity? To determine whether DAP10 and DAP12 activate ERK1/2, we analyzed ERK1/2 phosphorylation in lysates from WT, DAP10ko, and DAP12ko NK cells stimulated via NKG2D at day 7 of IL-2 culture. Both NKG2D/DAP10 and NKG2D/DAP12 signaling mediated ERK1/2 phosphorylation (Fig. 4 A). We also observed marked ERK1/2 phosphorylation in WT NK cells conjugated with CHO cells, which was partially reduced in DAP12ko NK cells. This provides further evidence that recognition of CHO cells most likely involves receptors other than Ly49D.
Figure 4.

Effect of Vav deficiency on DAP10- and DAP12-dependent activation of ERK. (A) NKG2D–DAP10 and NKG2D–DAP12 pathways activate ERK1/2. WT, DAP10ko, and DAP12ko NK cells were incubated at 37°C for the indicated times with anti-NKG2D mAb (left). WT and DAP12ko NK cells were also conjugated with fixed CHO cells (right). NK cell lysates were analyzed by immunoblotting with antibody against active ERK1/2 (pERK1/2) and anti-ERK1/2. (B) Impact of Vav deficiency on activation of ERK1/2. WT, Vav1ko, and Vav1,2,3ko NK cells (left) were stimulated via NKG2D as described in A. WT and Vav1,2,3ko NK cells were also conjugated with CHO cells (right). NK cell lysates were analyzed as indicated in A. (C) MEK inhibitor PD98059 reduces NKG2D-, FcγRIIIA-, and Ly49H-mediated cytotoxicity. IL-2–activated NK cells were preincubated at 37°C for 30 min with PD98059 (50 μM) or DMSO and tested for cytolytic activity against RMA-S, RMA-S/Rae1γ, EL-4 cells coated with anti-Thy1.2 mAb, Baf-3, and Baf-3/m157.
To determine whether Vav deficiency impairs DAP10- and DAP12-mediated ERK1/2 activation, we cultured WT, Vav1ko, and Vav1,2,3ko NK cells for 7 d in IL-2 to induce expression of both NKG2D-L and NKG2D-S and stimulated them with an antibody specific for NKG2D. ERK1/2 phosphorylation was slightly reduced in Vav1ko NK cells and completely abrogated in Vav1,2,3ko NK cells (Fig. 4 B). In contrast, no defect of ERK1/2 activation was observed when Vav1,2,3ko NK cells were conjugated with CHO cells (Fig. 4 B). Thus, there is a consistent correlation between Vav-dependent changes in ERK1/2 activation and cytotoxicity. Specifically, in Vav1,2,3ko NK cells, diminished cytotoxicity is paralleled by disruption of ERK1/2 activation. Conversely, lysis of CHO cells by Vav1,2,3ko NK cells corresponds with conserved ERK1/2 activation. The substantial preservation of ERK1/2 phosphorylation in Vav1ko NK cells most likely reflects ERK1/2 activation through the Vav2–Vav3 pathway, which mediates DAP12-mediated cytotoxicity.
To address the impact of impaired ERK1/2 activation on DAP10- and ITAM-mediated cytotoxicity, we assessed the effect of a pharmacological inhibitor of MEK on ADCC, lysis of Baf3-m157, and RMAS-Rae1γ by WT NK cells, which selectively express NKG2D-L/DAP10, at day 20 of IL-2 culture. Remarkably, blockade of ERK1/2 dramatically reduced NK cell–mediated cytotoxicity in all cases (Fig. 4 C). Identical results were obtained when we tested DAP10ko and DAP12ko NK cells against RMAS-Rae1γ in the presence of the ERK1/2 inhibitor (unpublished data). Together, these results suggest that ERK1/2 may be important in linking DAP10–Vav1 and ITAM–Vav2,3 signaling pathways with exocytosis of lytic granules. However, other downstream signaling mediators may be also implicated in DAP10/Vav1 and ITAM–Vav2,3-mediated cytotoxicity. For example, Vav1 may promote the recruitment of PLCγ2, which triggers Ca2+ mobilization (20). In addition, Vav proteins may cooperate with PI-3K, which initiates the PI-3K→ Rac1→PAK1→MEK→ERK cascade (13). Although Vav proteins mediate signals that regulate cell adhesion and organization of actin cytoskeleton, conjugation of NK cells with target cells was not affected by Vav deficiency, excluding a role of defective adhesion in impairment of NK cell–mediated lysis (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20031847/DC1).
In summary, we investigated the contribution of Vav1, Vav2, and Vav3 to NK cell cytotoxicity pathways triggered by individual activating receptors. Our results demonstrate that Vav1 is essential for DAP10-mediated cytotoxicity, whereas Vav2 and Vav3 are required for FcRγ and DAP12 signaling. Consistent with this conclusion, lack of Vav1 drastically reduced NKG2D-L/DAP10-mediated cytotoxicity of tumor cells expressing NKG2D ligands, but had no significant effect on ADCC and Ly49D/DAP12-, Ly49H/DAP12-, and NKG2D-S/DAP12-mediated cytotoxicity. In striking contrast, lack of both Vav2 and Vav3 abrogated ADCC and substantially reduced Ly49H/DAP12- and NKG2D-S/DAP12-mediated cytotoxicity. Because neither Vav2ko nor Vav3ko NK cells had obvious cytolitic defects, we suggest that there is functional redundancy between Vav2 and Vav3.
Early in vitro studies on human NK cells had indicated a role for Vav1 in natural cytotoxicity against tumor cells and ADCC (16, 17). Later in vivo studies using Vav1-deficient mice confirmed that Vav1 is important for natural cytotoxicity (21, 22). However, they did not conclusively determine whether Vav1 deficiency globally impairs NK cell–mediated cytotoxicity (21), or selectively affects natural cytotoxicity and maintains ADCC (22). Our observation that Vav1 is not required for ITAM-mediated cytotoxicity corroborates and extends the latter model. It has been proposed that Vav1 may be a crucial target for inhibitory NK cell receptors to prevent NK cell activation and cytotoxicity (25). Our data suggest that, although Vav1 dephosphorylation might inhibit the NKG2D–DAP10-dependent pathway, it would not be sufficient to block all NK cell–activating pathways. Previous studies showed that Vav2 is expressed in human NK cells and enhances their natural and FcγRIIIa-mediated cytotoxicity (24). Our analysis of Vav2,3ko NK cells confirms that Vav2 (and Vav3) performs an essential function in ADCC and extends it to ITAM signaling.
We also analyzed Vav1,2,3ko mice and demonstrated that lack of all three Vav proteins results in a profound reduction in NK cell–mediated cytotoxicity. However, we noted one remarkable exception: Vav1,2,3ko NK cells can efficiently lyse CHO cells. NK cell recognition of CHO cells is partially mediated by the Ly49D–DAP12 receptor complex, which binds the CHO class I molecule Hm1-C4 (4). It is possible that, in contrast with Ly49H/DAP12 and NKG2D-S/DAP12, Ly49D/DAP12 signaling is entirely Vav independent. However, a more plausible explanation is that NK cell recognition of CHO cells depends not only on signals triggered by Ly49D/DAP12, but on multiple signals transduced by distinct receptors, which integrate and synergize to reach a critical activating threshold (8). Some of these CHO cell–specific receptors may be Vav independent. Therefore, even if weakened by the absence of all Vav proteins, residual Ly49D/DAP12 signaling may reach the threshold required for cytotoxicity, if facilitated by concurrent activating signals transduced by Vav-independent receptors. In support of this model, we also observed that the defect of YAC-1 killing by Vav1,2,3ko NK cells goes beyond that expected based on the impairment of NKG2D–DAP10 and NKG2D–DAP12 signaling pathways alone, suggesting that additional YAC-1–specific receptors may integrate with NKG2D signaling to reach the threshold for the release of lytic granules.
The involvement of Vav1 in NKG2D/DAP10-mediated signaling in mice shown in this work is in agreement with the recent demonstration that NKG2D/DAP10 activates Vav1 in human NK cells (20). It remains to be determined how DAP10 selectively recruits Vav1, whereas ITAM-containing adapters mainly signal through Vav2 and Vav3. Although DAP10 recruits PI-3K, it has been shown that activation of Vav1 through NKG2D/DAP10 occurs independently of PI-3K in human NK cells (20). Thus, DAP10 may recruit Vav1 through adapters such as Grb2 and SLP76 (20). Once recruited, Vav1 may trigger the Rac1→PAK1→ MEK→ERK cascade that elicits cytotoxicity in human NK cells (13). In addition, Vav1 may facilitate formation of the SLP76–PLCγ complex, PLC-γ activation and subsequent Ca2+ mobilization, as previously observed in T cells (15). DAP12 and FcRγ trigger Syk and PI-3K (7), which represent the most likely mediators of Vav2 and Vav3 activation. Moreover, Syk allows recruitment of the molecular adaptor LAT (26), which can recruit PLCγ2 independently of Vav1, possibly explaining why Vav1 is not critical for DAP12 and FcRγ signaling.
Despite the different requirements for Vav proteins, both DAP10 and DAP12 signaling pathways promoted ERK1/2 activation (Fig. 4 and references 7, 8, 11). ERK1/2 may be an important link between Vavs and DAP10- and DAP12-mediated cytotoxicity. Consistent with this, we showed that deficiency of Vav proteins reduces ERK1/2 activation and pharmacological inhibition of ERK1/2 activation reduces both DAP10- and DAP12-mediated cytotoxicities. Whether Vav proteins differ in their capacity to recruit additional molecules linking DAP10 and ITAM with cytotoxicity, such as PLCγ (20, 27), remains to be determined. It is also possible that ITAM→Vav2/Vav3 signaling differs from DAP10→Vav1 signaling in strength and kinetics. The former may be more sustained than the latter, allowing not only for changes in actin cytoskeleton, polarization, and exocytosis of cytotoxic granules, but also for transcriptional activation of cytokine genes and cytokine secretion (28). In agreement with this model, it has been shown that NKG2D can trigger IFN-γ secretion only through DAP12, not DAP10 (29) and that Vav1 is not required for NK cell secretion of IFN-γ (21). It is not yet known whether DAP12 and ITAMs require Vav2 and Vav3 to trigger IFN-γ secretion.
Finally, our work illustrates a unique role for the Vav proteins in NK cells in comparison to T and B cells. Although lack of Vav proteins leads to a profound impairment of T and B cell maturation and function (23), Vav proteins are dispensable for NK cell development, but are required for NK cell effector functions. This result suggests a fundamental difference in function of the ITAM-containing signaling adapters, DAP12 and FcRγ in NK cells, CD3ζ in T cells, and Igα and Igβ in B cells.
Acknowledgments
We thank S. Gilfillan for reading the manuscript.
This work was supported by National Institutes of Health (NIH) grant no. 5R01AI056139-03. W.M. Yokoyama is supported by NIH grants and is an investigator of the Howard Hughes Institute.
The authors have no conflicting financial interests.
M. Cella and K. Fujikawa contributed equally to this work.
Abbreviations used in this paper: ADCC, antibody-dependent, cell-mediated cytotoxicity; CHO, Chinese hamster ovary; ERK, extracellular signal-regulated kinase; ITAM, immunoreceptor tyrosine-based activation motif; MEK, mitogen-activated protein kinase; nt, nucleotide; PAK1, p21-activated kinase 1; PI-3K, phosphatidylinositol-3-kinase; PLC, phospholipase C; RPA, RNase protection assay; SLP, Src homology 2 domain-containing leukocyte phosphoprotein.
References
- 1.Raulet, D.H. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781–790. [DOI] [PubMed] [Google Scholar]
- 2.Cerwenka, A., and L.L. Lanier. 2001. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1:41–49. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura, M.C., P.A. Linnemeyer, E.C. Niemi, L.H. Mason, J.R. Ortaldo, J.C. Ryan, and W.E. Seaman. 1999. Mouse Ly-49D recognizes H-2Dd and activates natural killer cell cytotoxicity. J. Exp. Med. 189:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furukawa, H., K. Iizuka, J. Poursine-Laurent, N. Shastri, and W.M. Yokoyama. 2002. A ligand for the murine NK activation receptor Ly-49D: activation of tolerized NK cells from beta 2-microglobulin-deficient mice. J. Immunol. 169:126–136. [DOI] [PubMed] [Google Scholar]
- 5.Arase, H., E.S. Mocarski, A.E. Campbell, A.B. Hill, and L.L. Lanier. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 296:1323–1326. [DOI] [PubMed] [Google Scholar]
- 6.Smith, H.R., J.W. Heusel, I.K. Mehta, S. Kim, B.G. Dorner, O.V. Naidenko, K. Iizuka, H. Furukawa, D.L. Beckman, J.T. Pingel, et al. 2002. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA. 99:8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McVicar, D.W., and D.N. Burshtyn. 2001. Intracellular signaling by the killer immunoglobulin-like receptors and Ly49. Sci. STKE. 2001:RE1. [DOI] [PubMed]
- 8.Tomasello, E., M. Blery, F. Vely, and E. Vivier. 2000. Signaling pathways engaged by NK cell receptors: double concerto for activating receptors, inhibitory receptors and NK cells. Semin. Immunol. 12:139–147. [DOI] [PubMed] [Google Scholar]
- 9.Diefenbach, A., E. Tomasello, M. Lucas, A.M. Jamieson, J.K. Hsia, E. Vivier, and D.H. Raulet. 2002. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat. Immunol. 3:1142–1149. [DOI] [PubMed] [Google Scholar]
- 10.Gilfillan, S., E.L. Ho, M. Cella, W.M. Yokoyama, and M. Colonna. 2002. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat. Immunol. 3:1150–1155. [DOI] [PubMed] [Google Scholar]
- 11.Wu, J., Y. Song, A.B. Bakker, S. Bauer, T. Spies, L.L. Lanier, and J.H. Phillips. 1999. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. 285:730–732. [DOI] [PubMed] [Google Scholar]
- 12.Perussia, B. 2000. Signaling for cytotoxicity. Nat. Immunol. 1:372–374. [DOI] [PubMed] [Google Scholar]
- 13.Jiang, K., B. Zhong, D.L. Gilvary, B.C. Corliss, E. Hong-Geller, S. Wei, and J.Y. Djeu. 2000. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat. Immunol. 1:419–425. [DOI] [PubMed] [Google Scholar]
- 14.Turner, M., and D.D. Billadeau. 2002. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat. Rev. Immunol. 2:476–486. [DOI] [PubMed] [Google Scholar]
- 15.Reynolds, L.F., L.A. Smyth, T. Norton, N. Freshney, J. Downward, D. Kioussis, and V.L. Tybulewicz. 2002. Vav1 transduces T cell receptor signals to the activation of phospholipase C-γ1 via phosphoinositide 3-kinase–dependent and –independent pathways. J. Exp. Med. 195:1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Billadeau, D.D., K.M. Brumbaugh, C.J. Dick, R.A. Schoon, X.R. Bustelo, and P.J. Leibson. 1998. The Vav–Rac1 pathway in cytotoxic lymphocytes regulates the generation of cell-mediated killing. J. Exp. Med. 188:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galandrini, R., G. Palmieri, M. Piccoli, L. Frati, and A. Santoni. 1999. Role for the Rac1 exchange factor Vav in the signaling pathways leading to NK cell cytotoxicity. J. Immunol. 162:3148–3152. [PubMed] [Google Scholar]
- 18.Riteau, B., D.F. Barber, and E.O. Long. 2003. Vav1 phosphorylation is induced by β2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J. Exp. Med. 198:469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mainiero, F., A. Soriani, R. Strippoli, J. Jacobelli, A. Gismondi, M. Piccoli, L. Frati, and A. Santoni. 2000. RAC1/P38 MAPK signaling pathway controls beta1 integrin-induced interleukin-8 production in human natural killer cells. Immunity. 12:7–16. [DOI] [PubMed] [Google Scholar]
- 20.Billadeau, D.D., J.L. Upshaw, R.A. Schoon, C.J. Dick, and P.J. Leibson. 2003. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat. Immunol. 4:557–564. [DOI] [PubMed] [Google Scholar]
- 21.Colucci, F., E. Rosmaraki, S. Bregenholt, S.I. Samson, V. Di Bartolo, M. Turner, L. Vanes, V. Tybulewicz, and J.P. Di Santo. 2001. Functional dichotomy in natural killer cell signaling: Vav1-dependent and -independent mechanisms. J. Exp. Med. 193:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan, G., T. Hanke, and K.D. Fischer. 2001. Vav-1 regulates NK T cell development and NK cell cytotoxicity. Eur. J. Immunol. 31:2403–2410. [DOI] [PubMed] [Google Scholar]
- 23.Fujikawa, K., A.V. Miletic, F.W. Alt, R. Faccio, T. Brown, J. Hoog, J. Fredericks, S. Nishi, S. Mildiner, S.L. Moores, et al. 2003. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J. Exp. Med. 198:1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billadeau, D.D., S.M. Mackie, R.A. Schoon, and P.J. Leibson. 2000. The Rho family guanine nucleotide exchange factor Vav-2 regulates the development of cell-mediated cytotoxicity. J. Exp. Med. 192:381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stebbins, C.C., C. Watzl, D.D. Billadeau, P.J. Leibson, D.N. Burshtyn, and E.O. Long. 2003. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol. Cell. Biol. 23:6291–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang, W., J. Sloan-Lancaster, J. Kitchen, R.P. Trible, and L.E. Samelson. 1998. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell. 92:83–92. [DOI] [PubMed] [Google Scholar]
- 27.Ting, A.T., L.M. Karnitz, R.A. Schoon, R.T. Abraham, and P.J. Leibson. 1992. Fc γ receptor activation induces the tyrosine phosphorylation of both phospholipase C (PLC)-γ 1 and PLC-γ 2 in natural killer cells. J. Exp. Med. 176:1751–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doody, G.M., D.D. Billadeau, E. Clayton, A. Hutchings, R. Berland, S. McAdam, P.J. Leibson, and M. Turner. 2000. Vav-2 controls NFAT-dependent transcription in B- but not T-lymphocytes. EMBO J. 19:6173–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zompi, S., J.A. Hamerman, K. Ogasawara, E. Schweighoffer, V.L. Tybulewicz, J.P. Di Santo, L.L. Lanier, and F. Colucci. 2003. NKG2D triggers cytotoxicity in mouse NK cells lacking DAP12 or Syk family kinases. Nat. Immunol. 4:565–572. [DOI] [PubMed] [Google Scholar]