

Abstract
Tumor necrosis factor (TNF) is a potent cytokine exerting critical functions in the activation and regulation of immune and inflammatory responses. Due to its pleiotropic activities, the amplitude and duration of TNF function must be tightly regulated. One of the mechanisms that may have evolved to modulate TNF function is the proteolytic cleavage of its cell surface receptors. In humans, mutations affecting shedding of the p55TNF receptor (R) have been linked with the development of the TNFR-associated periodic syndromes, disorders characterized by recurrent fever attacks and localized inflammation. Here we show that knock-in mice expressing a mutated nonsheddable p55TNFR develop Toll-like receptor–dependent innate immune hyperreactivity, which renders their immune system more efficient at controlling intracellular bacterial infections. Notably, gain of function for antibacterial host defenses ensues at the cost of disbalanced inflammatory reactions that lead to pathology. Mutant mice exhibit spontaneous hepatitis, enhanced susceptibility to endotoxic shock, exacerbated TNF-dependent arthritis, and experimental autoimmune encephalomyelitis. These results introduce a new concept for receptor shedding as a mechanism setting up thresholds of cytokine function to balance resistance and susceptibility to disease. Assessment of p55TNFR shedding may thus be of prognostic value in infectious, inflammatory, and autoimmune diseases.
Keywords: TRAPS, arthritis, EAE, chronic hepatitis, LPS toxicity
Introduction
TNF is a potent cytokine exerting critical beneficial activities in immune regulation and host defense, as well as hazardous proinflammatory and cytotoxic functions. TNF mediates its pleiotropic activities via two cell surface receptors, p55TNFR (TNFRI, CD120a) and p75TNFR (TNFRII, CD120b). The two TNFRs are ubiquitously expressed and display structurally similar extracellular domains but signal through distinct intracellular regions, with p55TNFR containing a death domain that is not present in the p75TNFR (1). Signaling through the p55TNFR leads to the activation of the IκB kinase complex and mitogen-activated protein kinase pathways, and to the initiation of apoptotic cell death (for review see references 2 and 3). Previous studies in p55TNFR-deficient mice established the requirement of this receptor in the organization of secondary lymphoid organ structure and function (4), in host defense against various microorganisms (3, 5–8), in the induction and maintenance of various chronic inflammatory pathologies (9), and the toxicity of TNF during endotoxic shock (10–12).
TNFRs are initially synthesized as membrane-anchored proteins, which can subsequently be released from the cell surface by proteolysis, forming soluble molecules capable of binding the TNF ligand. Soluble TNFRs are constitutively released in the circulation (13), and their levels increase in the course of various disease states (14) and after TNF stimulation (15, 16). In cell culture systems, soluble receptors are rapidly produced in response to various stimuli such as TNF (17), LPS (18), PMA, and IL-10, or after T cell (19) and neutrophil activation (20). Shedding and the resultant acute decrease in the number of receptor molecules on the cell surface may serve to transiently desensitize cells to the TNF action. In addition, the pool of soluble forms produced could function as physiological attenuators of the TNF activity, competing for the ligand with the cell surface receptors. Moreover, it has been proposed that soluble receptors can stabilize and preserve circulating soluble TNF and thus function as TNF agonists (21).
Up until now, a variety of approaches have been used to determine the biological functions of membrane-bound p55TNFR. These include experiments using receptor-specific antibodies (22), receptor-specific ligands (23), and p55TNFR-deficient mice (10–12). However, the biological significance of p55TNFR shedding has remained unclear. In humans, impaired p55TNFR clearance from the cell surface has recently been proposed as a novel mechanism of disease. Mutations on the human p55TNFR gene causing reduced levels of shedding were proposed to be responsible for a newly defined class of autoinflammatory diseases, termed TNFR1-associated periodic syndromes (TRAPS; reference 24).
To investigate the in vivo role of p55TNFR shedding, we have used a knock-in approach to generate mutant mice expressing nonsheddable p55TNFRs. We show here that similarly to the TRAPS syndrome in humans, defective shedding of the p55TNFR in mice leads to a persistent expression of the receptor on the cell surface and acts as a dominant genetic trait to provoke spontaneous autoinflammatory reactions. We further show that defective p55TNFR shedding causes innate immune hyperresponsiveness, resulting in enhanced host defenses to bacterial infections as well as to the sensitization of the host to the in vivo toxicity of TNF and LPS. Notably, defective TNFR shedding leads to exacerbated clinical outcomes in mouse models of inflammatory diseases, demonstrating the significance of receptor shedding in setting up thresholds of TNF function and balancing resistance and susceptibility to disease.
Materials and Methods
Generation of p55ΔNS Mice.
A mutation deleting 15 nucleotides encoding for amino acids 202–206 was introduced in exon 6 of the mp55TNFR gene by PCR mutagenesis on a 2-kb Sma/HindIII subcloned genomic fragment containing exons 6 and 7 (36). For the targeting vector, a 4.6-kb XbaI/SmaI genomic fragment was used as the left arm of homology and a 4-kb Sma/HindIII genomic fragment containing the p55 ΔNS mutation was used as the right homology arm. A loxP-flanked neo selection cassette (L2neo) was introduced into an SmaI site in intron 5 of the p55TNFR gene between the two arms of homology and a PGK-tk− selection marker was introduced at the 3′ of the targeting vector. The targeting vector was linearized by NotI before electroporation into CCE mouse embryonic stem cells derived from 129Sv mice (37). Transfection, selection, expansion, and screening of recombinant clones, and generation of germline-transmitting chimeras was performed as described previously (28). For the excision of the loxP-flanked neo selection cassette, fertilized oocytes from mice carrying the targeted neo-containing p55 ΔNS gene were used to introduce the pMC-Cre circular plasmid vector using pronuclear DNA microinjection. Pups born from these oocytes were tested and shown to have excised the loxP-flanked neo cassette.
Genotyping was performed by Southern blot using the SmaI-BamHI probe shown in Fig. 1 A. PCR genotyping was performed with the following primers: 5′-GTGTATGAAGTTGTGCCTACC-3′ and 5′-CTCAGCCTAGAGTCCTTAACC-3′, producing fragments of 425 and 410 bp for the wild-type and the mutant allele, respectively.
Figure 1.
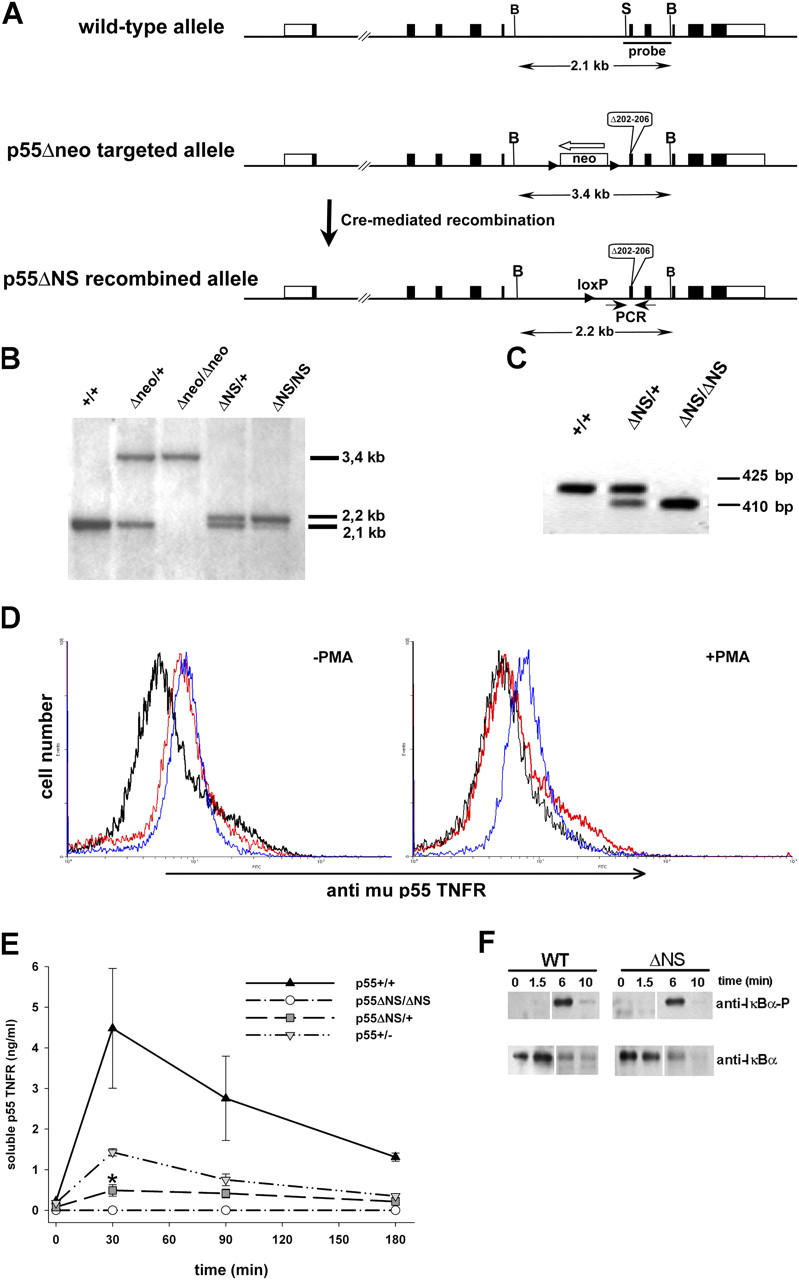
Generation and initial characterization of p55 ΔNS mice. (A) Schematic representation of the murine p55TNFR genomic locus (top), the p55 Δneo targeted locus generated by homologous recombination in embryonic stem cells (middle), and the p55 ΔNS locus generated after Cre-mediated excision of the loxP-flanked neo cassette (bottom). Exons are indicated as closed boxes with open boxes representing 5′ and 3′ untranslated regions. The p55 ΔNS mutation that deletes amino acids 202–206 in the mature protein is indicated. The white arrow shows the transcriptional orientation of the neo cassette and arrowheads indicate loxP sites. B and S indicate BamHI and SmaI restriction enzyme sites, respectively. (B) Southern blot analysis of BamHI-digested tail DNA from mice with the indicated genotypes using the SmaI-BamHI probe. (C) PCR analysis of tail DNA from mice with the indicated genotypes using PCR primers (small arrows in A) that amplify fragments of 425 and 410 bp for the wild-type and the mutant alleles, respectively. (D) Cell surface p55TNFR levels on peritoneal exudate cells from p55TNFR-deficient (black), wild-type (red), and homozygous p55 ΔNS/ΔNS mice (blue) analyzed for mp55TNFR expression before or after PMA activation. Data are representative of two independent experiments. (E) ELISA analysis of soluble p55TNFR levels in sera from mice with the indicated genotypes before and after LPS stimulation. Data are representative of three independent experiments. (*, P < 0.05). (F) Western blot analysis of IκBα phosphorylation and degradation in wild-type (wt) and homozygous p55 ΔNS/ΔNS (ΔNS) MEFs stimulated with human TNF and harvested at the indicated time points. All mice were in a B6,129 genetic background. White lines indicate that intervening lanes have been spliced out.
Mice.
p55 ΔNS mice were maintained on a C57BL/6 × 129Sv genetic background (referred to as B6,129), or backcrossed to C57BL/6 mice for at least five generations. Strains deficient in TNF (28), RAG-1 (The Jackson Laboratory; reference 38), p55TNFR (10), p75TNFR (39), and lpr/lpr (The Jackson Laboratory) were maintained on a mixed C57BL/6 × 129Sv genetic background. Tg197 mice (30) were maintained on a CBA × C57BL/6 background. All mice were kept under specific pathogen-free conditions.
Use of Mouse Embryonic Fibroblasts (MEFs) to Assess Signaling through p55ΔNS Receptor.
MEFs (2 × 106 cells) from wild-type and p55 ΔNS/ΔNS mice were cultured in DMEM (GIBCO BRL) supplemented with 10% heat-inactivated FCS (GIBCO BRL), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, and were either left untreated or stimulated with 100 ng/ml human TNF (Genentech, Inc.). Whole cell lysates were harvested with a lysis buffer containing 1% Igepal CA-630 (Sigma-Aldrich) and the proteins were separated in 12% SDS-polyacrylamide gels and blotted onto nitrocellulose membrane. The following antibodies were used: phospho-specific IκBα monoclonal antibody (catalog no. 9246; Cell Signaling Technology) and rabbit anti-peptide antibody directed against IκBα (catalog no. sc-371; Santa Cruz Biotechnology, Inc.).
Flow Cytometry.
Peritoneal exudate cells were adjusted to 2 × 106 cells/ml in DMEM (GIBCO BRL) supplemented as described above and stimulated with 100 ng/ml PMA (Sigma-Aldrich) or left untreated for 20 min at 37°C, 5% CO2. Cells were then washed and incubated with primary monoclonal rat anti-mup55 TNFR IgG antibody (provided by W. Buurman, University of Limburg, Diepenbeek, Netherlands) and secondary goat anti–rat IgG FITC (catalog no. 3052-02; Southern Biotechnology Associates, Inc.) for 30 min at 4°C, and analyzed by flow cytometry.
ELISA from Murine p55TNFR, p75TNFR, and TNF.
Serum was collected via tail vein exsanguinations before or 30, 90, and 180 min after intraperitoneal injection of 100 μg LPS (Salmonella enteritidis, L-6011; Sigma-Aldrich) and assayed using specific ELISA assays (based on the same anti-mup55 TNFR antibody used for flow cytometry as above; provided by W. Buurman) as described previously (28).
Histopathology.
Tissues from freshly dissected mice were immersion fixed overnight in neutral buffered formalin and embedded in paraplast (BDH Laboratory Supplies). Sections were cut and stained with hematoxylin and eosin according to standard procedure, dehydrated, and mounted in DPX (BDH Laboratory Supplies).
Immunohistochemistry.
Mice were perfused via the aorta with 4% paraformaldehyde in 0.1 M phosphate buffer. Brains and spinal cords were removed and immersed in the same fixative for 12–24 h. Tissue blocks were routinely embedded in paraffin. 5-μm thick sections were stained with hematoxylin/eosin to visualize infiltrated inflammatory cells and with luxol fast blue for myelin. Immunocytochemistry was performed with a biotin/avidin technique with primary antibodies against T cells (CD3; Seralab) and activated macrophages/microglia (Mac-3; Serotec). An average inflammatory index was created by counting the number of inflamed blood vessels in 10–15 spinal cord cross sections and subsequent division by the number of cross sections.
TNF and LPS Administration and Listeria Infection.
Recombinant human TNF (Genentech, Inc.) was administered intravenously at 60 μg/mouse and LPS (S. enteritidis, L-6011; Sigma-Aldrich) was administered intraperitoneally at the indicated doses. Enbrel (etanercept; Immunex) was administered intraperitoneally 1 h before LPS administration. Listeria monocytogenes strain L028 (40) was administered intraperitoneally at the indicated doses. Survival was monitored for 10 d in all cases.
Northern Blot Analysis of Macrophage RNA.
10 μg total RNA, isolated as described previously (28), was resolved in 1.2% formaldehyde agarose gel, blotted onto Hybond N+ nylon membrane (Amersham Biosciences), and hybridized with a 0.45-kb 32P-labeled NarI-BglII genomic probe containing part of the first exon of the muTNF gene. Membranes were stripped and rehybridized with a β actin probe.
Measurement of Nitric Oxide (NO).
NO was measured in culture supernatants of thioglycollate-elicited peritoneal macrophages. Macrophages were isolated from wild-type and p55 ΔNS/ΔNS mice (n = 2–3 per group), cultured in flat-bottomed 96-well plates at a density of 5 × 105/well in DMEM (GIBCO BRL), supplemented as described above, and either left untreated or stimulated with 0.1, 1, 2, and 5 μg/ml LPS (S. enteritidis, L-6011; Sigma-Aldrich), or 1, 10, 50, and 100 μg/ml polyinosinic-polycytidylic acid (poly IC; Amersham Biosciences). Supernatants were collected 24 h later, and the presence of NO was determined by measuring the amount of nitrite, a metabolic product of NO (41).
Immunizations and Experimental Autoimmune Encephalomyelitis (EAE) Induction.
T cell priming was assayed in popliteal LN cells isolated from 7–11-wk-old mice 9 d after footpad immunization with 150 μg rat myelin oligodendrocyte glycoprotein (MOG) peptide (MOGp35-55, MEVGWYRSPFSRVVHLYRNGK; Sigma-Genosys) in CFA containing a total of 1 mg H37Ra (Difco Laboratories) per mouse. EAE was induced in 10–12-wk-old male mice after tail-base immunization with 150 μg rat MOG peptide in CFA containing a total of 1 mg H37Ra per mouse. At the time of immunization and 48 h later, mice received 40 ng of pertussis toxin (PTx; Sigma-Aldrich) intraperitoneally. Clinical signs of disease were scored on a scale of 0–5 as follows: 0, no disease; 1, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 4, hind limb plus forelimb paralysis; 5, moribund or dead animals. T cell “memory” was assayed in spleen cells of mice 20 d after induction of EAE. For histological evidence of EAE, brains and spinal cords were removed.
T Cell Proliferation Assay.
T cell proliferative responses were assessed at indicated time points in total LN or spleen cells. After erythrocyte lysis, cells were cultured in flat-bottomed 96-well plates at a density of 7 × 105 per well. Cultured medium consisted of DMEM (GIBCO BRL) supplemented with 5% heat-inactivated FCS, l-glutamine, and antibiotics. Triplicate cultures were incubated at 37°C in the presence or absence of indicated concentrations of rat MOG peptide. After 52 h, cultures were pulsed with 1 μCi [methyl-3H]TdR (Amersham Biosciences) for 18 h. Cells were harvested on glass fiber filters and incorporation of [methyl-3H]TdR was measured by liquid scintillation counting.
Results
Generation and Initial Characterization of p55TNFR Nonsheddable (p55ΔNS) Mice.
To investigate the biological function of p55TNFR shedding, we generated p55TNFR nonsheddable mutant mice (referred to as p55ΔNS) using gene targeting by homologous recombination in embryonic stem cells (Fig. 1, A–C). To examine the expression levels and assess the shedding of the mutant p55TNFR from the cell surface, we analyzed freshly isolated peritoneal exudate cells by flow cytometry using an antibody specific for the murine p55TNFR. Cells were analyzed before and 20 min after treatment with PMA, an agent widely used to induce proteolytic cleavage of several cell surface proteins including p55TNFR (25). Before treatment, the expression levels of the mutant receptor in p55 ΔNS/ΔNS cells were similar to those in control cells, indicating that the p55 ΔNS mutation does not interfere with the correct regulation of p55TNFR expression. However, p55TNFR levels persisted in p55 ΔNS/ΔNS cells after activation with PMA, in contrast to wild-type cells where the receptors were completely cleared from the cell surface (Fig. 1 D), indicating that the p55 ΔNS mutation abolishes receptor shedding. Defective shedding of the mutated receptor was also confirmed by the complete absence of soluble p55TNFR protein in sera from LPS-stimulated p55 ΔNS/ΔNS mice (Fig. 1 E). Interestingly, the levels of soluble p55TNFR in sera from heterozygous p55 ΔNS/+ mice were as low as 15% of wild-type levels, significantly lower than those in p55 +/− mice (∼40% of wild-type), suggesting that the p55 ΔNS mutation functions in a dominant fashion to also suppress shedding of the wild-type protein. Taken together, these results show that in resting cells, the cell surface expression of the mutant p55ΔNS protein is correctly regulated and that after appropriate stimulation, inability to shed the mutant receptor leads to its persistent expression on the cell surface.
To address whether the nonsheddable mutant receptor remains functional and to investigate possible alterations in its signaling capacity, we analyzed the activation of the nuclear factor κB signaling pathway, which occurs within a few minutes after p55TNFR stimulation. MEFs from wild-type and p55 ΔNS/ΔNS mutant mice were treated with recombinant human TNF, which is known to signal exclusively through the mouse p55TNFR (26), and IκBα phosphorylation and degradation were measured by Western blot analysis (Fig. 1 F). Rapid appearance of phosphorylated IκBα with concomitant IκBα degradation occurs with similar kinetics in mutant and control MEFs, indicating that the p55 ΔNS mutation does not influence the signaling capacity of the mutant receptor. Because it is known that a physiological function of the p55TNFR is to regulate the correct structural organization of the spleen (27), to further investigate the in vivo functionality of the p55 ΔNS receptor, we analyzed the splenic microarchitecture of p55 ΔNS/ΔNS mice. The spleens of p55 ΔNS/ΔNS mice showed a normal microarchitecture and efficient germinal center formation after immunization with the thymus-dependent antigen sheep red blood cells (not depicted), suggesting that the p55 ΔNS mutation does not interfere with the physiological function of the p55TNFR.
Spontaneous Development of Chronic Active Hepatitis in p55ΔNS Mice.
Impaired shedding of the p55TNFR is thought to be responsible for recurrent attacks of generalized inflammation in human TRAPS patients. To examine the potential development of spontaneous pathologies in p55 ΔNS mice, we performed histopathological evaluation of tissue sections from several organs of both young and adult homozygous and heterozygous animals. No obvious abnormalities could be observed in the lung, spleen, pancreas, kidneys, and joints (not depicted). However, histopathological analysis of the liver revealed the spontaneous development of a mild form of active chronic hepatitis, characterized by focal parenchymal inflammation with infiltration of lymphocytes, polymorphonuclear cells, and other leukocytes, and by the presence of apoptotic hepatocytes in the majority of the inflammatory foci. Liver pathology was already apparent at 3–4 wk of age, persisted throughout adulthood, and was most profound in homozygous p55 ΔNS/ΔNS mutant mice, which showed larger and more numerous inflammatory foci in comparison to heterozygous p55 ΔNS/+ mice (Fig. 2, A and B). Hepatitis developed with 100% phenotypic penetrance in both homozygous and heterozygous mutant mice, whereas no liver pathology could be detected in littermate controls.
Figure 2.
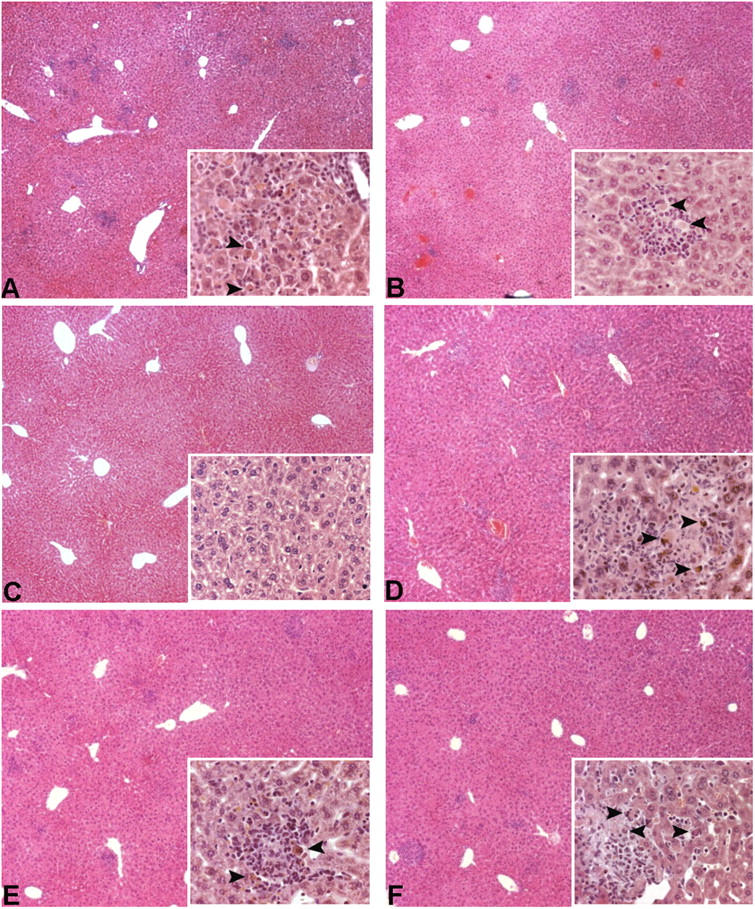
Active chronic hepatitis in p55 ΔNS mice. Histologic examination of liver sections from B6,129 p55 ΔNS mice at 8 wk in different experimental groups. (A and B) Focal inflammation in liver parenchyma characterized by infiltration of lymphocytes and polymorphonuclear cells and by the presence of apoptotic hepatocytes (inserts). Disease develops in homozygous p55 ΔNS/ΔNS (A) as well as in heterozygous p55 ΔNS/+ (B) mutant mice, albeit with reduced severity. (C) Liver sections from p55 ΔNS/ΔNS .TNF −/− mice present no signs of pathology. Inflammatory foci and presence of apoptotic hepatocytes persist in the liver of p55 ΔNS/ΔNS .RAG-1 −/− (D), p55 ΔNS/ΔNS .lpr/lpr (E), or p55 ΔNS/ΔNS .p75TNFR −/− (F) double mutant mice. Magnification, 40 and 400 for the insets.
To evaluate the molecular and cellular mechanisms of this liver pathology, we generated a series of intercrosses with strains of mice that are genetically deficient in the expression of TNF, RAG-1, Fas, or the p75TNFR. TNF expression is essential for hepatitis development because p55 ΔNS/ΔNS .TNF −/− mice do not show this phenotype (Fig. 2 C). Interestingly, T and B lymphocytes are not required for the development of liver inflammation and the induction of hepatocyte apoptosis in this model, as shown by the development of hepatic lesions including apoptotic hepatocytes in the T and B cell–deficient p55 ΔNS/ΔNS .RAG-1 −/− mice (Fig. 2 D). Finally, histological examination of liver sections from Fas-deficient p55 ΔNS/ΔNS .lpr/lpr or p75TNFR-deficient p55 ΔNS/ΔNS .p75TNFR −/− mice showed that the liver pathology develops independently of the function of Fas or the p75TNFR in this system (Fig. 2, E and F). These results demonstrate that defective p55TNFR shedding triggers the spontaneous development of a chronic autoinflammatory reaction in the liver, which leads to the establishment of a TNF-dependent, yet Fas- and T cell–independent, chronic active hepatitis. Interestingly, this phenotype also develops in heterozygous mutant animals, indicating that the p55 ΔNS mutation acts in a dominant fashion to induce liver inflammation.
Defective p55TNFR Shedding Increases Susceptibility of Mice to the Systemic Toxicity of TNF and LPS.
To examine the potential involvement of p55TNFR shedding in TNF toxicity in vivo, we investigated the susceptibility of homozygous p55 ΔNS/ΔNS and heterozygous p55 ΔNS/+ mutant mice to normally sublethal doses of exogenously administered recombinant human TNF, which can only bind to the murine p55TNFR (26). Intravenous administration of 60 μg of recombinant human TNF to normal mice resulted in the death of 1 out of 9 mice (11% lethality). Notably, the same dose of human TNF resulted in significantly increased lethality in both homozygous (100%) and heterozygous (71%) p55 ΔNS mice (Table I), suggesting that p55TNFR shedding is important for protection from TNF toxicity in vivo.
Table I.
Measurements of Human Recombinant TNF– or LPS-induced Lethality in p55ΔNS Mice
| p55+/+ | p55ΔNS/ΔNS | p55ΔNS/+ | p55ΔNS/ΔNS | |
|---|---|---|---|---|
| (deaths/total) | (deaths/total) | (deaths/total) | (deaths/total) | |
| rhTNF | 1/9 | 8/8a | 5/7b | — |
| (60 μg/mouse) | (11%) | (100%) | (71%) | |
| LPS | 7/19 | 17/18a | 10/10a | — |
| (340 μg/25 g) | (37%) | (94%) | (100%) | |
| LPS | 0/11 | 11/11 | — | 2/11c |
| (300 μg/25 g) | (0%) | (100%) | (18%) |
C57BL/6 p55 Δ NS/ Δ NS, p55 Δ NS/ + littermates and age-matched C57BL/6 controls (8–12 wk of age) were injected with the indicated amounts of human recombinant TNF (intravenously) or LPS (intraperitoneally; S. enteritidis, L-6011; Sigma-Aldrich). Enbrel (etanercept; Immunex) was administered intraperitoneally 1 h before LPS injections at a dose of 2 mg/mouse. Survival was monitored for 10 d. P-values by Fisher's exact test:
P < 0.004.
P < 0.04 (p55 Δ NS/ Δ NSor p55 Δ NS/ + vs. p55 +/+).
P = 0.0002 (p55 Δ NS/ Δ NSwith Enbrel vs. p55 Δ NS/ Δ NSwithout Enbrel administration).
Previous studies have shown that the function of the TNF–p55TNFR system is not required for LPS toxicity, suggesting that pathways different from TNF–p55TNFR are critical in this model of endotoxic shock (10, 12, 28). To address the involvement of p55TNFR shedding in LPS toxicity, we measured the susceptibility of mutant mice to normally sublethal doses of LPS. Administration of 340 or 300 μg LPS (per 25 g of body weight) resulted in 37 and 0% lethality, respectively, in wild-type mice, but caused significantly increased lethality (nearly 100%) in both homozygous and heterozygous p55 ΔNS mutant mice (Table I). Interestingly, anti-TNF treatment protected p55 ΔNS/ΔNS mice from LPS toxicity, demonstrating that the increased susceptibility of these mice to LPS is absolutely dependent on TNF function (Table I). These results demonstrate that p55TNFR shedding plays a crucial role in the desensitization of the host from the deleterious activities of TNF during LPS-induced endotoxic shock.
Increased Resistance of p55ΔNS Mice to L. monocytogenes Infections.
TNF–p55TNFR signaling plays a critical role in antibacterial host defense and TNF- or p55TNFR-deficient mice are highly susceptible to infection with the intracellular bacterium L. monocytogenes (10, 11, 28). To investigate the effect of impaired p55TNFR shedding in host defense mechanisms, we measured the survival of p55 ΔNS/ΔNS and control mice after infection with different doses of Listeria. p55 ΔNS/ΔNS mice showed increased resistance to Listeria infection and could tolerate 10-fold higher doses than control mice (Table II). These results show that impaired shedding of the p55TNFR enables enhanced antibacterial host defenses, suggesting that induction of p55TNFR shedding might be a mechanism used by pathogenic bacteria to escape a neutralizing immune response.
Table II.
Measurements of Lethality after L. monocytogenes Infections
| CFU | p55+/+(deaths/total) | p55 ΔNS/ Δ NS(deaths/total) |
|---|---|---|
| 108 | 15/15 | 10/15a |
| 107 | 15/15 | 4/15b |
| 106 | 5/15 | 0/15a |
| 105 | 0/15 | 0/15 |
B6,129 p55 Δ NS/ Δ NSand age-matched B6,129 p55 +/+ mice (10–12 wk of age) were infected intraperitoneally with the indicated doses of L. monocytogenes and survival was monitored for 10 d. P values by Fisher's exact test:
P = 0.042.
P = 0.00005.
Absence of p55TNFR Shedding Leads to Increased Innate Responsiveness.
Administration of LPS to mice induces a rapid increase in circulating levels of TNF and soluble TNFRs (29). We have followed the kinetics of the systemic release of TNF and the soluble p75TNFR in p55 ΔNS/ΔNS and control mice before and after LPS administration. Levels of both soluble proteins before LPS stimulation were comparable in mutant and control mice. After LPS stimulation, however, the peak concentration of both TNF and p75TNFR were found to be significantly enhanced in sera of mutant mice as compared with control animals (Fig. 3 A).
Figure 3.

Innate immune hyperresponsiveness in p55 ΔNS mice. (A) ELISA analysis of soluble TNF and p75TNFR levels in sera of wild-type (p55 +/+) and homozygous p55 ΔNS/ΔNS mice (n = 3 per group) before and after intraperitoneal injection of LPS. Data are representative of three independent experiments. (B) Northern blot analysis of TNF mRNA in thioglycollate-elicited peritoneal macrophages obtained from wild-type (p55 +/+) and p55 ΔNS/ΔNS mice before or after stimulation with LPS. The membrane was stripped and rehybridized with a β actin probe. (C) NO production in culture supernatants of wild-type (p55 +/+) and p55 ΔNS/ΔNS thioglycollate-elicited peritoneal macrophages in response to poly IC or LPS stimulation. All mice were in a B6,129 genetic background. Data are representative of three independent experiments.
Macrophages are central regulators of the inflammatory response and when activated they are able to produce a variety of effector molecules. Moreover, macrophages are considered to constitute the most prominent source of TNF production in vivo. To investigate whether macrophages expressing the nonsheddable p55TNFR show increased expression of TNF, we isolated thioglycollate-elicited peritoneal macrophages from p55 ΔNS/ΔNS and wild-type mice, and we compared the kinetics and the levels of TNF mRNA production after LPS induction using Northern analysis. TNF mRNA was expressed at a low level in resting macrophages of both types of mice. After LPS induction, mRNA levels in wild-type macrophages rapidly increase to a peak at 1 h and slowly return to baseline between 3 and 6 h. Although p55 ΔNS/ΔNS macrophages showed similar kinetics of TNF mRNA production as wild-type cells, they displayed substantially increased levels of TNF mRNA at all time points measured. At 1 or 3 h after LPS induction, we could measure a 1.5–2-fold increased production of TNF mRNA in p55 ΔNS/ΔNS cells. Moreover, at 6 h after LPS induction, TNF mRNA levels in wild-type macrophages had returned to baseline but were readily detectable and at a fivefold higher level in p55 ΔNS macrophages (Fig. 3 B). These data show that persistent expression of cell surface p55TNFR results in increased TNF mRNA accumulation upon LPS stimulation, indicating the presence of a positive feedback loop operating between cell surface p55TNFR and TNF mRNA transcription and/or stabilization. Apparently, induced shedding of the receptor seems to be important for the disruption of this positive regulatory loop and the negative regulation of TNF production.
To further validate the innate immune hyperresponsiveness of stimulated p55 ΔNS/ΔNS macrophages, we have determined their capacity to release NO. Thioglycollate-elicited peritoneal macrophages were stimulated in vitro with different doses of LPS or poly IC. Macrophages from p55 ΔNS/ΔNS mice responded to these innate immune activators with 1.5–3-fold higher NO production than normal macrophages (Fig. 3 C). Taken together, these results show that after innate immune stimulation, defective p55TNFR shedding promotes immune hyperresponsiveness, characterized by enhanced and sustained production of innate effectors such as TNF or NO.
Increased Susceptibility of p55ΔNS Mice to Chronic Arthritis and EAE.
To address the potential impact of impaired p55TNFR shedding on inflammatory disease induction and progression, we have examined the responses of p55 ΔNS mice in two models of human disease. First, p55 ΔNS mice were crossed to transgenic mice expressing a 3′ modified human TNF transgene (Tg197 line) and developing a TNF-dependent chronic inflammatory polyarthritis that resembles human rheumatoid arthritis (30). Histological examination of joint sections from 4-wk-old Tg197.p55 ΔNS/ΔNS and Tg197.p55 +/+ littermate controls revealed accelerated development of arthritis in mice carrying the p55 ΔNS mutation. All histological features of the disease, including hyperplasia of the synovial membrane and polymorphonuclear and lymphocytic inflammatory infiltrates of the synovial space, were much more profound in joints from Tg197.p55 ΔNS/ΔNS mice in comparison to controls. Moreover, histological characteristics such as pannus formation, articular cartilage destruction, and bone erosion that appear only in the advanced stages of disease in 7–8-wk-old Tg197.p55 +/+ mice, were already evident in joints of 4-wk-old Tg197.p55 ΔNS/ΔNS mice (Fig. 4, A and B).
Figure 4.

Increased susceptibility of p55 ΔNS mice to chronic arthritis. Histologic examination of ankle joints from Tg197.p55 +/+ (A) and Tg197.p55 ΔNS/ΔNS (B) littermates at 4 wk of age. Increased formation of the inflammatory pannus and areas of cartilage and bone erosion are evident in B, whereas mild hyperplasia of the synovial membrane and onset of infiltration of polymorphonuclear and lymphocytic inflammatory cells in the synovial space is seen in A. All mice were littermates in a mixed CBA,B6,129 background.
Next, we assessed the susceptibility of p55 ΔNS mice to the development of EAE, a widely used model of human multiple sclerosis. EAE was induced by immunization with a MOG peptide and by PTx coadministration, and disease was monitored by both clinical examination and histopathological analysis. Using an immunization protocol with a low dose of PTx (40 ng), mild disease develops in ∼40% of control mice. In contrast, p55 ΔNS/ΔNS mice develop disease with an almost 100% incidence (18 out of 19 p55 ΔNS/ΔNS mice vs. 8 out of 19 p55 +/+ controls; P = 0.001 by Fisher's exact test) and with significantly increased severity in comparison to controls (Fig. 5 A). Neuropathological evaluation was performed on a limited number (two per experimental group) of animals on day 20 after induction of EAE. Inflammatory indices, measured in 10–15 cross sections of the spinal cord and a parameter for the degree of inflammation, were higher in the p55 ΔNS animals than in the wild-type animals (p55 ΔNS/ΔNS: 2.4 and 4.5 vs. wild-type: 0.6 and 2.3). Demyelination in all animals was very limited being <5% of the spinal cord white matter. Immunohistochemistry revealed higher numbers of inflammatory T cells and activated macrophages/microglia in spinal cord sections of p55 ΔNS/ΔNS mice compared with normal mice (Fig. 5 B). These results verify the clinical differences observed between p55 ΔNS/ΔNS and wild-type controls.
Figure 5.
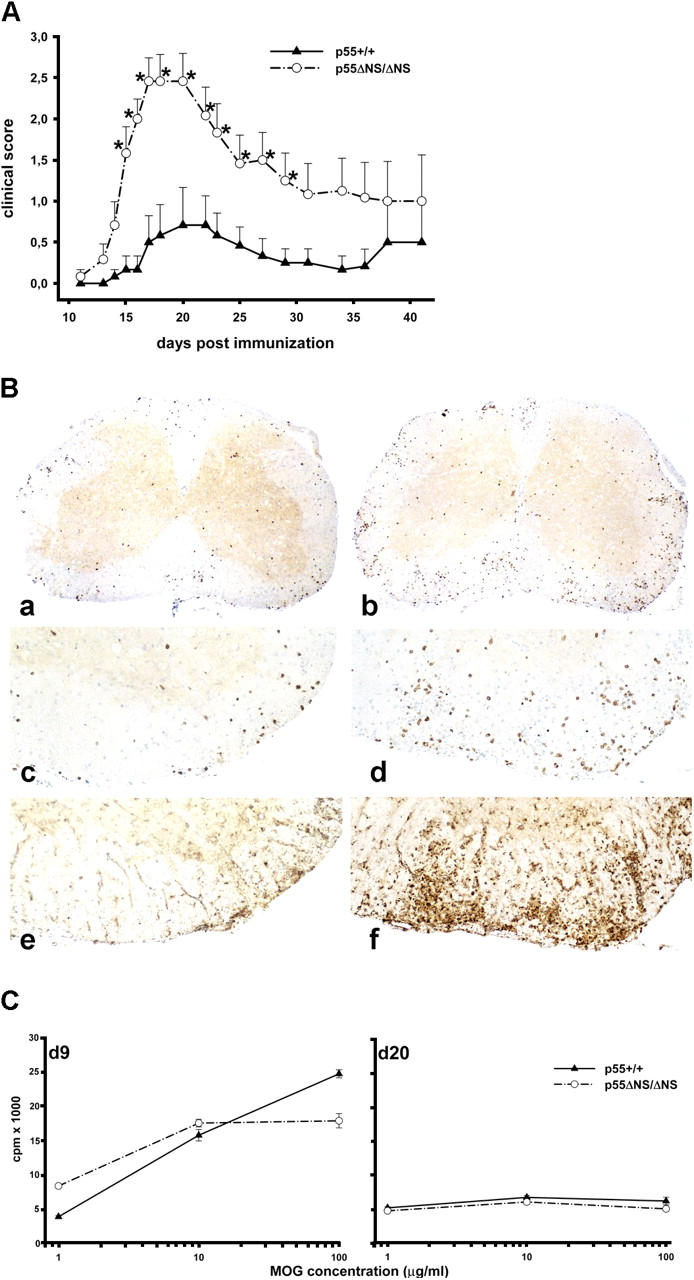
Increased susceptibility of p55ΔNS mice to EAE. (A) EAE was induced in C57BL/6 p55 ΔNS/ΔNS and age-matched C57BL/6 controls (10–12 wk of age; n = 6 per group), by MOG p35–55 immunization as described in Materials and Methods. Data presented are means ± SEM of disease scores. *, P < 0.05 as determined by Student's t test. Data are representative of four separate experiments. (B) Immunohistochemistry from C57BL/6 controls (a, c, and e) and C57BL/6 p55 ΔNS/ΔNS (b, d, and f) mice. In both cases the animals with the highest inflammatory index and clinical signs were taken. (a and b) CD3 staining in spinal cord sections showing lymphocyte infiltration. Inflammation in the p55 ΔNS/ΔNS mouse is more prominent (magnification, 37). Higher magnifications (80) of panels a and b are shown in panels c and d, respectively. In addition to higher numbers of inflammatory T cells, the p55 ΔNS/ΔNS mouse (f; magnification, 80) also reveals more pronounced expression of mac-3+ macrophages than the wild-type control (e; magnification, 80). (C) Similar MOG p35–55–specific T cell responses in p55 ΔNS/ΔNS and p55 +/+ mice. T cell priming was measured in LN cells isolated from C57BL/6 p55 ΔNS/ΔNS and age-matched C57BL/6 control mice (7–11 wk of age) 9 d after immunization with MOG peptide as described in Materials and Methods. MOG-specific memory responses were examined in total spleen cells of C57BL/6 p55 ΔNS/ΔNS and age-matched C57BL/6 controls 20 d after EAE induction. Proliferation was determined by [3H]thymidine incorporation in T cell proliferation assays in which T cells were incubated with the indicated concentrations of MOG peptide. For each curve, two to three mice of each genotype were pooled together. One representative set of experiments out of three performed is shown.
To determine whether the absence of p55TNFR shedding could alter T cell responsiveness to MOG peptide, thus explaining the observed differential susceptibility to EAE, mice were immunized with the MOG p35–55 peptide, and then analyzed for peptide-induced T cell proliferation. Both T cell priming (examined 9 d after immunization) and MOG-specific memory responses (examined 20 d after immunization) were comparable between p55 ΔNS/ΔNS and control mice (Fig. 5 C).
Taken together, these results show that persistent expression of a nonsheddable p55TNFR on the cell surface lowers the thresholds for the induction of chronic inflammatory disease and results in exacerbated clinical outcome.
Discussion
TNF activity is controlled by multiple regulatory mechanisms that maintain a balance between beneficial effects in immune regulation and host defense, and potentially hazardous cytotoxic and proinflammatory functions. The activation-induced shedding of the TNFRs is thought to provide a means for the regulation of TNF activity; however, the in vivo role and significance of receptor shedding has remained unclear. In humans, different missense mutations on the p55TNFR gene that associate with impaired receptor shedding are linked to a group of dominantly inherited autoinflammatory syndromes named TRAPS (24). To date, >30 different mutations have been linked to TRAPS, the majority of which represent single amino acid substitutions in the first two cysteine-rich domains of the extracellular part of the receptor (31). More recently, a novel TRAPS-associated mutation has been reported that substitutes isoleucine 199, an amino acid located within the core of the p55TNFR cleavage site (32). Characteristic clinical features of TRAPS include recurrent episodes of fever, myalgia, rash, abdominal pain, arthritis, and conjunctivitis, which are thought to be primarily caused by a dysregulation of the innate immune system in these patients. Some of the p55TNFR mutations associated with TRAPS do not affect shedding, suggesting that alternative mechanisms may contribute to disease pathogenesis in these patients (33).
Here we investigate in vivo the physiological role of p55TNFR shedding by using a knock-in approach to generate mice expressing nonsheddable p55TNFR. The p55 ΔNS mutation does not affect the functionality of the receptor, but completely abolishes both constitutive and stimulation-induced proteolytic shedding, resulting in the persistent expression of the p55TNFR on the cell surface. Interestingly, heterozygous expression of the mutant receptor results in strong reduction of soluble p55TNFR release upon LPS stimulation, suggesting that the p55 ΔNS mutation has a dominant effect, reproducing the dominant nature of the TRAPS mutations in humans. Although the specific mechanism of this dominant interference of the nonsheddable receptor is still unclear, it might be hypothesized that defective shedding might be due to the formation of noncleavable heterooligomers consisting of wild-type and mutant receptors (34). Alternatively, if proteolytic shedding plays a role in the physiological turnover and regulation of the number of p55TNFR molecules present on the cell surface, the nonsheddable receptors may preferentially accumulate on the surface of heterozygous cells and cause the dominant effect observed.
We show that interference with p55TNFR shedding in mice leads to the development of a spontaneous autoinflammatory reaction manifesting as a mild form of active chronic hepatitis involving local polymorphonuclear cell activation and hepatocyte apoptosis. This pathology develops in both homozygous and heterozygous mutant mice, indicating yet again the dominant nature of the mutation. With reference to potential similarities of the p55 ΔNS mice to human TRAPS, it is worth noting that amyloidosis that is occasionally observed in the human patients might be explained by such chronic TNF-dependent mild liver pathology. Notably, anti-TNF (etanercept) administration to TRAPS patients results in dramatic improvement in both clinical and laboratory parameters, and decreases the severity, duration, and frequency of inflammatory symptoms (35). Genetic elimination of TNF expression prevents the development of the liver phenotype in p55 ΔNS mice, demonstrating that TNF-induced p55TNFR signaling is critical for disease pathogenesis in this model, similarly to human TRAPS patients. In addition, these liver lesions develop in the absence of FasL expression, showing that Fas/FasL signaling does not play an essential role for disease pathogenesis in p55 ΔNS mice. Furthermore, the hepatic lesions develop with similar severity in mice lacking both B and T lymphocytes, suggesting that antigen-specific immune responses are not required for the liver pathology in this model. Therefore, it seems that the liver lesions in p55 ΔNS mice are caused by an autoinflammatory reaction driven by the innate immune system.
Our results show that inability to shed the p55TNFR from the cell surface results in enhanced macrophage activation and increased release of effector molecules in response to stimulation of Toll-like receptor 4 and Toll-like receptor 3 using LPS and double stranded RNA, respectively. This hyperactivity can be explained at least in part by the finding that macrophages expressing nonsheddable p55TNFR express increased levels of TNF in response to LPS, suggesting that TNF expression is augmented through a positive regulatory loop requiring the presence of surface p55TNFR. The presence of significantly increased levels of TNF in the serum of LPS-injected p55 ΔNS mice compared with controls also shows that in vivo, TNF accumulates at higher levels in the absence of p55TNFR shedding. Therefore, it seems that the defective shedding of p55TNFR potentiates TNF function in the p55 ΔNS mice.
We further show that both the beneficial and the hazardous effects of TNF are exacerbated in mice expressing nonsheddable p55TNFR. p55 ΔNS mice show increased resistance to infection with L. monocytogenes, demonstrating that lack of p55TNFR shedding potentiates antibacterial host defense mechanisms controlled by TNF. On the other hand, p55 ΔNS mice are more susceptible to acute and chronic inflammatory diseases. Impaired shedding of the p55TNFR sensitizes mice to the toxicity of LPS by transforming an otherwise TNF-independent and sublethal challenge to a TNF-dependent lethal endotoxic shock reaction. Therefore, conceivably, reduced shedding of p55TNFR in humans could lead to an increased susceptibility to inflammatory complications including acute septic shock and severe sepsis. The lack of p55TNFR shedding in the p55 ΔNS model also led to exacerbated clinical outcome in two models of chronic inflammatory diseases. The expression of nonsheddable p55TNFR led to aggravated disease in a genetic model of TNF-dependent arthritis and to increased incidence and more severe clinical course of autoimmune demyelination in EAE. Although the detailed mechanisms underlying these exacerbations remain to be studied, it might be predicted that cellular hyperactivation due to sustained expression of the p55TNFR on the cell surface, coupled with enhanced and sustained production of TNF and perhaps also the absence of soluble p55TNFR at the time of response to TNF, alters the thresholds of immune activation and tips the balance toward more aggressive inflammatory reactions. Indirect effects of the mutation may also be at play, potentially enhancing susceptibility to proinflammatory stimuli due to an already established inflammatory state.
Taken together, our findings demonstrate that the proteolytic shedding of the p55TNFR constitutes a critically important mechanism for the regulation of TNF activity in vivo. Although impaired receptor shedding is beneficial for host defense to bacterial pathogens, it inevitably leads to increased susceptibility to acute and chronic inflammatory conditions. Thus, p55TNFR shedding is a mechanism that evolved to balance the two opposing faces of TNF action. Given the potential selective advantage that mutations affecting shedding of the p55TNFR may offer to antibacterial host defenses, it might be anticipated that such mutations are conserved and widespread in the human population. Therefore, assessment of TNFR shedding in humans might be of important prognostic value for susceptibility to a variety of chronic inflammatory and autoimmune diseases.
Acknowledgments
We wish to thank Spiros Lalos for technical assistance in histopathology, Wim Buurman for murine TNF and receptor-specific ELISAs, and George Kontogiorgos and Katerina Kontoyiannis for consultation in histopathology.
This work was supported by European Commission grants QLG1-CT1999-00202, QLK6-1999-02203, QLRT-CT-2001-01407, and QLRT-2001-00422 to G. Kollias.
The authors have no conflicting financial interests.
S. Xanthoulea and M. Pasparakis contributed equally to this work.
C. Brakebusch's present address is Max Planck Institute for Biochemistry, Dept. of Molecular Medicine, Martinsried 82152, Germany.
Abbreviations used in this paper: EAE, experimental autoimmune encephalomyelitis; MEF, mouse embryonic fibroblast; MOG, myelin oligodendrocyte glycoprotein; NO, nitric oxide; poly IC, polyinosinic-polycytidylic acid; PTx, pertussis toxin; TRAPS, TNFR1-associated periodic syndromes.
References
- 1.Smith, C.A., T. Farrah, and R.G. Goodwin. 1994. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 76:959–962. [DOI] [PubMed] [Google Scholar]
- 2.Wallach, D., E.E. Varfolomeev, N.L. Malinin, Y.V. Goltsev, A.V. Kovalenko, and M.P. Boldin. 1999. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 17:331–367. [DOI] [PubMed] [Google Scholar]
- 3.Wajant, H., K. Pfizenmaier, and P. Scheurich. 2003. Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. [DOI] [PubMed] [Google Scholar]
- 4.Pasparakis, M., S. Kousteni, J. Peschon, and G. Kollias. 2000. Tumor necrosis factor and the p55TNF receptor are required for optimal development of the marginal sinus and for migration of follicular dendritic cell precursors into splenic follicles. Cell. Immunol. 201:33–41. [DOI] [PubMed] [Google Scholar]
- 5.Steinshamn, S., M.H. Bemelmans, L.J. van Tits, K. Bergh, W.A. Buurman, and A. Waage. 1996. TNF receptors in murine Candida albicans infection: evidence for an important role of TNF receptor p55 in antifungal defense. J. Immunol. 157:2155–2159. [PubMed] [Google Scholar]
- 6.Deckert-Schluter, M., H. Bluethmann, A. Rang, H. Hof, and D. Schluter. 1998. Crucial role of TNF receptor type 1 (p55), but not of TNF receptor type 2 (p75), in murine toxoplasmosis. J. Immunol. 160:3427–3436. [PubMed] [Google Scholar]
- 7.Castanos-Velez, E., S. Maerlan, L.M. Osorio, F. Aberg, P. Biberfeld, A. Orn, and M.E. Rottenberg. 1998. Trypanosoma cruzi infection in tumor necrosis factor receptor p55-deficient mice. Infect. Immun. 66:2960–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Brien, D.P., D.E. Briles, A.J. Szalai, A.H. Tu, I. Sanz, and M.H. Nahm. 1999. Tumor necrosis factor alpha receptor I is important for survival from Streptococcus pneumoniae infections. Infect. Immun. 67:595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kollias, G., E. Douni, G. Kassiotis, and D. Kontoyiannis. 1999. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol. Rev. 169:175–194. [DOI] [PubMed] [Google Scholar]
- 10.Rothe, J., W. Lesslauer, H. Lotscher, Y. Lang, P. Koebel, F. Kontgen, A. Althage, R. Zinkernagel, M. Steinmetz, and H. Bluethmann. 1993. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 364:798–802. [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer, K., T. Matsuyama, T.M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P.S. Ohashi, M. Kronke, and T.W. Mak. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 73:457–467. [DOI] [PubMed] [Google Scholar]
- 12.Peschon, J.J., D.S. Torrance, K.L. Stocking, M.B. Glaccum, C. Otten, C.R. Willis, K. Charrier, P.J. Morrissey, C.B. Ware, and K.M. Mohler. 1998. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 160:943–952. [PubMed] [Google Scholar]
- 13.Pinckard, J.K., K.C. Sheehan, C.D. Arthur, and R.D. Schreiber. 1997. Constitutive shedding of both p55 and p75 murine TNF receptors in vivo. J. Immunol. 158:3869–3873. [PubMed] [Google Scholar]
- 14.Diez-Ruiz, A., G.P. Tilz, R. Zangerle, G. Baier-Bitterlich, H. Wachter, and D. Fuchs. 1995. Soluble receptors for tumour necrosis factor in clinical laboratory diagnosis. Eur. J. Haematol. 54:1–8. [DOI] [PubMed] [Google Scholar]
- 15.Lantz, M., S. Malik, M.L. Slevin, and I. Olsson. 1990. Infusion of tumor necrosis factor (TNF) causes an increase in circulating TNF-binding protein in humans. Cytokine. 2:402–406. [DOI] [PubMed] [Google Scholar]
- 16.Jansen, J., T. van der Poll, M. Levi, H. ten Cate, H. Gallati, J.W. ten Cate, and S.J. van Deventer. 1995. Inhibition of the release of soluble tumor necrosis factor receptors in experimental endotoxemia by an anti-tumor necrosis factor-alpha antibody. J. Clin. Immunol. 15:45–50. [DOI] [PubMed] [Google Scholar]
- 17.Lantz, M., U. Gullberg, E. Nilsson, and I. Olsson. 1990. Characterization in vitro of a human tumor necrosis factor-binding protein. A soluble form of a tumor necrosis factor receptor. J. Clin. Invest. 86:1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leeuwenberg, J.F., M.A. Dentener, and W.A. Buurman. 1994. Lipopolysaccharide LPS-mediated soluble TNF receptor release and TNF receptor expression by monocytes. Role of CD14, LPS binding protein, and bactericidal/permeability-increasing protein. J. Immunol. 152:5070–5076. [PubMed] [Google Scholar]
- 19.Leeuwenberg, J.F., T.M. Jeunhomme, and W.A. Buurman. 1994. Slow release of soluble TNF receptors by monocytes in vitro. J. Immunol. 152:4036–4043. [PubMed] [Google Scholar]
- 20.Porteu, F., and C. Nathan. 1990. Shedding of tumor necrosis factor receptors by activated human neutrophils. J. Exp. Med. 172:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aderka, D. 1996. The potential biological and clinical significance of the soluble tumor necrosis factor receptors. Cytokine Growth Factor Rev. 7:231–240. [DOI] [PubMed] [Google Scholar]
- 22.Sheehan, K.C., J.K. Pinckard, C.D. Arthur, L.P. Dehner, D.V. Goeddel, and R.D. Schreiber. 1995. Monoclonal antibodies specific for murine p55 and p75 tumor necrosis factor receptors: identification of a novel in vivo role for p75. J. Exp. Med. 181:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Ostade, X., P. Vandenabeele, B. Everaerdt, H. Loetscher, R. Gentz, M. Brockhaus, W. Lesslauer, J. Tavernier, P. Brouckaert, and W. Fiers. 1993. Human TNF mutants with selective activity on the p55 receptor. Nature. 361:266–269. [DOI] [PubMed] [Google Scholar]
- 24.McDermott, M.F., I. Aksentijevich, J. Galon, E.M. McDermott, B.W. Ogunkolade, M. Centola, E. Mansfield, M. Gadina, L. Karenko, T. Pettersson, et al. 1999. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 97:133–144. [DOI] [PubMed] [Google Scholar]
- 25.Mullberg, J., F.H. Durie, C. Otten-Evans, M.R. Alderson, S. Rose-John, D. Cosman, R.A. Black, and K.M. Mohler. 1995. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J. Immunol. 155:5198–5205. [PubMed] [Google Scholar]
- 26.Lewis, M., L.A. Tartaglia, A. Lee, G.L. Bennett, G.C. Rice, G.H. Wong, E.Y. Chen, and D.V. Goeddel. 1991. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA. 88:2830–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumoto, M., S. Mariathasan, M.H. Nahm, F. Baranyay, J.J. Peschon, and D.D. Chaplin. 1996. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 271:1289–1291. [DOI] [PubMed] [Google Scholar]
- 28.Pasparakis, M., L. Alexopoulou, V. Episkopou, and G. Kollias. 1996. Immune and inflammatory responses in TNF α–deficient mice: a critical requirement for TNF α in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 184:1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bemelmans, M.H., D.J. Gouma, and W.A. Buurman. 1993. LPS-induced sTNF-receptor release in vivo in a murine model. Investigation of the role of tumor necrosis factor, IL-1, leukemia inhibiting factor, and IFN-gamma. J. Immunol. 151:5554–5562. [PubMed] [Google Scholar]
- 30.Keffer, J., L. Probert, H. Cazlaris, S. Georgopoulos, E. Kaslaris, D. Kioussis, and G. Kollias. 1991. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 10:4025–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarrauste de Menthiere, C., S. Terriere, D. Pugnere, M. Ruiz, J. Demaille, and I. Touitou. 2003. INFEVERS: the registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res. 31:282–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriegel, M.A., U. Huffmeier, E. Scherb, C. Scheidig, T. Geiler, J.R. Kalden, A. Reis, and H.M. Lorenz. 2003. Tumor necrosis factor receptor-associated periodic syndrome characterized by a mutation affecting the cleavage site of the receptor: implications for pathogenesis. Arthritis Rheum. 48:2386–2388. [DOI] [PubMed] [Google Scholar]
- 33.Hull, K.M., E. Drewe, I. Aksentijevich, H.K. Singh, K. Wong, E.M. McDermott, J. Dean, R.J. Powell, and D.L. Kastner. 2002. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore). 81:349–368. [DOI] [PubMed] [Google Scholar]
- 34.Chan, F.K., H.J. Chun, L. Zheng, R.M. Siegel, K.L. Bui, and M.J. Lenardo. 2000. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 288:2351–2354. [DOI] [PubMed] [Google Scholar]
- 35.Galon, J., I. Aksentijevich, M.F. McDermott, J.J. O'Shea, and D.L. Kastner. 2000. TNFRSF1A mutations and autoinflammatory syndromes. Curr. Opin. Immunol. 12:479–486. [DOI] [PubMed] [Google Scholar]
- 36.Brakebusch, C., E.E. Varfolomeev, M. Batkin, and D. Wallach. 1994. Structural requirements for inducible shedding of the p55 tumor necrosis factor receptor. J. Biol. Chem. 269:32488–32496. [PubMed] [Google Scholar]
- 37.Robertson, E., A. Bradley, M. Kuehn, and M. Evans. 1986. Germ-line transmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature. 323:445–448. [DOI] [PubMed] [Google Scholar]
- 38.Spanopoulou, E., C.A. Roman, L.M. Corcoran, M.S. Schlissel, D.P. Silver, D. Nemazee, M.C. Nussenzweig, S.A. Shinton, R.R. Hardy, and D. Baltimore. 1994. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. Genes Dev. 8:1030–1042. [DOI] [PubMed] [Google Scholar]
- 39.Erickson, S.L., F.J. de Sauvage, K. Kikly, K. Carver-Moore, S. Pitts-Meek, N. Gillett, K.C. Sheehan, R.D. Schreiber, D.V. Goeddel, and M.W. Moore. 1994. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 372:560–563. [DOI] [PubMed] [Google Scholar]
- 40.Cossart, P., M.F. Vicente, J. Mengaud, F. Baquero, J.C. Perez-Diaz, and P. Berche. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57:3629–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stuehr, D.J., and C.F. Nathan. 1989. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 169:1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]