

Abstract
Tumor-specific CD8+ T cells can potentially be activated by two distinct mechanisms of major histocompatibility complex class I–restricted antigen presentation as follows: direct presentation by tumor cells themselves or indirect presentation by professional antigen-presenting cells (APCs). However, controversy still exists as to whether indirect presentation (the cross-priming mechanism) can contribute to effective in vivo priming of tumor-specific CD8+ T cells that are capable of eradicating cancer in patients. A clinical trial of vaccination with granulocyte macrophage–colony stimulating factor–transduced pancreatic cancer lines was designed to test whether cross-presentation by locally recruited APCs can activate pancreatic tumor-specific CD8+ T cells. Previously, we reported postvaccination delayed-type hypersensitivity (DTH) responses to autologous tumor in 3 out of 14 treated patients. Mesothelin is an antigen demonstrated previously by gene expression profiling to be up-regulated in most pancreatic cancers. We report here the consistent induction of CD8+ T cell responses to multiple HLA-A2, A3, and A24-restricted mesothelin epitopes exclusively in the three patients with vaccine-induced DTH responses. Importantly, neither of the vaccinating pancreatic cancer cell lines expressed HLA-A2, A3, or A24. These results provide the first direct evidence that CD8 T cell responses can be generated via cross-presentation by an immunotherapy approach designed to recruit APCs to the vaccination site.
Keywords: immunotherapy, antigen, cancer vaccine, epitopes, T lymphocytes
Introduction
A major goal of vaccine development is to design immunization strategies that activate CD8+ T cells. CD8+ T cells are the effector cells most capable of directly recognizing and lysing their target, whether it be a virally infected cell or a tumor cell. Activation of CD8+ T cells requires target cell presentation of antigen on MHC class I (1). In a host with cancer, tumor cells can present endogenous MHC class I–restricted antigens to CD8+ T cells by direct presentation. However, the APCs of the host, rather than the tumor cells themselves, can also process and present acquired tumor antigens captured from the tumor's microenvironment, to prime CD8+ T cells. The mechanism of transferring exogenously acquired antigens from the APC endocytic processing and presentation pathway into the cytosol for processing and presentation via the proteosome (the endogenous processing and presentation pathway) is referred to as cross-priming (2–10).
Many preclinical studies have demonstrated that APCs have the ability to capture dying cells, and process and present captured antigens expressed by these cells to CD8+ T cells (5, 8, 11–16). Several groups have shown that the DC, in particular, exhibits efficient cross-priming in both human and mouse models in vitro (6, 17). Although cross-priming has been confirmed as a mechanism by which CD8+ T cells can be primed in vitro, controversy still exists concerning the efficiency of this mechanism at priming CD8+ T cells in vivo (18). In several murine tumor models, Zinkernagel et al. found that, whereas MHC class II–restricted antigens were efficiently cross-presented, CTL activation occurred exclusively via direct presentation of MHC class I–restricted antigens by the tumor (18). A better understanding of the role of the cross-priming mechanism in the induction of CD8+ T cells in vivo will have important implications for future vaccine development.
Several cancer vaccine approaches under clinical development specifically aim to recruit and activate DCs as a first step in priming both CD4+ and CD8+ T cells (19, 20). The unique capacity of DCs to stimulate tumor-reactive T cell lines from cancer patients emphasizes the importance of the cell type in recruiting cryptic populations of tolerant or low affinity T cells into an antitumor response (21). In particular, whole cell vaccine approaches have already demonstrated that the APCs of the host, rather than the vaccinating tumor cells themselves, can prime both CD4+ and CD8+ T cells that are capable of generating systemic antitumor immunity against transplanted murine tumors in vivo (14, 22, 23). It has been more difficult to prove that cross-priming is involved in the induction of clinically meaningful CD8+ T cell responses in patients. The major impediment has been the lack of correlation of immunization and the induction of T cell responses in reported studies.
In a recently completed phase I trial, a vaccine consisting of two allogeneic, GM-CSF–secreting pancreatic tumor cell lines induced a dose-dependent delayed-type hypersensitivity (DTH) response to autologous tumor cells in 3 out of 14 patients (24). A whole cell tumor vaccine approach allows for polyvalent immunizations under circumstances where relevant tumor rejection antigens have not yet been identified. This allogeneic GM-CSF–secreting pancreatic tumor vaccine was specifically designed to test whether GM-CSF can recruit APCs, in particular DCs, to the site of vaccination and subsequently prime CD8+ T cells by the cross-priming mechanism. To determine whether this vaccine induced CD8+ T cell responses and to study the mechanism of activation of these responses, we have developed a functional genomic approach that uses immunized lymphocytes from vaccinated patients to identify immunologically recognized tumor-associated antigens from among genes overexpressed in the relevant tumor type. Here, we identify a pancreatic tumor-associated antigen, mesothelin, as a relevant target of vaccine-induced CD8+ T cell responses. We use these responses to directly evaluate the capacity of the GM-CSF–transduced vaccines to induce cross-priming in pancreatic cancer patients.
Materials and Methods
Identification of Candidate Genes and Epitope Selection.
Serial analysis of gene expression (SAGE) was used to identify mesothelin as one of the genes overexpressed in pancreatic cancer cell lines and fresh tissue (25, 26). Two computer algorithms “BIMAS” (27) and “SYFPEITHI” (28) that are available to the general public and accessible through the internet were used to predict peptides that bind to HLA-A2, A3, and A24 molecules.
Peptides and T2 Cell Lines.
All peptides were purified to >95% purity and synthesized by Macromolecular Resources according to the following published sequences: M1 peptide(58–66) GILGFVFTL, derived from influenza matrix protein (29); mesothelin A2(20–28) peptide SLLFLLFSL; mesothelin A2 (530–538) peptide VLPLTVAEV, identified using the available databases; and HIV-gag A2 peptide SLYNTVATL(75–83) (30), which contains an HLA-A2 binding motif. Mesothelin A3(83–91) peptide ELAVALAQK, mesothelin A3(225–233) peptide ALQGGGPPY, and HIV-NEF A3(94–103) peptide QVPLRPMTYK (31) contain an HLA-A3 binding motif. Mesothelin A24(435–443) peptide FYPGYLCSL, mesothelin A24(475–483) peptide LYPKARLAF, and tyrosinase peptide AFLPWHRLF(206–214) (32) contain an HLA-A24 binding motif. The mesothelin A1(309–317) peptide EIDESLIFY was used as a negative control peptide and contains an HLA-A1 binding motif. Stock solutions (10 mg/ml) of peptides were prepared in 100% DMSO (JTBaker) and further diluted in cell culture medium to yield a final peptide concentration of 10 ng/ml for each assay. The T2 cells are a human B and T lymphoblast hybrid that only expresses the HLA-A*0201 allele (33). T2 cells are TAP deficient and, therefore, fail to transport newly processed HLA class I binding epitopes from the cytosol into the endoplasmic reticulum, where these epitopes would normally bind to nascent HLA molecules and stabilize them for expression on the cell surface (33). The T2-A3 are T2 cells genetically modified to express the HLA-A*0301 allele and were a gift from W. Storkus (University of Pittsburgh, Pittsburgh, PA; reference 34). The T2-A24 are T2 cells genetically modified to express the HLA-A24 allele. The HLA-A24 gene was a gift from P. Robbins (National Cancer Institute, Bethesda, MD; reference 32). T2 cells were grown in suspension culture in RPMI 1640 (GIBCO BRL), 10% FBS (Hyclone) supplemented with 200 μM l-glutamine (JRH Biosciences), 50 U/μg/ml of Pen/Strep (Sigma-Aldrich), 1% NEAA (Sigma-Aldrich), and 1% Na-Pyruvate (Sigma-Aldrich) in 5% CO2 at 37°C.
Peptide/MHC Binding Assays.
T2 cells expressing the HLA molecule of interest were resuspended in AimV serum-free media (GIBCO BRL) to a concentration of 106 cells/ml and pulsed with β-2 microglobulin (β2-M) plus peptide at concentrations ranging from 0 to 225 μg/ml of peptide at room temperature overnight. The level of stabilized MHC on the cell surface of the T2 and T2-A24 cells was analyzed by direct staining of cell samples with unlabeled anti–class I mAb W6/32 and a goat anti–mouse FITC-labeled IgG2a secondary antibody. The level of stabilized MHC on the cell surface of the T2-A3 cells was analyzed by direct staining of cell samples with unlabeled anti–HLA-A3 mAb GAPA3 and a goat anti–mouse FITC-labeled IgG2a secondary antibody. Viable cells, as determined by exclusion of propidium iodide, were analyzed by flow cytometry on a dual laser FACSCalibur™ (Becton Dickinson) using FlowJo analysis software (Treestar). Data are expressed as an increase in mean fluorescence intensity (ΔMFI) of cells with each peptide compared with that determined for cells without peptide or a negative control peptide.
PBLs and Donors.
Peripheral blood (100 cc prevaccination and 28 d after each vaccination) were obtained from all 14 patients who received an allogeneic GM-CSF–secreting pancreatic tumor vaccine as part of a previously reported phase I vaccine analysis (24). Informed consent for banking lymphocytes to be used for this antigen identification study was obtained at the time of patient enrollment into the study. Pre- and postvaccine PBLs were isolated by density gradient centrifugation using Ficoll-Hypaque (Amersham Biosciences). Cells were washed twice with serum-free RPMI 1640. PBLs were stored frozen at −140°C in 90% AIM-V media containing 10% DMSO.
Enrichment of PBLs for CD8+ T Cells.
CD8+ T cells were isolated from thawed PBLs using magnetic cell sorting of human leukocytes as per the manufacturer's directions (MACS; Miltenyi Biotec). Cells were fluorescently stained with CD8-PE antibody (Becton Dickinson) to confirm that the positive population contained CD8+ T cells and analyzed by flow cytometry. This procedure consistently yielded >95% CD8+ T cell purity.
CD8+ M1-specific T Cell Lines.
M1-specific T cell lines were generated by repeated in vitro stimulation of HLA-A*0201+ PBLs initially with irradiated autologous dendritic cells followed by irradiated autologous EBV-transformed B cells, both pulsed with the HLA-A*0201–restricted epitope. T cells were stimulated at a 1:2 T cell/EBV cell ratio in T cell media consisting of RPMI 1640, 10% human serum (pooled serum collected at the Johns Hopkins University Hemapheresis Unit), 200 μM l-Glutamine, 50 U/μg/ml Pen/Strep, 10 mM Hepes (GIBCO BRL) supplemented with 60 international units IL-2/ml (R&D Systems), and 10 ng/well IL-7 (R&D Systems). This line was used as a positive control T cell line in all assays.
ELISPOT Assay.
Multiscreen 96-well filtration plates (Millipore) were coated overnight at 4°C with 60 μl/well of 10 μg/ml anti–hIFN-γ mouse Mab 1-D1K (Mabtech). Wells were washed three times each with PBS and blocked for 2 h with T cell media. 105 T2 cells pulsed with 10 ng/ml of peptide in 100 μl of T cell media were incubated overnight with 105 thawed PBLs that were purified to select CD8+ T cells in 100 μl T cell media on the ELISPOT plates in replicates of six. The plates were incubated overnight at 37°C in 5% CO2. Cells were removed from the ELISPOT plates by washing six times with PBS + 0.05% Tween 20 (Sigma-Aldrich). Wells were incubated for 2 h at 37°C in 5% CO2 using 60 μl/well of 2 μg/ml of biotinylated Mab anti–hIFN-γ 7-B6-1 (Mabtech). The avidin peroxidase complex (Vectastain ELITE ABC kit; Vector Laboratories) was added after washing six times with PBS/Tween 0.05% at 100 μl/well and incubated for 1 h at room temperature. AEC substrate solution (3-amino-9-ethylcarbazole) was added at 100 μl/well and incubated for 4–12 min at room temperature. Color development was stopped by washing with tap water. Plates were dried overnight at room temperature, and colored spots were counted using an automated image system ELISPOT reader (Axioplan2; Carl Zeiss Microimaging, Inc.).
Flow Cytometry.
The cell lines were washed twice and resuspended in FACS® buffer (HBSS supplemented with 1% PBS, 2% FBS, and 0.2% sodium azide), stained with mouse monoclonal mesothelin (CAK1; Signet Laboratories) followed by FITC-labeled goat anti–mouse IgG1 (BD Biosciences) for flow analysis in a FACScan™ analyzer (BD Immunocytometry Systems).
In Vitro Generation of Tumor-reactive CTLs.
Purified monocytes were cultured for 4 d in the presence of 100 ng/ml recombinant human GM-CSF (R&D Systems) and 10 ng/ml rhIL-4 (R&D Systems) in complete RPMI 1640 medium (35). The monocytes were activated overnight by incubation with 0.5 μg/ml LPS (Sigma-Aldrich). The tissue culture–generated monocytes were pulsed with 30 μg/ml of synthetic peptides together with 3 μg/ml β2-M (Sigma-Aldrich) in PBS containing 1% human serum albumin (Sigma-Aldrich) for 4 h at room temperature. The peptide-pulsed DCs were washed twice, irradiated (4,200 rad), and mixed with autologous CD8+ T cells (purified with antibody-coated magnetic beads by positive selection; Miltenyi Biotec) at a 1:10 (DC/T cell) ratio. This medium was supplemented with 10 ng/ml rhIL-7 (R&D Systems). 1 d later, 60 international units/ml rhIL-2 (R&D Systems) were added to the cultures to increase the efficiency of CTL induction. Approximately every 10 d, the T cell cultures were restimulated with irradiated peptide-pulsed autologous DCs as previously mentioned, adding rhIL-7 and rhIL-2 on the same day. The cytotoxicity assays were performed after three rounds of peptide stimulation.
Chromium Release Assay.
106 target cells were labeled in 100 μl complete medium and 100 μCi 51Cr (Amersham Biosciences) at 37°C for 1–1.5 h (36). To determine the mesothelin-specific lysis from patient 13 CD8+ T cell line, 51Cr-labeled target cells (3 × 103) were added to varying concentrations of the CD8+ T cell line in a total of 200 μl in a v-bottom 96-well plate for 4 h at 37°C. Each data point was performed in triplicate and averaged. Data are expressed as percentage of specific lysis = (measured release − spontaneous release) − (maximum release − spontaneous release) × 100. The spontaneous release ranged between 10 and 15% of the total label incorporated into the cells. For HLA-blocking studies, either the pan-HLA antibody W6/32 (HB-95; American Type Culture Collection) or the isotype matched anti–Schistosoma mansoni antibody, MBL (HB-193; American Type Culture Collection) were added to the target cells (50 μg/ml) for 30 min at 37°C before adding the T cells.
Results
Vaccination with an Allogeneic Pancreatic Tumor Vaccine Induces Mesothelin-specific CD8+ T Cells.
To determine whether the cross-priming mechanism is functional and efficient in inducing CD8+ T cells in pancreatic cancer patients receiving an allogeneic, GM-CSF–secreting vaccine, we first needed to identify pancreatic tumor antigens against which vaccine-induced immune responses are elicited. A growing number of genes shown to be differentially expressed in pancreatic adenocarcinomas using SAGE have been tabulated and reported (25, 26, 37). We screened this SAGE analysis database to identify genes that can also serve as potential immune targets for the majority of pancreatic adenocarcinoma patients. We focused specifically only on those genes that were nonmutated, overexpressed by the majority of pancreatic cancer patients, and overexpressed by the vaccine cell lines (all being important requirements for evaluating CD8+ T cell cross-priming). One gene that met all three criteria was mesothelin (25), the focus of this paper. We used the combination of two public-use computer algorithms (27, 28, 35) to predict peptide nonamers that bind to three common HLA class I molecules. Both computerized algorithms score candidate epitopes based on amino acid sequences within a given protein that have similar binding motifs to previously published HLA-binding epitopes. We synthesized the top two ranking mesothelin epitopes for HLA-A2, HLA-A3, and HLA-A24 favored by both algorithms because at least one of these three HLA class I molecules is expressed by each of the 14 patients that were treated in the vaccine paper (24). The human T2 cell line, which expresses empty MHC class I molecules on its surface because it is TAP transporter deficient, was used to confirm epitope binding to MHC class I (33). Binding of these epitopes to their respective HLA class I molecule was confirmed by pulsing TAP-deficient T2 cells that expressed the corresponding HLA class I molecule (T2-A2, T2-A3, or T2-A24 cells). As shown in Fig. 1 A, pulsing of two mesothelin-derived epitopes predicted to bind to HLA-A2 allowed for detection of HLA-A2 on the cell surface of T2-A2 cells by flow cytometry after staining with the HLA class I–specific antibody, W6/32. In contrast, T2 cells pulsed with a mesothelin epitope predicted to bind to HLA-A1 do not stain with the same antibody. Binding of T2 cells pulsed with two candidate mesothelin-derived HLA-A3 and two candidate HLA-A24 epitopes demonstrated similar results (Fig. 1, B and C, respectively).
Figure 1.
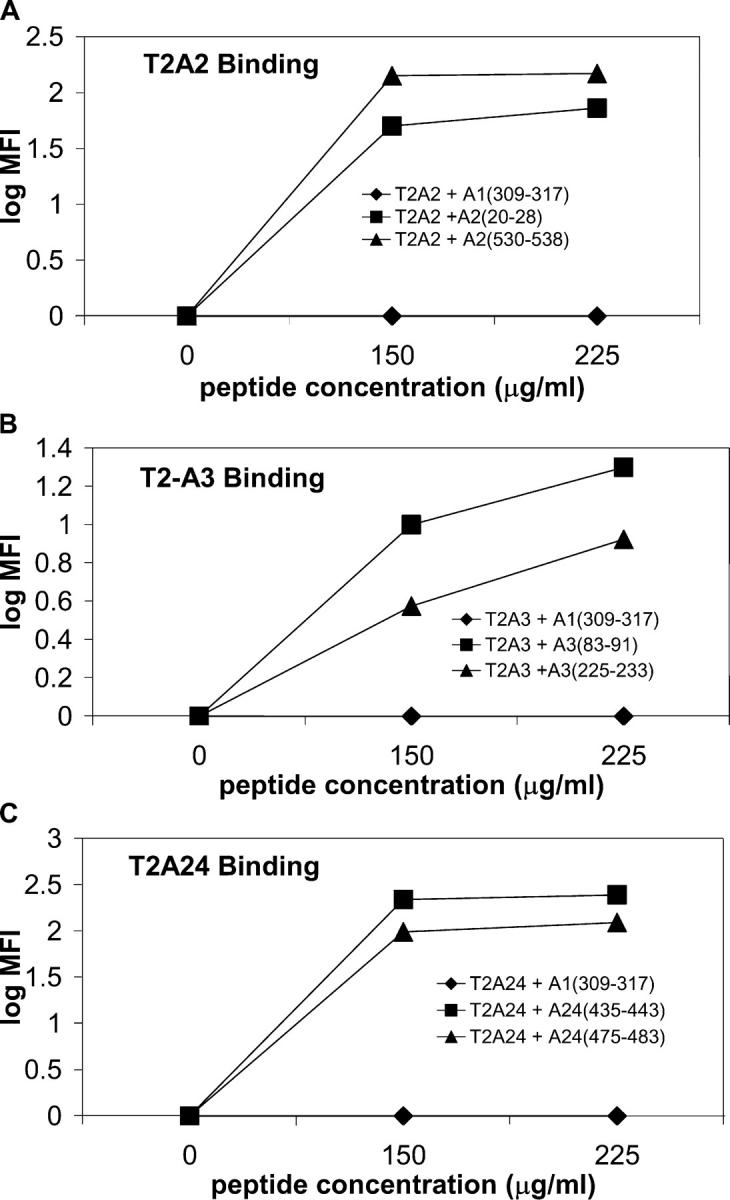
T2 binding assay identifies mesothelin protein-derived epitopes that bind to HLA-A2, A3, and A24 molecules. T2 cells were pulsed with 0–225 μg/ml of peptide overnight at room temperature before analysis by flow cytometry. (A) T2 cells expressing HLA-A2 and pulsed with either a mesothelin A1(309–317) peptide (closed diamond), mesothelin A2(20–28) (closed square), and mesothelin A2(530–538) (closed triangle). (B) T2 cells genetically modified to express HLA-A3 and pulsed with either mesothelin A1(309–317) peptide (closed diamond), mesothelin A3(83–91) (closed square), and mesothelin A3(225–233) (closed triangle). (C) T2 cells genetically modified to express HLA-A24 and pulsed with either mesothelin A1(309–317) peptide (closed diamond), mesothelin A24(435–443) (closed square), and mesothelin A24(475–483) (closed triangle).
To determine if mesothelin is recognized by CD8+ T cells, we screened antigen-pulsed T2 cells in a quantitative ELISPOT-based assay using pre- and postvaccination CD8+ T cell–enriched PBLs from the 14 patients treated previously with the allogeneic, GM-CSF–secreting pancreatic tumor vaccine. Previously, we reported the association of in vivo postvaccination DTH responses to autologous tumor in three out of eight patients receiving the highest two doses of vaccine. PBLs obtained before vaccination and 28 d after the first vaccination were initially analyzed. T2-A3 cells pulsed with the two A3 binding epitopes were incubated overnight with CD8+ T cell–enriched lymphocytes isolated from the peripheral blood of patients 11 (an A3 non-DTH responder) and 13 (an A3 DTH responder), and analyzed using an IFN-γ ELISPOT assay. The ELISPOT assay was chosen because it requires relatively few lymphocytes, is among the most sensitive in vitro assays for quantitating antigen-specific T cells, and correlates the number of antigen-specific T cells with function (cytokine expression; references 38–40). Induction of mesothelin-specific T cells was detected 28 d after vaccination in patient 13, a DTH responder, but not in patient 11, a non-DTH responder (Fig. 2 A). Similarly, postvaccination induction of mesothelin-specific CD8+ T cells was also observed in the two other disease-free DTH responders (patient 8 [HLA-A2/A3] and patient 14 [HLA-A24]), but not in other non-DTH responders when tested with T2-A2/A3 (patient 2) and T2-A24 (patient 7) cells pulsed with the A2, A3 (Fig. 2 B), and A24 (Fig. 2 C) binding epitopes, respectively. A summary of the ELISPOT results evaluating vaccine-induced mesothelin-specific CD8+ T cell responses for all 14 patients treated with one allogeneic vaccination in this analysis is shown in Fig. 2 D. This difference in detection of mesothelin-specific T cell responses is statistically significant at a P < 0.001 by the Fisher exact test. These data suggest that there is a direct correlation between observed postvaccination in vivo DTH responses to autologous tumor and postvaccination in vivo mesothelin-specific CD8+ T cell responses for patients treated with an allogeneic vaccine in this paper. Specifically, each of the three DTH responders demonstrated a postvaccination induction in CD8+ T cell responses to two different mesothelin peptides that matched their respective HLA type, whereas only 1 out of 11 DTH nonresponders had an increased postvaccination mesothelin-specific CD8+ T cell response and only to a single peptide.
Figure 2.
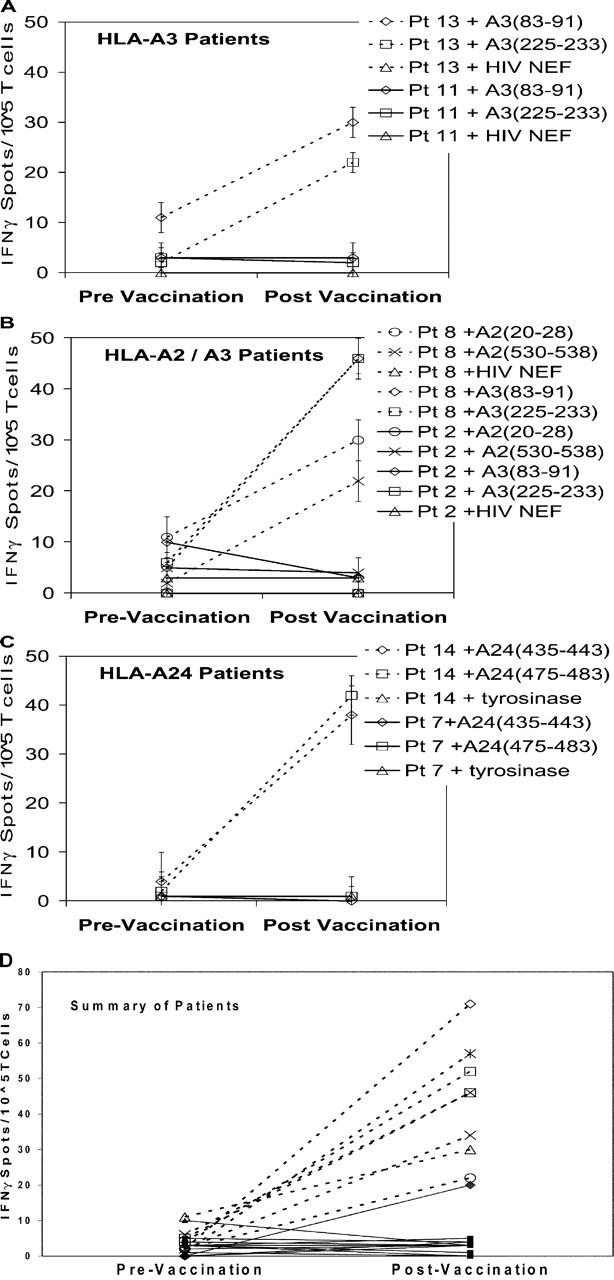
ELISPOT analysis of CD8+ T cells from PBMCs demonstrates postvaccination induction of mesothelin-specific T cells in three DTH responders. (A) ELISPOT analysis of PBLs from two patients who were HLA-A3+. (B) ELISPOT analysis of PBLs from two patients who were HLA-A-2 and HLA-A3+. (C) ELISPOT analysis of PBLs from two patients who were HLA-A24+. (D) ELISPOT analysis of PBLs from all 14 patients who were treated with the vaccine (reference 24). ELISPOT analysis for IFN-γ–expressing cells was performed using PBMCs that were isolated on the day before vaccination or 28 d after the first vaccination. T2-A3 cells were pulsed with the two mesothelin-derived epitopes MesoA3(83–91) (squares), MesoA3(225–233) (X), and HIV-NEF(94–102) (not shown). T2-A2 cells were pulsed with the two mesothelin-derived epitopes MesoA2(20–28) (triangles), MesoA2(530–538) (circles), and HIV-gag(77–85) (not shown). T2-A24 cells were pulsed with the two mesothelin-derived epitopes MesoA24(435–443) (asterisks), MesoA24 (475–483) (diamonds), and tyrosinase A24(206–214) (not shown). All DTH responders are represented by dotted lines and open symbols, and DTH nonresponders are represented by solid lines and closed symbols. For the detection of nonspecific background, the number of IFN-γ spots for CD8+ T cells specific for the irrelevant control peptides were counted. The HLA-A2 binding HIV-gag protein-derived epitope (SLYNTVATL), the HLA-A3 binding HIV-NEF protein-derived epitope (QVPLRPMTYK), and the HLA-A24 binding tyrosinase protein-derived epitope (AFLPWHRLF) were used as negative control peptides in these assays. Background was minimal to negative control peptides, ranging from zero to three spots total (data included in each graph). Data represent the mean of each condition assayed in triplicate, and standard deviations were <5%. The number of human IFN-γ spots per 105 CD8+ T cells is plotted. Analysis of each patient's PBLs was performed at least twice.
Poor Immune Status Does Not Explain the Failure to Measure Vaccine-induced, Mesothelin-specific CD8+ T Cell Responses in DTH Nonresponders.
The data in Fig. 2 correlate in vivo DTH responses to autologous tumor with the postvaccination induction of mesothelin-specific CD8+ T cell responses. However, it is possible that this correlation represents generalized differences in overall immune function between the DTH responder and nonresponder patients, rather than a vaccine-specific induction of T cell responses to mesothelin in the DTH responder patients. To demonstrate that the postvaccination induction of mesothelin-specific CD8+ T cells is tumor antigen specific, we evaluated each HLA-A2+ patient for T cell responses to the HLA-A2 binding influenza matrix peptide, M1 (29). We chose the influenza M1 peptide because most patients on the vaccine analysis had received yearly influenza vaccines as a standard of care before enrollment. As shown in Fig. 3, all HLA-A2+ patients demonstrated similar pre- and postvaccination T cell responses to the M1 peptide. Prevaccination responses ranged from 19 to 50 IFN-γ spots per 105 total CD8 T cells, and postvaccination responses remained approximately the same in each patient, unaffected by immunization with the pancreatic tumor vaccine (Fig. 3). A similar analysis confirmed that the HLA-A3 and HLA-A24 patients have detectable CD8+ T cell responses to influenza and EBV peptides (27, 28, 41), respectively, and that these responses are unaltered by immunization with the allogeneic pancreatic tumor vaccine (unpublished data).
Figure 3.

ELISPOT analysis of CD8+ T cells from PBMCs demonstrates similar pre- and postvaccination responses to the influenza matrix protein HLA-A2 binding epitope M1 (GILGFVFTL) in all HLA-A2+ patients This analysis was performed on the same PBL samples described in Fig. 2. The DTH responders are represented by dotted lines, and the DTH nonresponders are represented by solid lines. For the detection of nonspecific background, the number of IFN-γ spots for CD8+ T cells specific for the irrelevant control peptides were counted. The HLA-A2 binding HIV-gag protein-derived epitope (SLYNTVATL), the HLA-A3 binding HIV-NEF protein-derived epitope (QVPLRPMTYK), and the HLA-A24 binding melanoma tyrosinase protein-derived epitope (AFLPWHRLF) were used as negative control peptides in these assays. Data represent the mean of each condition assayed in triplicate, and standard deviations were <5%. The number of human IFN-γ spots per 105 CD8+ T cells is plotted. Analysis of each patient's PBLs was performed at least twice, and all ELISPOT assays were performed in a blinded fashion.
In Vivo Cross-priming Explains the Induction of Mesothelin-specific CD8+ T Cells in Patients Vaccinated with the Allogeneic Pancreatic Tumor Vaccine.
Vaccine-induced in vivo priming of host T cells by allogeneic tumor cells can occur by one of two mechanisms. The allogeneic tumor cells might directly prime the host's CD8+ T cells if the immunizing cells express HLA class I molecules in common with the host's HLA type. If not, transfer of the tumor antigen from the allogeneic cells to professional APCs would be required. Murine studies have already demonstrated that both mechanisms can contribute to the induction of systemic antitumor immunity (42). However, controversy still remains as to whether vaccination can result in the induction of antigen-specific CD8+ T cells via the cross-priming mechanism that are efficient enough and in large enough quantities to treat actively growing cancer in patients. In this paper, we have evaluated the induction of HLA-A locus-restricted, mesothelin-specific CD8+ T cells in patients who received an allogeneic vaccine that is mismatched at the HLA-A locus (Table I). Specifically, we have demonstrated the induction of mesothelin-specific CD8+ T cells to HLA-A2, A3, and A24 mesothelin-derived peptides in patients receiving a mixture of two allogeneic pancreatic tumor vaccines. Both of these vaccine lines overexpress mesothelin (Fig. 4). However, neither vaccine line expresses HLA-A2, A3, or A24 (Table I). Therefore, these data provide direct evidence at the epitope level that allogeneic vaccine cells can activate CD8+ T cells against shared pancreatic tumor antigens, and that these CD8+ T cell responses are associated with other measures of in vivo immune responses. Because the vaccine cells are HLA mismatched with the three DTH responders' at the HLA-A locus, CD8+ T cell activation must occur by transfer of MHC class I antigens from the tumor cells to professional APCs, where they are processed and presented on MHC class I molecules via cross-priming.
Table I.
HLA Mismatch with the Vaccine Cells at the HLA-A Locus Provide Direct Human Evidence for MHC Class I Antigen Cross-Priming by APCs
| Vaccine line Panc 10.05 |
Vaccine line Panc 6.03 |
Patient 8 | Patient 13 | Patient 14 | |
|---|---|---|---|---|---|
| HLA class I expression at the A locusa | A1, A19 | A1, A1 | A2, A3 | A3, A23 | A1, A24 |
HLA typing was performed serologically and confirmed molecularly.
Figure 4.

Expression of surface mesothelin of both the Panc 6.03 and Panc 10.05 vaccine and on cell lines used as targets for CTL assays. Panc 6.03, Panc 10.05, autologous EBV, autologous EBV transduced with mesothelin, Panc 2.5 (HLA-A3+), Panc 3.014 (HLA-A3+), and Panc 3.11 (HAL-A3−) were analyzed by flow cytometry for their levels of surface mesothelin using the mesothelin-specific monoclonal antibody CAK1 as the primary antibody and goat anti–mouse IgG1 FITC as the secondary antibody. The dotted line represents the isotype control, and the solid line represents mesothelin staining.
Cross-Priming by Vaccine Recruited APCs Results in the Generation of Mesothelin-specific CD8+ T Cells Capable of Lysing Mesothelin-expressing Tumor Cells.
The most important role of vaccine-induced CD8+ T cells in vivo is the ultimate lysis of antigen-expressing tumors. Currently, the best measure of this aspect of CD8+ T cell function is the ability of isolated CD8+ T cells to lyse antigen-expressing tumors in vitro. In an effort to correlate IFN-γ release with lytic activity in response to mesothelin, we analyzed the reactivity of a patient-derived T cell line to a panel of HLA-A3+ and HLA-A3− tumor cell lines in a 4-h chromium release assay. The level of mesothelin expression of these tumor lines is shown in Fig. 4. Autologous DCs pulsed with the HLA-A3 mesothelin peptide 6293 were used to expand patient 13 CD8+ mesothelin-specific T cells in vitro. Cytolytic activity was assessed after three in vitro stimulations against HLA-A3+ and HLA-A3− mesothelin-expressing target cells. As shown in Fig. 5 A, mesothelin-specific CD8+ T cells were able to lyse autologous EBV-transformed B cells transduced with the mesothelin gene, T2-A3 cells pulsed with the HLA-A3 mesothelin peptide 6293, and an HLA-A3+, mesothelin-expressing allogeneic tumor cell line, Panc 3.014. In a second study, lysis of the Panc 3.014 line could be blocked by the pan-HLA blocking antibody W6/32, but not by the isotype matched antibody against S. mansoni (unpublished data). In contrast, mesothelin-specific, patient-derived CD8+ T cells did not lyse well the mesothelin-expressing HLA-A3− tumor cell line Panc 3.11, nor the HLA-A3-expressing, mesothelin-negative cell lines Panc 2.5 and the nonmesothelin-expressing autologous EBV-transformed B cells. After repeated in vitro stimulation, the T cell line lysed the Panc 3.014 mesothelin and HLA-A3–expressing line well (which was genetically modified to express HLA-A3), but not the original HLA-A3−, mesothelin-expressing Panc 3.014 tumor cell line (Fig. 5 B). These studies have been repeated six times with similar lysis results. These data confirm that an allogeneic vaccine can induce mesothelin-specific CD8+ T cell responses via the cross-priming mechanism, and that these CD8+ T cells are capable of lysing mesothelin-expressing cell line.
Figure 5.

A mesothelin-specific CTL line derived from patient 13 PBL lyses HLA-A3+, mesothelin-expressing cells. Patient-derived CD8+ T cells stimulated with an HLA-A3 mesothelin peptide (peptide 6293) were tested for their capability to recognize and kill mesothelin-expressing tumor and EBV cell lines shown in Fig. 4. 51Cr-labeled target cells (3 × 103) were mixed with varying concentrations of patient 13 CD8+ T cell line (starting with 9 × 104) in a total of 200 μl in a v-bottom 96-well plate. Percent lysis was calculated after 4 h at 37°C. Results are expressed as the percentage of specific lysis of triplicate samples.
Discussion
These data describing CD8+ T cell responses induced by an allogeneic GM-CSF–secreting pancreatic tumor vaccine support the following two conclusions. First, these findings provide direct human evidence that allogeneic vaccine cells induce CD8+ T cell responses by a cross-priming mechanism that requires transfer of antigen from the vaccine cells to professional APCs. Second, mesothelin is a new candidate pancreatic tumor antigen that can be used to analyze immune responses induced by whole cell pancreatic tumor vaccines.
The identification of shared, biologically relevant tumor antigens provides the opportunity to study the mechanisms by which vaccines induce antitumor immune responses. An important result of this paper is the direct demonstration of cross-priming at the MHC class I epitope level. Controversy still exists as to whether cross-priming is a clinically important mechanism for in vivo priming of CD8+ T cells (18). Previously published murine studies evaluating whole cell vaccines have shown that the professional APCs of the host can prime both CD4+ and CD8+ T cells, both of which are required for generating systemic antitumor immunity (14, 22, 23). Furthermore, Jung et al. have previously shown that cross-priming is relevant in vivo because depletion of CD11C+ DC abrogated effective immunization in mice (43). Cross-priming has also been shown to play an important role in generating CD8+ T cell responses to infectious diseases (44–46). Furthermore, human tumor studies have demonstrated that both macrophages and DCs can take up antigens in vitro and prime naive CD8+ T cells by the cross-priming mechanism (5, 8, 11–16, 44–46). However, other studies suggest that the mechanism of cross-priming is inefficient at inducing CD8+ T cell responses in vivo in vaccinated healthy subjects (47). Several factors may explain the differences in results between these studies. First, the efficiency of cross-priming in vivo may be influenced by several factors, including the following: the route of APC exposure to the antigen (44), whether the APC is a macrophage or DC (6, 17, 47), the maturation status of the APCs (48), and the form of antigen taken up by the APCs (4, 11). The form of the antigen that is presented to the APCs has been of particular interest because several recent studies suggest that apoptotic tumors are more efficiently processed and presented by an APC than necrotic tumor or soluble protein (8, 11). Furthermore, we have reported previously that irradiated GM-CSF–secreting vaccine cells are much more efficient at inducing systemic immune responses than unirradiated vaccine cells or vaccine cells that are inactivated by nonapoptotic inducing mechanisms (49). In addition, several studies have used the ovalbumin antigen system, which is a strong foreign antigen that may be processed and presented differently than naturally occurring tumor-associated antigens (4, 6).
The biologic importance of these data in demonstrating CD8+ T cell cross-priming is strongly supported by our data correlating these responses with in vivo postvaccination DTH responses to autologous tumor cells. Until now, observed postvaccination DTH responses against autologous tumor cells has provided the best evidence in support of a vaccine-induced, T cell–mediated antitumor immunity in patients treated in clinical trials (24, 50–57). Three recent papers have linked antibody responses to clinical responses in patients receiving a melanoma vaccine (58, 59) and a human chorionic gonadotropin–based vaccine (60). However, this paper also demonstrates postvaccination in vitro antigen-specific T cell responses that correlate with in vivo evidence of immune induction (DTH responses to autologous tumor cells). In addition, the use of uncultured lymphocytes rather than T cell lines and clones that have been in long-term culture demonstrates immune responses that are more closely associated with human in vivo T cell function. Unfortunately, it is difficult to demonstrate direct pancreatic cell killing without several rounds of in vitro CD8+ T cell expansion. However, after three rounds of stimulation with autologous DCs pulsed with the A3 mesothelin peptide, these T cells can lyse a pancreatic tumor line and other mesothelin-expressing cell lines, providing evidence that mesothelin can serve as a tumor rejection target of T cells. Only a small panel of tumor lines were tested due to the significant challenge in generating in vitro pancreatic tumor lines, including the three DTH responder patients. However, the fact that the T cells lyse mesothelin-expressing HLA-A3+ tumor cells, but not mesothelin-expressing HLA-A3− lines, demonstrates the mesothelin and HLA-restricted specificity of the T cell activity.
In this paper, we also demonstrate that mesothelin-specific T cells can be induced against at least six different peptides presented by three different HLA-A locus alleles. T cell responses determined by ELISPOT were comparable for each of the epitopes. This finding provides further support that mesothelin can serve as a shared antigen. This finding also provides further evidence that cross-priming is an efficient mechanism for antigen processing and presentation onto MHC class I. It is interesting to point out that the highest-ranking antigenic epitopes predicted to be the best HLA-A allele binding epitopes based on their motif, bound to their respective HLA alleles and were also recognized by mesothelin-specific T cells. Papers analyzing other tumor antigens have found that the highest ranking epitopes do not necessarily correlate with optimal recognition by T cells (35). We also performed the computer algorithms on two melanoma antigens, tyrosinase and MAGE 1, to determine how their published HLA-A2 binding peptides rank by this method (61, 62). We found that our HLA-A2 binding mesothelin epitopes were given similar scores as the known tyrosinase and MAGE 1 HLA-A2 binding epitopes. This was also true for the published HLA-A2 HIV-gag and HLA-A3 HIV-NEF epitopes that were used as control antigens in our analyses (30, 31). Choosing epitopes that rank high by both algorithms appears to be an important predictor of the probability of binding to the respective HLA molecule. However, the likelihood of successfully predicting HLA binding epitopes will probably be determined in part by the antigen being studied. Studies aimed at addressing this question are underway.
In conclusion, we have directly demonstrated cross-priming at the MHC class I epitope level. These studies were facilitated through the development of a functional genomic approach that identified mesothelin as a new candidate pancreatic tumor antigen recognized by CD8+ T cells. The correlation of in vitro T cell responses with in vivo measures of immunologic response validates the biologic importance of this approach. Because we detected evidence for cross presentation in the three patients with a better clinical course, we suspect that cross-presentation may have clinical relevance, but larger studies are required to investigate this.
Acknowledgments
We would like to thank Dr. D. Pardoll for his critique of this paper. We also want to acknowledge L. Metzler and the flow cytometry core.
This work was supported by a Specialized Program of Research Excellence grant in Gastrointestinal Cancer from the National Cancer Institute (no. CA62924), by a grant from the Lustgarten Foundation for Pancreatic Cancer Research, and in part by a generous donation from the Goodwin Family. Dr. Jaffee is the Dana and Albert Broccoli Professor of Oncology.
Under a licensing agreement between Cell Genesys and the Johns Hopkins University, the University is entitled to milestone payments and royalty on sales of the vaccine product described in this presentation. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies. The vaccine trial was supported by grant no. RO1CA71806 from the National Cancer Institute.
Abbreviations used in this paper: DTH, delayed-type hypersensitivity; SAGE, serial analysis of gene expression.
References
- 1.Townsend, A., and H. Bodmer. 1989. Antigen recognition by class I-restricted T lymphocytes. Annu. Rev. Immunol. 7:601–624. [DOI] [PubMed] [Google Scholar]
- 2.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation in viral immunity and self-tolerance. Annu. Rev. Immunol. 1:126. [DOI] [PubMed] [Google Scholar]
- 3.Heath, W.R., and F.R. Carbone. 2001. Cross-presentation, dendritic cells, tolerance, and immunity. Annu. Rev. Immunol. 19:47. [DOI] [PubMed] [Google Scholar]
- 4.Norbury, C.C., M.F. Princiotta, I. Bacik, R.R. Brutkewicz, P. Wood, T. Elliott, J.R. Bennink, and J.W. Yewdell. 2001. Multiple antigen-specific processing pathways for activating näive CD8+ T cells in vivo. J. Immunol. 166:4355–4362. [DOI] [PubMed] [Google Scholar]
- 5.Kovacsovics-Bankowski, M., and K.L. Rock. 1995. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 267:243–246. [DOI] [PubMed] [Google Scholar]
- 6.den Haan, J.M.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuurhuis, D.H., A. Ioan-Facsinay, B. Nagelkerken, J.J. van Schipp, C. Sedlik, C.J. Melief, J.S. Verbeek, and F. Ossendorp. 2002. Antigen-antibody immune complexes empower dendritic cells to efficiently prime specific CD8+ CTL responses in vivo. J. Immunol. 168:2240–2246. [DOI] [PubMed] [Google Scholar]
- 8.Nowak, A.K., R.A. Lake, A.L. Marzo, B. Scott, W.R. Heath, E.J. Collins, J.A. Frelinger, and B.W.S. Robinson. 2003. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J. Immunol. 170:4905–4913. [DOI] [PubMed] [Google Scholar]
- 9.Yu, P., M.T. Spiotto, Y. Lee, H. Schreiber, and Y.X. Fu. 2003. Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J. Exp. Med. 197:985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohashi, P.S., and A.L. DeFranco. 2002. Making and breaking tolerance. Curr. Opin. Immunol. 14:744–759. [DOI] [PubMed] [Google Scholar]
- 11.Bellone, M., I. Giandomenica, P. Rovere, G. Galati, A. Ronchetti, M.P. Protti, J. Davoust, C. Rugarli, and A.A. Manfredi. 1997. Processing of engulfed apoptotic bodies yields T cell epitopes. J. Immunol. 159:5391–5399. [PubMed] [Google Scholar]
- 12.Nouri-Shirazi, M., J. Banchereau, D. Bell, S. Burkeholder, E.T. Kraus, J. Davoust, and K.A. Palucka. 2000. Dendritic cells capture killed tumor cells and present their antigens to elicit tumor-specific immune responses. J. Immunol. 165:3797–3808. [DOI] [PubMed] [Google Scholar]
- 13.Huang, A.Y., A.T. Bruce, D.M. Pardoll, and H.I. Levitsky. 1996. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity. 4:349–355. [DOI] [PubMed] [Google Scholar]
- 14.Huang, A.Y.C., P. Golumbek, M. Ahmadzeadeh, E.M. Jaffee, D.M. Pardoll, and H. Levitsky. 1994. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 264:961–965. [DOI] [PubMed] [Google Scholar]
- 15.Falo, L.D., Jr., M. Kovacsovics-Bankowski, K. Thompson, and K.L. Rock. 1995. Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat. Med. 1:649–653. [DOI] [PubMed] [Google Scholar]
- 16.Dai, J., B. Liu, M.M. Caudill, H. Zheng, Y. Qiao, E.R. Podack, and Z. Li. 2003. Cell surface expression of heat shock protein gp96 enhances cross-presentation of cellular antigens and the generation of tumor-specific T cell memory. Cancer Immun. 3:1–11. [PubMed] [Google Scholar]
- 17.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 18.Zinkernagel, R.M. 2002. On cross-priming of MHC class I-specific CTL: rule or exception? Eur. J. Immunol. 32:2385–2392. [DOI] [PubMed] [Google Scholar]
- 19.Pardoll, D.M. 2002. Spinning molecular immunology into successful immunotherapy. Nat. Rev. Immunol. 2:227–238. [DOI] [PubMed] [Google Scholar]
- 20.Greten, T.F., and E.M. Jaffee. 1999. Cancer vaccines. J. Clin. Oncol. 17:1047–1060. [DOI] [PubMed] [Google Scholar]
- 21.Dhodapkar, M.V., J. Krasovsky, and K. Olson. 2002. T cells from the tumor microenvironment of patients with progressive myeloma can generate strong, tumor-specific cytolytic responses to autologous, tumor-loaded dendritic cells. Proc. Natl. Acad. Sci. USA. 99:13009–13013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dranoff, G., E. Jaffee, A. Lazenby, P. Golumbek, H. Levitsky, K. Brose, V. Jackson, H. Hamada, D. Pardoll, and R.C. Mulligan. 1993. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA. 90:3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas, M.C., T.F. Greten, D.M. Pardoll, and E.M. Jaffee. 1998. Enhanced tumor protection by granulocyte-macrophage colony-stimualting factor expression at the site of an allogeneic vaccine. Hum. Gene Ther. 9:835–843. [DOI] [PubMed] [Google Scholar]
- 24.Jaffee, E.M., R.H. Hruban, B. Biedrzycki, D. Laheru, K. Schepers, P.R. Sauter, M. Goemann, J. Coleman, L. Grochow, R.C. Donehower, et al. 2001. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: phase I trial of safety and immune activation. J. Clin. Oncol. 19:145–156. [DOI] [PubMed] [Google Scholar]
- 25.Argani, P., C. Iacobuzio-Donahue, B. Ryu, C. Rosty, M. Goggins, R.E. Wilentz, S.R. Murugesan, S.D. Leach, E. Jaffee, C.J. Yeo, et al. 2001. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin. Cancer Res. 3862:3862–3868. [PubMed] [Google Scholar]
- 26.Argani, P., C. Rosty, R.E. Reiter, R.E. Wilenz, S.R. Murgesan, S.D. Leach, B. Ryu, H.G. Skinner, M. Goggins, E.M. Jaffee, et al. 2001. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 61:4320–4324. [PubMed] [Google Scholar]
- 27.Parker, K.C., M.A. Bednarek, and J.E. Coligan. 1994. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side chains. J. Immunol. 152:163–175. [PubMed] [Google Scholar]
- 28.Rammensee, H., J. Bachmann, N.P. Emmerich, O.A. Bachor, and S. Stevanovic. 1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 50:213–219. [DOI] [PubMed] [Google Scholar]
- 29.Bednarek, M.A., S.Y. Sauma, M.C. Gammon, G. Porter, S. Tamhanker, A.R. Williamson, and H.J. Zweerink. 1991. The minimum peptide epitope from the influenza matrixprotein. J. Immunol. 147:4047–4053. [PubMed] [Google Scholar]
- 30.Altman, J.D., P.A. Moss, P.J. Goulder, D.H. Barouch, M.G. McHeyzer-Williams, J.I. Bell, A.J. McMichael, and M.M. Davis. 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science. 274:94–96. [DOI] [PubMed] [Google Scholar]
- 31.Propato, A., E. Schiaffella, E. Vicenzi, V. Francavilla, L. Baloni, M. Paroli, L. Finocchi, N. Tanigaki, S. Ghezzi, R. Ferrara, et al. 2001. Spreading of HIV-specific CD8+ T-cell repertoire in long-term nonprogressors and its role in the control of viral load and disease activity. Hum. Immunol. 62:561–576. [DOI] [PubMed] [Google Scholar]
- 32.Kang, X., Y. Kawankami, M. el-Gamil, R. Wang, K. Sakaguchi, J.R. Yannelli, E. Appella, S.A. Rosenberg, and P.F. Robbins. 1995. Identification of a tyrosinase epitope recognized by HLA-A24 restricted, tumor-infiltrating lymphocytes. J. Immunol. 155:1343–1348. [PubMed] [Google Scholar]
- 33.Salter, R.D., D.N. Howell, and P. Cresswell. 1985. Genes regulatiing HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 21:235–246. [DOI] [PubMed] [Google Scholar]
- 34.Anderson, K.S., J. Alexander, M. Wei, and P. Cresswell. 1993. Intracellular transport of class I MHC molecules in antigen processing mutant cell lines. J. Immunol. 151:3407–3419. [PubMed] [Google Scholar]
- 35.Lu, J., and E. Celis. 2000. Use of two predictive algorithms of the world wide web for the identification of tumor-reactive T-cell epitopes. Cancer Res. 60:5223–5227. [PubMed] [Google Scholar]
- 36.Ercolini, A.M., J.P. Machiels, Y.C. Chen, J.E. Slansky, M. Giedlen, R.T. Reilly, and E.M. Jaffee. 2003. Identification and characterization of the immunodominant rat HER-2/neu MHC class I epitope presented by spontaneous mammary tumors from HER-2/neu-transgenic mice. J. Immunol. 170:4273–4280. [DOI] [PubMed] [Google Scholar]
- 37.Ryu, B., J. Jones, N.J. Blades, G. Parmigiani, M.A. Hollingsworth, R.H. Hruban, and S.E. Kern. 2002. Relationships and differentially expressed genes among pancreatic cancers examined by large-scale serial analysis of gene expression. Cancer Res. 62:819–826. [PubMed] [Google Scholar]
- 38.Miyahira, Y., K. Murata, D. Rodriguez, J.R. Rodriguez, M. Esteban, M.M. Rodrigues, and F. Zavala. 1995. Quantification of antigen specific CD8+ T cells using an ELISPOT assay. J. Immunol. Methods. 181:45–54. [DOI] [PubMed] [Google Scholar]
- 39.McCutcheon, M., N. Wehner, A. Wensky, M. Kushner, S. Doan, L. Hsiao, P. Calabresi, T. Ha, T.V. Tran, K.M. Tate, et al. 1997. A sensitive ELISPOT assay to detect low-frequency human T lymphocytes. J. Immunol. Methods. 210:149–166. [DOI] [PubMed] [Google Scholar]
- 40.Schmittel, A., U. Keilholz, and C. Scheibenbogen. 1997. Evaluation of the interferon-y ELISPOT-assay for the quantification of peptide specific T lymphocytes from peripheral blood. J. Immunol. Methods. 210:167–174. [DOI] [PubMed] [Google Scholar]
- 41.DiBrino, M., T. Tsuchida, R.V. Turner, K.C. Parker, J.E. Coligan, and W.E. Biddison. 1993. HLA-A1 and HLA-A3 T cell epitopes derived from influenza virus proteins predicted from peptide binding motifs. J. Immunol. 121:5930–5935. [PubMed] [Google Scholar]
- 42.Wolkers, M.C., G. Stoetter, F.A. Vyth-Dreese, and T.N.M. Schumacher. 2001. Redundancy of direct priming and cross-priming in tumor-specific CD8+ T cell responses. J. Immunol. 167:3577–3584. [DOI] [PubMed] [Google Scholar]
- 43.Jung, S., D. Unutmaz, P. Wong, G.I. Sano, K. De los Santos, T. Sparwasser, S. Wu, S. Vuthoori, K. Ko, F. Zavala, et al. 2002. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 17:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen, X., S.B.J. Wong, C.B. Buck, J. Zhang, and R.F. Siliciano. 2002. Direct priming and cross-priming contribute differentially to the induction of CD8+ CTL following exposure to vaccinia virus via different routes. J. Immunol. 169:4222–4229. [DOI] [PubMed] [Google Scholar]
- 45.Sigal, L.J., R. Crotty, R. Andino, and K.L. Rock. 1999. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 398:77–80. [DOI] [PubMed] [Google Scholar]
- 46.Sigal, L.J., and K.L. Rock. 2000. Bone marrow–derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J. Exp. Med. 192:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maecker, H.T., S.A. Ghanekar, M.A. Suni, X.S. He, L.J. Picker, and V.C. Maino. 2001. Factors affecting the efficiency of CD8+ T cell cross-priming with exogenous antigens. J. Immunol. 166:7268–7275. [DOI] [PubMed] [Google Scholar]
- 48.Albert, M.L., M. Jegathesan, and R.B. Darnell. 2001. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat. Immunol. 2:1010–1017. [DOI] [PubMed] [Google Scholar]
- 49.Scheffer, S.R., H. Nave, F. Korangy, K. Schlote, R. Pabst, E.M. Jaffee, M.P. Manns, and T.F. Greten. 2003. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int. J. Cancer. 103:205–211. [DOI] [PubMed] [Google Scholar]
- 50.Simons, J.W., E.M. Jaffee, C.E. Weber, H.I. Levitsky, W.G. Nelson, M.A. Carducci, A.J. Lazenby, L.K. Cohen, C.C. Finn, S.M. Clift, et al. 1997. Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte-macrophage colony-stimulating factor gene transfer. Cancer Res. 57:1537–1546. [PMC free article] [PubMed] [Google Scholar]
- 51.Soiffer, R., T. Lynch, M. Mihm, K. Jung, C. Rhuda, C. Jan, F. Schmollinger, S. Hodi, L. Liebster, P. Lam, et al. 1998. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA. 95:13141–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sokol, J.E. 1995. Measurement of delayed skin test responses. N. Engl. J. Med. 29:501–503. [DOI] [PubMed] [Google Scholar]
- 53.Oren, M.E., and R.B. Herberman. 1977. Delayed cutaneous hypersensitivity reactions to membrane extracts of human tumour cells. Clin. Exp. Immunol. 9:45–56. [PMC free article] [PubMed] [Google Scholar]
- 54.McCune, C.S., R.W. O'Donnell, D.M. Marquis, and D.M. Sahasrabudhe. 1990. Renal cell carcinoma treated by vaccines for active specific immunotherapy: correlation of survival with skin testing by autologous tumor cells. Cancer Immunol. Immunother. 32:62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoover, H.C., Jr., M. Surdyke, R.B. Dangel, L.C. Peters, and M.G. Hanna, Jr. 1984. Delayed cutaneous hypersensitivity to autologous tumor cells in colorectal cancer patients immunized with an autologous tumor cell: bacillus calmette-guerin vaccine. Cancer Res. 44:1671–1676. [PubMed] [Google Scholar]
- 56.Disis, M.L., K. Schiffman, T.A. Gooley, D.G. McNeel, K. Rinn, and K.L. Knutson. 2000. Delayed-type hypersensitivity response is a predictor of peripheral blood T-cell immunity after HER-2/neu peptide immunization. Clin. Cancer Res. 6:1347–1350. [PubMed] [Google Scholar]
- 57.Berd, D., H.C. Maguire, Jr., and M.J. Mastrangelo. 1986. Induction of cell-mediated immunity to autologous melanoma cells and regression of metastases after treatment with a melanoma cell vaccine preceded by cyclophoshamide. Cancer Res. 46:2572–2577. [PubMed] [Google Scholar]
- 58.DiFronzo, L.A., R.K. Gupta, R. Essner, L.J. Foshag, S.J. O'Day, L.A. Wanek, S.L. Stern, and D.L. Morton. 2002. Enhanced humoral immune response correlates with improved disease-free and overall survival in American Joint Committee on Cancer stage II melanoma patients receiving adjuvant polyvalent vaccine. J. Clin. Oncol. 20:3242–3248. [DOI] [PubMed] [Google Scholar]
- 59.Hodi, F.S., J.C. Schmollinger, R.J. Soiffer, R. Saliga, T. Lynch, J. Ritz, A. E.P., J. Yang, D. Neuberg, M. Mihm, and G. Dranoff. 2002. ATP6S1 elicits potent humoral responses associated with immune-mediated tumor destruction. Proc. Natl. Acad. Sci. USA. 99:6919–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moulton, H.M., P.H. Yoshihara, D.H. Mason, P.L. Iverson, and P.L. Triozzi. 2002. Active specific immunotherapy with a β-human chorionic gonadotropin peptide vaccine in patients with metastatic colorectal cancer: antibody response is associated with improved survival. Clin. Cancer Res. 8:2044–2051. [PubMed] [Google Scholar]
- 61.Skipper, J.C., R.C. Hendrickson, P.H. Gulden, V. Brichard, A. Van Pel, Y. Chen, J. Shabanowitz, T. Wolfel, C.L. Slingluff, Jr., T. Boon, D.F. Hunt, and V.H. Engelhard. 1996. An HLA-A2–restricted tyrosinase antigen on melanoma cells results from posttranslational modification and suggests a novel pathway for processing of membrane proteins. J. Exp. Med. 183:527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pascolo, S., M. Schirle, B. Guckel, T. Dumrese, S. Stumm, S. Kayser, A. Moris, D. Wallwiener, H.G. Rammensee, and S. Stevanovic. 2001. A MAGE-A1 HLA-A A*0201 epitope identified by mass spectrometry. Cancer Res. 61:4072–4077. [PubMed] [Google Scholar]