

Abstract
CD83 is up-regulated on the surface of dendritic cells (DCs) during maturation and has been widely used as a marker for mature DCs. Recently, we reported the recombinant expression of the extracellular immunoglobulin domain of human CD83 (hCD83ext). Using this soluble form of CD83, allogeneic as well as specific cytotoxic T lymphocyte proliferation could be blocked in vitro. Here we report the functional analysis of soluble CD83 in vivo, using murine experimental autoimmune encephalomyelitis (EAE) as a model. Strikingly, only three injections of soluble CD83 prevented the paralysis associated with EAE almost completely. In addition, even when the EAE was induced a second time, CD83-treated mice were protected, indicating a long-lasting suppressive effect. Furthermore, soluble CD83 strongly reduced the paralysis in different therapeutic settings. Most important, even when the treatment was delayed until the disease symptoms were fully established, soluble CD83 clearly reduced the paralyses. In addition, also when EAE was induced a second time, soluble CD83-treated animals showed reduced disease symptoms. Finally, hCD83ext treatment almost completely reduced leukocyte infiltration in the brain and in the spinal cord. In summary, this work strongly supports an immunosuppressive role of soluble CD83, thereby indicating its therapeutic potential in the regulation of immune disorders in vivo.
Keywords: CD83, dendritic cells, EAE, T cells, proliferation
Introduction
Human CD83 is a 45-kD glycoprotein and a member of the immunoglobulin superfamily (1–3). CD83 expression is induced by inflammatory cytokines and the up-regulation of CD83 together with the costimulatory molecules such as CD80 and CD86 during DC maturation, suggest an important role of CD83 in the induction of specific immune responses. For a long time, CD83 has been used as the best surface marker for mature DCs (4). DCs are the most potent APCs of the immune system and act as “nature's adjuvant” in inducing T cell–mediated immunity (5). In their immature stage, DCs reside in peripheral tissues and are specialized in antigen uptake. During maturation they change their phenotype and migrate to lymphoid organs, were they trigger the antigen-specific T cell responses (6).
The cDNA sequence of human CD83 has been known for quite some time and recently the cloning of murine CD83, which shares an amino acid identity of 63% with the human CD83, hinting at a conserved function, has been reported (7, 8). However, the functional importance of CD83 was unknown.
The first hints of a possible biological role for CD83 came from experiments where DCs were virus infected. In this respect, we reported the lysosomal degradation of CD83 in HSV-1–infected DCs that correlated with a reduced allostimulatory capacity of these virus-infected DCs (9). Others reported similar effects with measles virus– (10) and vaccinia virus–infected DCs (11, 12). In addition, the inhibition of CD83 mRNA export from the nucleus into the cytoplasm, thereby blocking CD83 protein expression, also led to a reduced T cell stimulatory capacity (13). In combination, these data suggested a crucial role for CD83. However, this is only based on circumstantial evidence because it is well known that viruses interfere with several cellular aspects, and also the inhibition of an mRNA export pathway is not CD83 specific. The first functional role for CD83 was reported in vitro using a soluble CD83 molecule. This soluble CD83 protein completely inhibited DC-mediated T cell stimulation in a concentration-dependent manner (3, 14). These data were confirmed by Scholler et al. (15), using a CD83–Ig fusion protein. These findings gain further biological importance when viewed with the fact that a soluble form of CD83 was detected in normal human serum and is also released from in vitro–cultured DCs and B lymphocytes (16). Furthermore, it has been found that the soluble form of CD83 is present at elevated levels in a number of hematological disorders including patients with chronic lymphocytic leukemia and mantle cell lymphoma (17). Furthermore, recently it has been reported that CD83 is essential for the development of CD4 single positive thymocytes, and consequently also for the generation of peripheral CD4+ T cells (18). Indeed, the lack of CD83 resulted in a selective 75–90% reduction of peripheral CD4+ T cells. Taken together, the data described above indicate that CD83 plays an important role for T cells as well as for DCs.
Here we investigated the effect of soluble CD83 on the murine experimental autoimmune encephalomyelitis (EAE), a CD4+ T cell–mediated disease model for the early inflammatory stage of human multiple sclerosis. Interestingly, only three injections of soluble CD83 inhibited the paralysis associated with EAE almost completely. Strikingly, this suppressive effect was also long-lasting because mice in which EAE was induced for a second time were still protected, whereas untreated mice were again strongly paralyzed. Furthermore, soluble CD83 also strongly reduced the paralysis in a therapeutic setting. Finally, hCD83ext treatment almost completely reduced CD45+ cell infiltration in the brain and in the spinal cord.
Materials and Methods
Allogeneic T Cell Proliferation.
BM-derived DCs (BM-DCs) were generated from C57BL/6 mice as described previously (19). In Brief, BM cells were seeded at a density of 2 × 106/10 ml in R10 cell culture medium. R10 culture medium was composed of RPMI 1640 (GIBCO BRL) supplemented with 100 U/ml penicillin (Sigma-Aldrich), 100 μg/ml streptomycin (Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich), 50 μM 2-mercaptoethanol (Sigma-Aldrich), and 10% heat-inactivated FCS (PAA Laboratories GmbH). GM-CSF supernatant (1:10) from a cell line transfected with the murine GM-CSF gene was used to obtain mature DCs. At day 10 of the BM-DC culture, 4 × 106 T cells, which were purified from pooled BALB/c lymph node cells, were seeded together with titrated numbers of BM-DCs into a 96-well flat-bottom plate (Falcon) for 72 h. BM-DCs were preincubated with different concentrations of the soluble human CD83 molecule (hCD83ext) or the murine CD83–Ig (mCD83–Ig) fusion protein (Biocarta). Glutathione S-transferase (GST) was used as negative control. Cell cultures of this allogeneic MLR were then pulsed with 1 μCi/well [3H]methyl-thymidine (Amersham Biosciences) overnight for 16 h. The plates were harvested with an IH-110 harvester (Innotech) and filters were counted in a 1450 Microplate Counter (Wallac).
EAE Induction and Treatment with hCD83ext.
Female C57Bl/6 mice (Charles River Laboratories) were immunized s.c. with 50 μg myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 (Sigma-Genosys) in 50 μl H2O emulsified in 50 μl CFA that was enriched with 10 μg/ml Mycobacterium tuberculosis (H37Ra; Difco/BD Biosciences) at day 0 to induce EAE. In addition, 200 ng pertussis toxin (Pt; List/Quadratech) was administered i.p. at days 0 and 2. EAE paralysis of mice was scored as follows: 0, no disease; 1, tail weakness; 2, paraparesis; 3, paraplegia; 4, paraplegia with forelimb weakness; 5, moribund or dead animals. Soluble hCD83ext (100 μg/injection) was injected i.p. on days −1, 1, and 3. One group of mice received BSA (100 μg/injection of Pentex-BSA; Bayer) as control protein. In the control group, the EAE was induced without any treatment. Three mice were treated in each group. In the therapeutic setting, soluble hCD83ext was administered at different time points after the EAE induction (day 3, 10, or 17), and was then given more often (every second day or every day; see Fig. 5, A–C).
Figure 5.
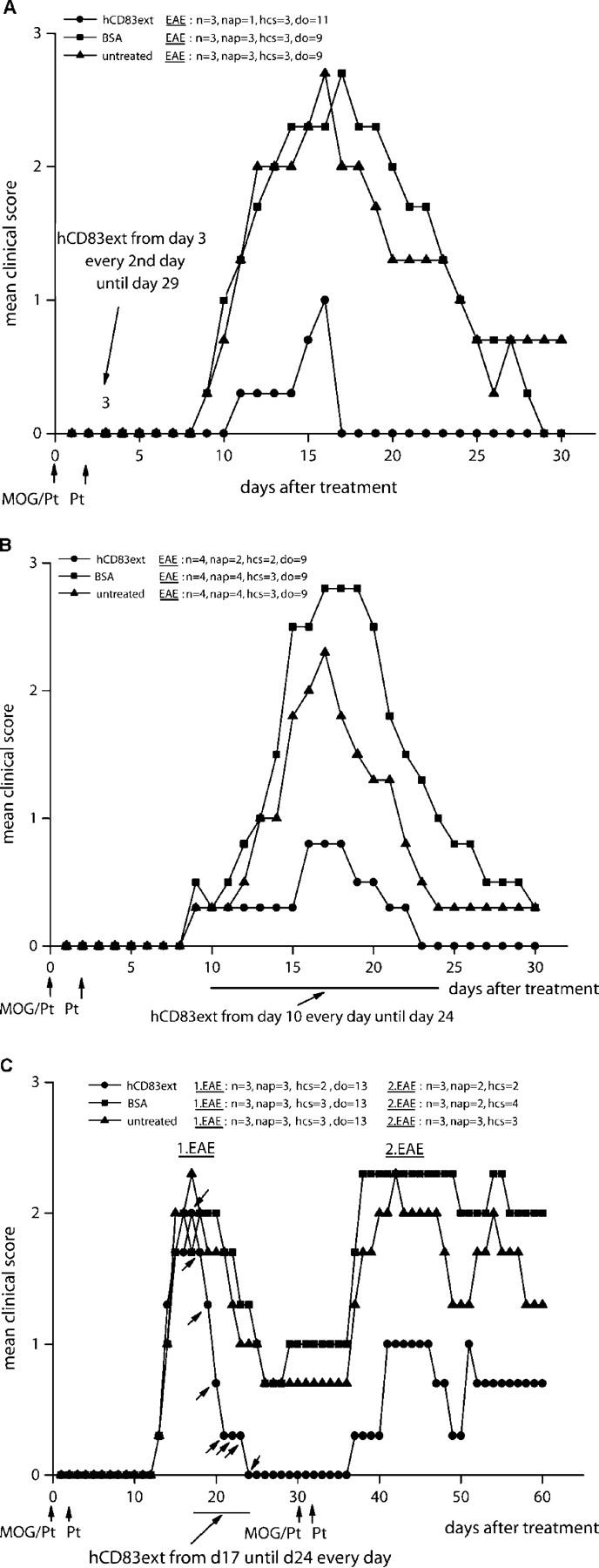
Soluble CD83 also protects mice from EAE in a therapeutic setting. (A) EAE was induced as shown in Fig. 2. Starting from day 3, 100 μg hCD83ext (or BSA as control) was injected i.p. every second day until day 29. hCD83ext almost completely inhibited the paralysis. (P < 0.05). In contrast, BSA-treated animals and untreated mice developed strong disease symptoms. (B) EAE was induced as described above, however, hCD83ext treatment started only on day 10 and was then given every day until day 24. Again soluble CD83 strongly reduced the paralysis in this therapeutic setting (P < 0.05). BSA-treated animals and untreated mice developed strong disease symptoms. These experiments were performed at least three times. Data presented here represent a typical experiment. (C) Treatment with soluble CD83 was delayed until day 17, when disease symptoms were fully established, and was then applied every day until day 24. Soluble CD83 clearly reduced the paralysis (P < 0.05). In contrast, BSA-treated animals and untreated mice developed strong, longer-lasting symptoms. On day 30, EAE was induced a second time by immunizing the mice with MOG peptide as described above. Strikingly, mice that were pretreated with hCD83ext from day 17 until 24 showed reduced disease symptoms, whereas untreated animals and BSA-treated mice showed strong disease symptoms. These experiments were performed three times. Data presented here represent a typical experiment. n, total number of animals used in this particular experiment; nap, total number of animals with paralysis in this particular experiment; hcs, highest clinical score observed in this particular experiment; do, day of onset.
The statistical significance of differences in clinical index (day 21) between groups was analyzed using Student's t test. Significance was accepted if P < 0.05.
Restimulation.
30, or alternatively 60, d after immunization of mice with MOG, spleens were removed for restimulation assays. Cells were cultured in HL-1 serum-free medium supplemented with 100 U/ml penicillin (Sigma-Aldrich), 100 μg/ml streptomycin (Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich), and 50 μM 2-mercaptoethanol (Sigma-Aldrich). MOG-specific cells were analyzed by incubating 4 × 105 spleen cells with different concentrations of MOG peptide in 200 μl HL-1/well in a 96-well tissue culture plate. Additionally, as a control, 4 × 105 spleen cells were stimulated with 500 U/ml IL-2 (Proleukin). As a negative control, unstimulated cultures were used. After 72 h, cultures were pulsed with 0.4 Ci/mmol [3H]thymidine (TRA-20; Amersham Biosciences). 12 h later, thymidine incorporation was measured using a microplate counter (Wallac).
Cytokine Assays.
To determine the ex vivo cytokine production, harvested splenocytes (day 12, 30, or 60) were stimulated with different concentrations of the MOG peptide. Culture supernatants were taken after 96 h and tested (either immediately or after freezing) using commercially available sandwich ELISA kits for INF-γ, IL-2, IL-4, and IL-10 (BD Biosciences).
Neuropathology.
For visualization of inflammatory infiltrates, brains and spinal cords from mice (day 12 after EAE induction) were removed, fixed in liquid nitrogen, and stored at −80°C. Sections were cryoprotected in Tissue-Tek® (Sakura) and sliced sequentially with a thickness of 7 μm with a cryotome (Kryocut CM 2000; Leica). Acetone-fixed cryostat sections were stained with Mayer's hemalaun solution (Merck) and examined by light microscopy (Leica). Immunohistological staining was performed using an immunoperoxidase detection system in a wet chamber. Acetone-fixed sections were incubated in PBS. Endogenous peroxidase was blocked by incubating sections in 3% H2O2. The primary antibody was an anti–mouse CD45 antibody (clone 30G12; provided by L. Sorokin, Lund University, Lund, Sweden). As secondary antibody, biotinylated goat anti–rat IgG and streptavidin horseradish peroxidase (Biocare Medical) were used. To visualize antigens, sections were incubated with AEC Chromogene Substrate (Vector Laboratories), mounted in Aqua Tex (Roth), and covered with a coverslip.
Results
Inhibition of DC-mediated Murine Allogeneic T Cell Proliferation Using Human or Murine CD83.
To test whether or not soluble human CD83 is able to inhibit murine DC-mediated T cell proliferation, hCD83ext was compared with the murine CD83–Ig fusion protein. On day 10 of culture, mouse BM-DCs were either incubated with hCD83ext or with the mCD83Ig, and their stimulatory capacity was tested in an allogeneic MLR. As shown in Fig. 1, both the human as well as the murine CD83 molecule were able to completely abrogate allogeneic murine T cell proliferation, suggesting a similar functional capacity. In contrast, DCs cultured in the presence of GST (as negative control) did not suppress T cell proliferation. These results clearly show that one can use soluble human CD83 to inhibit murine DC-mediated T cell responses.
Figure 1.

Soluble human CD83 as well as murine CD83–Ig inhibit murine allogeneic T cell proliferation. 4 × 106 T cells were seeded together with titrated numbers of BM-DCs into a 96-well flat-bottom plate for 72 h. BM-DCs were preincubated with different concentrations of the soluble human CD83 molecule (hCD83ext) or the murine CD83–Ig (mCD83–Ig) fusion protein. Cell cultures of this allogeneic MLR were then pulsed with 1 μCi/well [3H]methyl-thymidine for 16 h. The plates were harvested and the filters were counted. The data show that human (hCD83ext) as well as murine CD83 (mCD83–Ig) completely block murine T cell proliferation. GST, which was purified in the same way as hCD83ext, was used as a control protein. These experiments were performed at least five times. Data presented here represent a typical experiment.
Soluble CD83 Inhibits EAE In Vivo.
The murine EAE model was used to determine the influence of hCD83ext in vivo. Therefore, we injected hCD83ext (100 μg/i.p. injection) on day −1, 1, and 3, and EAE was induced on day 0 by injecting MOG/Pt, with a second injection of Pt on day 2. Strikingly, hCD83ext almost completely inhibited the MOG-induced paralysis, whereas BSA-treated and untreated control mice developed strong paralysis, reaching a maximum level at days 18–24 (Fig. 2 A, left). Furthermore, when EAE was induced for a second time on day 28, hCD83ext-treated mice were still completely protected, whereas BSA-treated and untreated mice developed paralysis symptoms once again (Fig. 2 A, right). This strongly suggests that the inhibitory effects induced by soluble CD83 are long-lasting in vivo.
Figure 2.
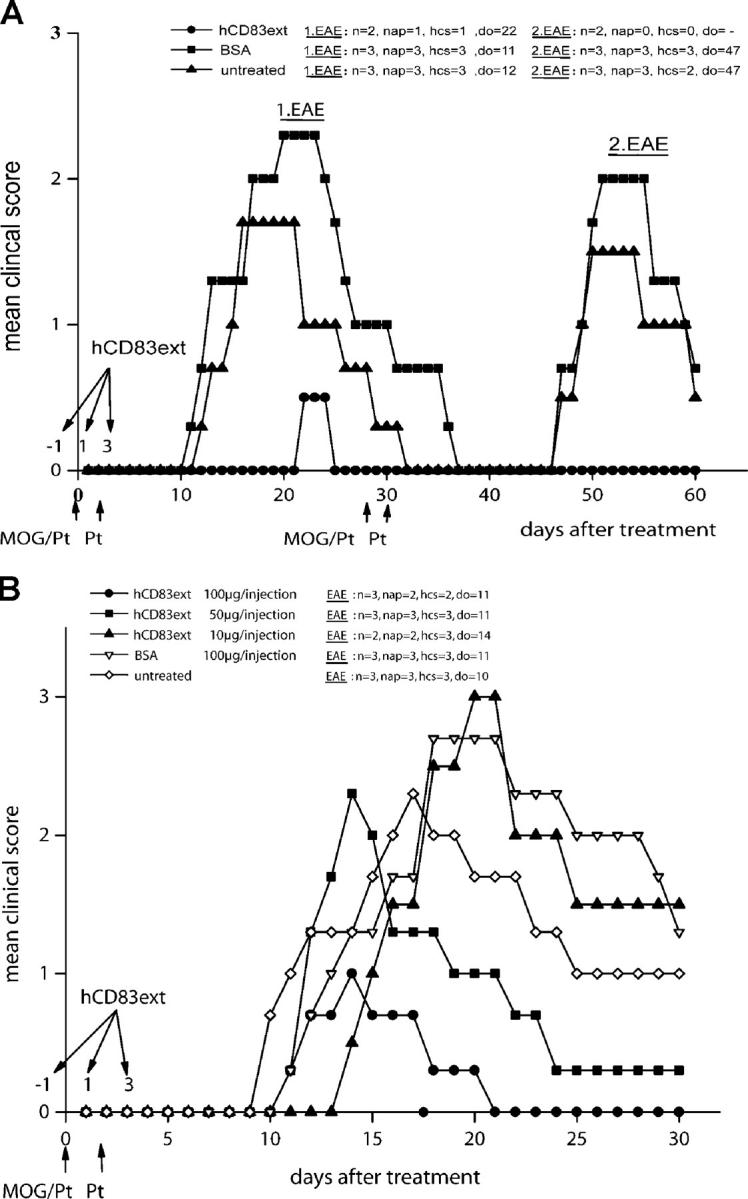
Three doses of soluble CD83 protect mice from EAE. (A, left) 100 μg hCD83ext (or BSA as control) were injected i.p. on days −1, 1, and 3. EAE was induced by s.c. injection of MOG peptide emulsified in CFA enriched with M. tuberculosis at day 0 as described in Materials and Methods. In addition, 200 ng Pt was administered i.p. on days 0 and 2. hCD83ext almost completely inhibited the paralysis. In contrast, BSA-treated and untreated mice developed strong disease symptoms. (A, right) On day 28, EAE was induced a second time by immunizing the mice with MOG peptide as described above. Strikingly, mice which were treated only three times with hCD83ext were completely protected, whereas untreated and BSA-treated mice were paralyzed. (B) Inhibitory effect is dose dependent. hCD83ext was administered at three different doses. The best inhibition was observed at a dose of 100 μg/injection (P < 0.05). These experiments were performed at least five times. Data presented here represent a typical experiment. n, total number of animals used in this particular experiment; nap, total number of animals with paralysis in this particular experiment; hcs, highest clinical score observed in this particular experiment; do, day of onset.
The Inhibitory Effect of Soluble CD83 Is Dose Dependent.
To establish whether or not this inhibitory effect is dose dependent, different concentrations of soluble CD83 were administered. As shown in Fig. 2 B, the effect of soluble CD83 is clearly dose dependent. The best inhibition was clearly observed at a concentration of 100 μg/injection. No inhibition was observed at a dose of 10 μg/injection, whereas 50 μg/injection showed an intermediate effect.
Soluble CD83-treated Mice Show a Strongly Inhibited Cytokine Production and a Reduced T Cell Proliferation.
To gain mechanistic insights into the inhibitory effects of soluble CD83, spleens were removed from treated and untreated mice on days 12, 30 (Fig. 2 A, 1. EAE), and 60 (Fig. 2 A, 2. EAE), and spleen cells were restimulated with MOG peptide at different concentrations for 72 h to determine their cytokine production and their antigen-specific proliferation. As shown in Fig. 3 A, IFN-γ production was strongly reduced in hCD83-treated mice (harvested on day 12). IL-2, IL-4, and IL-10 levels were very low (<40 pg/ml) or under the detection limit (not depicted).
Figure 3.
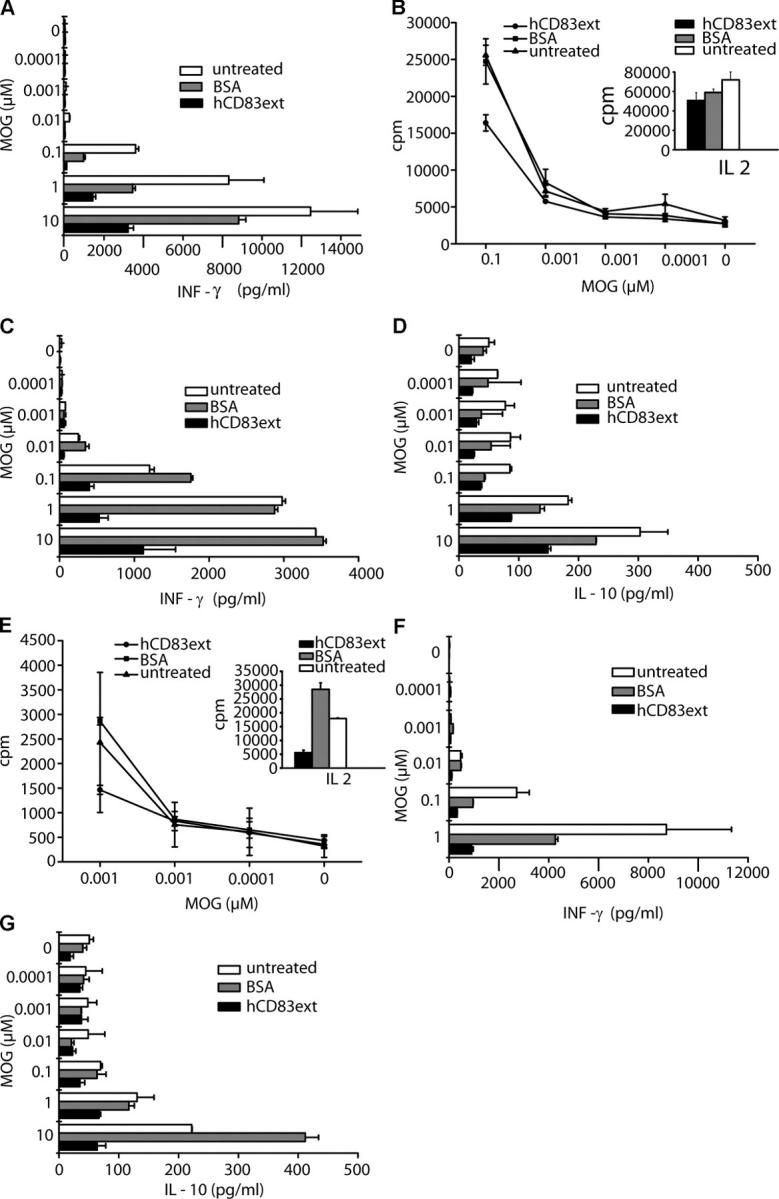
Soluble CD83 inhibits cytokine production and restimulation. To determine the ex vivo cytokine production, splenocytes, derived from hCD83ext-treated, BSA-treated, or untreated mice, where EAE was induced once, were harvested on day 12. MOG-specific cells were analyzed by incubating 4 × 105 spleen cells with different concentrations of MOG peptide as described in Materials and Methods. Culture supernatants were taken after 96 h and analyzed. (A) hCD83ext-treated cells are strongly inhibited in their IFN-γ production. (B) Restimulation of spleen cells, harvested on day 30 and derived from hCD83 ext-treated, BSA-treated, or un-treated mice, where EAE was induced once. MOG-specific cells were analyzed by incubating 4 × 105 spleen cells with different concentrations of MOG peptide as described in Materials and Methods. Spleen cells derived from hCD83ext-treated mice show a clearly reduced proliferation. Additionally, as a control, 4 × 105 spleen cells were stimulated with 500 U/ml IL-2. Also, hCD83ext-treated cells are still able to proliferate in response to IL-2 (see inset at right). (C and D) Ex vivo cytokine production of these day 30 harvested splenocytes. (C) hCD83ext-treated cells are strongly inhibited in their IFN-γ production. (D) Also, the IL-10 production is clearly reduced. (E) Restimulation of spleen cells harvested on day 60 and derived from hCD83ext-treated, BSA-treated, or untreated mice, where EAE was induced twice. hCD83ext-treated mice show reduced proliferation capacity. They also poorly proliferate in response to IL-2 (see inset at right). (F and G) Ex vivo cytokine production of these day 60 harvested splenocytes. (F) IFN-γ production is strongly inhibited. (G) The same is true for the IL-10 production. These experiments were performed at least three times. Data presented here represent a typical experiment.
Next, the proliferation capacity of day 30 harvested and then restimulated cells was analyzed. As shown in Fig. 3 B, the proliferation capacity of cells derived from hCD83ext-treated animals, where the EAE was induced only once, is clearly reduced when compared with BSA-treated or untreated mice. Interestingly, however, hCD83ext-treated spleen cells still proliferated in response to IL-2, indicating that soluble CD83 does not induce a complete deletion of T cells in general (Fig. 3 B, inset). Next, the cytokine production of these day 30 restimulated cells was analyzed. As shown in Fig. 3 C, IFN-γ production was strongly reduced in hCD83-treated mice. The same was true for the IL-10 production (Fig. 3 D), indicating that soluble CD83 does not induce IL-10–producing regulatory T cells. IL-2 and IL-4 production was again very low (<40 pg/ml) or undetectable (not depicted).
Next, spleen cells harvested on day 60 and derived from animals in which EAE was induced for a second time, were restimulated in vitro. Poor, reduced proliferation could be observed for all conditions, and hCD83ext-treated cells responded to a much lesser degree to IL-2 (Fig. 3 E). Also in these experiments, cells derived from CD83-treated animals produced much less IFN-γ and IL-10 (Fig. 3, F and G). As seen before, IL-2 and IL-4 production was very low (<40 pg/ml) or not detectable (unpublished data), indicating that soluble CD83 does not lead to a skewing of the MOG-specific immune response from a Th1 cell to a Th2 cell response during the memory phase.
Soluble CD83 Inhibits CD45+ Cell Infiltrates in the Brain and the Spinal Cord.
To investigate the effect of soluble CD83 on pathologic expression of EAE during the acute phase of the disease (day 12), histopathological analyses were performed. As show in Fig. 4, soluble CD83 strongly inhibits CD45+ leukocyte infiltration in the brain (Fig. 4 C) as well as in the spinal cord (Fig. 4 F). This was comparable to naive, untreated mice (Fig. 4, A and D). In sharp contrast, mock-treated EAE mice showed strong infiltrates in both the brain (Fig. 4 B) as well as the spinal cord (Fig. 4 E).
Figure 4.

Soluble CD83 inhibits CD45+ cell infiltrates in the brain and the spinal cord. A and D represent naive, untreated mice as a control where EAE was not induced. B and E represent untreated mice where EAE was induced. There is a strong CD45+ cell infiltrate visible in the brain (B) as well as in the spinal cord (E). In contrast, hCD83ext-treated mice show no or only very minor infiltrations in the brain (C) and the spinal cord (F). Mice were analyzed on day 12 after EAE induction. These experiments were performed at least three times. Data presented here represent a typical experiment.
Soluble CD83 Protects Mice from EAE also in a Therapeutic Setting.
In the experimental setting shown in Fig. 2, we clearly demonstrated that soluble CD83 has a great prophylactic potential to inhibit EAE symptoms. To determine whether or not soluble CD83 also has a therapeutic potential, EAE was induced as described above on day 0. However, the administration of soluble CD83 was postponed until day 3 (Fig. 5 A), or even longer until day 10 (Fig. 5 B). Strikingly, in both cases, soluble CD83 clearly reduced the paralysis when compared with the control animals. Furthermore, to “mimic” realistic therapeutic circumstances, treatment with soluble CD83 was delayed until day 17, i.e., when disease symptoms were fully established, and was then applied every day until day 24. As shown in Fig. 5 C, left, soluble CD83 clearly reduced the disease symptoms. In addition, EAE was induced a second time by immunizing the mice with MOG peptide on day 30. Strikingly, mice that were pretreated with soluble CD83 from day 17 until 24 showed strongly reduced disease symptoms when compared with BSA-treated and untreated mice (Fig. 5 C, right).
Discussion
DCs act as “nature's adjuvant” in inducing T cell–mediated immunity. However, to induce potent immune responses, DCs have to mature. During this maturation step, CD83 is rapidly up-regulated together with the costimulatory molecules CD80 and CD86. Although CD83 is strongly up-regulated on mature DCs, the function of this molecule was not known. To elucidate the mode of action of CD83, we previously reported the recombinant expression of the extracellular Ig domain of human CD83 (14, 20). Using this soluble CD83 molecule, we could show that DC-mediated T cell stimulation is completely abrogated in vitro (3, 14, 21). Furthermore, when soluble CD83 was added to immature DCs, it inhibited DC maturation and even when soluble CD83 was added to already fully matured DCs, it still led to a down-modulation of CD83 itself and to a block of T cell stimulation. These data strongly indicated a functional role for CD83 in DC biology.
Here we report the function of soluble CD83 in vivo using the murine EAE as a model disease for the early inflammatory stage of human multiple sclerosis. First, we showed that soluble, human CD83 is also able to inhibit murine T cell stimulation in vitro. Both human and murine CD83 molecules blocked allogeneic murine T cell stimulation in a concentration-dependent manner (Fig. 1), thus a similar mode of action has been postulated for both CD83 molecules. Consequently, human soluble CD83 was used for the mouse in vivo studies.
Strikingly, only three injections of soluble CD83 inhibited the EAE symptoms almost completely (Fig. 2 A). This effect was concentration dependent (Fig. 2 B). The effect was even more pronounced when the EAE was induced for a second time in the same mice. In this case, the soluble CD83-treated mice were completely protected, whereas control mice were once again paralyzed. This indicated a long-lasting effect of soluble CD83, which is unlikely to be purely due to the presence of CD83. Thus, the stimulatory capacity of antigen-specific spleen cells was analyzed in peptide-specific restimulation assays. After the first EAE induction, there was a clear reduction of MOG-specific proliferation in the CD83-treated animals (Fig. 3 B). However, these cells were not deleted because they still responded to IL-2, although more weakly than when compared with cells derived from BSA-treated or untreated animals. Similarly, MOG-specific restimulation, after the induction of the second EAE, was also reduced in cells derived from CD83-treated animals when compared with control cells (Fig. 3 E). However, all cells, whether treated or untreated, responded to a much lower degree after the second EAE induction when compared with the restimulation after the first EAE induction. A clear difference was, however, observed when CD83-treated cells were restimulated with IL-2. In this case, CD83-treated cells responded rather poorly when compared with BSA-treated or untreated cells, indicating that those cells might be deleted. However, no signs of deletion were observed as judged from in vitro assays (14). Nevertheless, we cannot exclude partial deletion in vivo.
Furthermore, the cytokine production of these restimulated cells was analyzed, and a strong reduction of IFN-γ production could be observed in CD83-treated animals after the first EAE induction (Fig. 3, A and C). This effect was even more pronounced after the second EAE induction (Fig. 3 F). A similar reduction was observed for the IL-10 production, excluding the induction of IL-10–producing Tr1 cells. (Fig. 3, D and G). IL-2 production was very low (<40 pg/ml) or undetectable in all samples. IL-4 production was also very low (<40 pg/ml) or undetectable in all samples and thus, a shift to Th2 cell polarization can be excluded as a mechanism of tolerogenic immune deviation. Taken together, these data clearly show that proliferation, IFN-γ, and IL-10 productions are inhibited in CD83-treated mice. Thus, the Th1 cell–specific immune response, responsible for the EAE-associated disease symptoms, has been suppressed. Taken together, none of the classical T cell tolerance mechanisms seem to be involved in the immunosuppression mediated by soluble CD83. However, this has to be further analyzed in detail and will be the topic of a different study. The striking effect observed in the initial experiments with three prophylactic applications of soluble CD83, which prevented the EAE symptoms, was very encouraging. Thus, the therapeutic effect of this molecule was also investigated. The data clearly show that when soluble CD83 was administered during the initial phase of EAE induction (day 3), which represents the immunological onset of the disease, or at a later period (day 10), which spans the onset of the first clinical symptoms, a clear reduction in paralysis was observed. These data strongly suggest that soluble CD83 is able to inhibit this autoimmune disorder at several different stages of the disease. First of all, therapeutic applications of soluble CD83 completely prevented paralysis (Fig. 2). Furthermore, and even more important, is the fact that soluble CD83 is also active in the therapeutic setting, be it at an early stage during the immune induction phase, or later when the first symptoms of paralyses were observed. Most impressive were the effects when soluble CD83 was administered at a very late stage, i.e., when disease symptoms were fully established (Fig. 5 C). Thus, therapeutic intervention with soluble CD83 has profound clinical effects. In addition, it is noteworthy to state that soluble CD83 also exhibits a very pronounced effect on inflammatory CD45+ cell infiltrates in the brain as well as in the spinal cord. There are almost no such infiltrates observed in soluble CD83-treated mice (Fig. 4). This is obviously a prerequisite to inhibit the associated pathology within the neuronal system. In summary, here we have shown for the first time that soluble CD83 inhibits pathological autoimmune disorders in vivo and that these data underline the therapeutic potential of this molecule in such disorders as well as in the inhibition of transplant rejection and T cell–mediated allergies.
Acknowledgments
We are very grateful to Ralph Steinman and Gerold Schuler for suggestions and critical discussions. We would like to thank Wolfgang Uter for the help with the statistical analyses.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 466, grant B5).
The authors have no conflicting financial interests.
Abbreviations used in this paper: BM-DC, BM-derived DC; EAE, experimental autoimmune encephalomyelitis; GST, glutathione S-transferase; MOG, myelin oligodendrocyte glycoprotein; Pt, pertussis toxin.
References
- 1.Zhou, L.J., R. Schwarting, H.M. Smith, and T.F. Tedder. 1992. A novel cell-surface molecule expressed by human interdigitating reticulum cells, Langerhans cells, and activated lymphocytes is a new member of the Ig superfamily. J. Immunol. 149:735–742. [PubMed] [Google Scholar]
- 2.Kozlow, E.J., G.L. Wilson, C.H. Fox, and J.H. Kehrl. 1993. Subtractive cDNA cloning of a novel member of the Ig gene superfamily expressed at high levels in activated B lymphocytes. Blood. 81:454–461. [PubMed] [Google Scholar]
- 3.Lechmann, M., S. Berchthold, J. Hauber, and A. Steinkasserer. 2002. CD83 on dendritic cells: more than just a marker for maturation. Trends Immunol. 23:273–275. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 5.Steinman, R.M. 1991. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9:271–296. [DOI] [PubMed] [Google Scholar]
- 6.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 7.Twist, C.J., D.R. Beier, C.M. Disteche, S. Edelhoff, and T.F. Tedder. 1998. The mouse Cd83 gene: structure, domain organization, and chromosome localization. Immunogenetics. 48:383–393. [DOI] [PubMed] [Google Scholar]
- 8.Berchtold, S., P. Mühl-Zürbes, C. Heufler, P. Winklehner, G. Schuler, and A. Steinkasserer. 1999. Cloning, recombinant expression and biochemical characterization of the murine CD83 molecule which is specifically upregulated during dendritic cell maturation. FEBS Lett. 461:211–216. [DOI] [PubMed] [Google Scholar]
- 9.Kruse, M., O. Rosorius, F. Kratzer, G. Stelz, C. Kuhnt, G. Schuler, J. Hauber, and A. Steinkasserer. 2000. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol. 74:7127–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fugier-Vivier, I., C. Servet-Delprat, P. Rivallier, M.C. Rissoan, Y.J. Liu, and C. Rabourdin-Combe. 1997. Measles virus suppresses cell-mediated immunity by interfering with the survival and functions of dendritic and T cells. J. Exp. Med. 186:813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelmayer, J., M. Larsson, M. Subklewe, A. Chahroudi, W.I. Cox, R.M. Steinman, and N. Bhardwaj. 1999. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J. Immunol. 163:6762–6768. [PubMed] [Google Scholar]
- 12.Jenne, L., C. Hauser, J.F. Arrighi, J.H. Saurat, and A.W. Hügin. 2000. Poxvirus as a vector to transduce human dendritic cells for immunotherapy: abortive infection but reduced APC function. Gene Ther. 7:1575–1583. [DOI] [PubMed] [Google Scholar]
- 13.Kruse, M., O. Rosorius, F. Kratzer, D. Bevec, C. Kuhnt, A. Steinkasserer, G. Schuler, and J. Hauber. 2000. Inhibition of CD83 cell surface expression during dendritic cell maturation by interference with nuclear export of CD83 mRNA. J. Exp. Med. 191:1581–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lechmann, M., D.J.E.B. Krooshoop, D. Dudziak, E. Kremmer, C. Kuhnt, C.G. Figdor, G. Schuler, and A. Steinkasserer. 2001. The extracellular domain of CD83 inhibits dendritic cell–mediated T cell stimulation and binds to a ligand on dendritc cells. J. Exp. Med. 194:1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scholler, N., M. Hayden-Ledbetter, A. Dahlin, I. Hellström, K.E. Hellström, and J.A. Ledbetter. 2002. CD83 regulates the development of cellular immunity. J. Immunol. 168:2599–2602. [DOI] [PubMed] [Google Scholar]
- 16.Hock, B.D., M. Kato, J.L. McKenzie, and D.N.J. Hart. 2001. A soluble form of CD83 is released from activated dendritic cells and B lymphocytes, and is detectable in normal human sera. Internat. Immunology. 13:959–967. [DOI] [PubMed] [Google Scholar]
- 17.Hock, B.D., L.F. Haring, A. Steinkasserer, K.G. Taylor, W.N. Patton, and J.L. McKenzie. 2004. The soluble form of CD83 is present at elevated levels in a number of hematological malignancies. Leuk. Res. 28:237–241. [DOI] [PubMed] [Google Scholar]
- 18.Fujimoto, Y., L. Tu, A.S. Miller, C. Bock, M. Fujimoto, C. Doyle, D.A. Steeber, and T.F. Tedder. 2002. CD83 expression influences CD4+ T cell development in the thymus. Cell. 108:755–767. [DOI] [PubMed] [Google Scholar]
- 19.Lutz, M.B., N. Kukutsch, A.L.J. Ogilvie, S. Rössner, F. Koch, N. Romani, and G. Schuler. 1998. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 223:77–92. [DOI] [PubMed] [Google Scholar]
- 20.Lechmann, M., E. Kremmer, H. Sticht, and A. Steinkasserer. 2002. Overexpression, purification, and biochemical characterization of the extracellular-human CD83 domain and generation of monoclonal antibodies. Protein Expr. Purif. 24:445–452. [DOI] [PubMed] [Google Scholar]
- 21.Lechmann, M., E. Zinser, A. Golka, and A. Steinkasserer. 2002. Role of CD83 in the immunomodulation of dendritic cells. Int. Arch. Allergy Immunol. 129:113–118. [DOI] [PubMed] [Google Scholar]