

Abstract
Human adult bone marrow–derived endothelial progenitors, or angioblasts, induce neovascularization of infarcted myocardium via mechanisms involving both cell surface urokinase-type plasminogen activator, and interactions between β integrins and tissue vitronectin. Because each of these processes is regulated by plasminogen activator inhibitor (PAI)-1, we selectively down-regulated PAI-1 mRNA in the adult heart to examine the effects on postinfarct neovascularization and myocardial function. Sequence-specific catalytic DNA enzymes inhibited rat PAI-1 mRNA and protein expression in peri-infarct endothelium within 48 h of administration, and maintained down-regulation for at least 2 wk. PAI-1 inhibition enhanced vitronectin-dependent transendothelial migration of human bone marrow–derived CD34+ cells, and resulted in a striking augmentation of angioblast-dependent neovascularization. Development of large, thin-walled vessels at the peri-infarct region was accompanied by induction of proliferation and regeneration of endogenous cardiomyocytes and functional cardiac recovery. These results identify a causal relationship between elevated PAI-1 levels and poor outcome in patients with myocardial infarction through mechanisms that directly inhibit bone marrow–dependent neovascularization. Strategies that reduce myocardial PAI-1 expression appear capable of enhancing cardiac neovascularization, regeneration, and functional recovery after ischemic insult.
Keywords: myocardial infarction, angiogenesis, bone marrow stem cells, myocardial regeneration, DNA enzyme
Introduction
We have recently isolated endothelial progenitor cells, or angioblasts, in human adult bone marrow that have phenotypic and functional characteristics of embryonic angioblasts (1), which are cells that are derived from the human ventral aorta and give rise to the definitive vascular network during early development (2–5). Intravenous administration of these cells to athymic rats who have undergone experimental myocardial infarction results in selective homing to ischemic myocardium, induction of infarct bed neovascularization, and significant improvement in myocardial function (1). Although the precise chemotactic factors regulating migration of bone marrow–derived angioblasts to the site of acute ischemia remain to be identified, transendothelial egress, extracellular matrix degradation, and neovascularization are thought to require activation of latent metalloproteinases by plasmin, which is derived from plasminogen through activation by urokinase-type plasminogen activator (u-PA) expressed on the surface of the infiltrating bone marrow–derived cells (6-8).
Plasmin generation is negatively regulated by local concentrations of the serpin plasminogen activator inhibitor (PAI)-1, which in its native state, is complexed to circulating and tissue vitronectin (9, 10). After reactivity with a proteinase such as u-PA, PAI-1 undergoes a rapid conformational change that causes it to dissociate from vitronectin and increase its affinity for the low density lipoprotein receptor (11), leading to its clearance and degradation. In addition, removal of PAI-1 from vitronectin exposes a cryptic epitope necessary for binding to the cell surface integrin alphavbeta3, which regulates cellular attachment and migration (9, 12).
Plasma levels of PAI-1 are increased in patients with myocardial infarction, atherosclerosis, and restenosis (13-16). Moreover, PAI-1 mRNA and protein expression are elevated in atherosclerotic human arteries and failed vein grafts (17–19), as well as in arterial walls and neointima formation in various animal models of arterial injury (20, 21). Despite the frequent associations between elevated PAI-1 expression and poor cardiovascular outcomes, a causal relationship has yet to be definitively established. Because both u-PA surface expression (8) and cellular interactions with tissue vitronectin are important components in new blood vessel growth (9, 22, 23), we hypothesized that increased PAI-1 expression after myocardial infarction might result in poor outcome by directly inhibiting the ability of bone marrow–derived angioblasts to induce neovascularization. This study examined the effect of PAI-1 down-regulation on postinfarction neovascularization and myocardial function recovery induced by bone marrow–derived angioblasts.
To develop an approach to inhibit PAI-1 expression that might have clinical applicability, we examined various potential strategies for inhibiting specific mRNA activity, including antisense oligonucleotides and ribozymes (24–26). Because these approaches are limited by sensitivity to chemical and enzymatic degradation and restricted target site specificity, we focused on the use of a new generation of catalytic nucleic acids containing DNA molecules with catalytic activity for specific RNA sequences (27–30). These DNA enzymes exhibit greater catalytic efficiency than hammerhead ribozymes, offer greater substrate specificity, are more resistant to chemical and enzymatic degradation, and are far cheaper to synthesize. In this study we created DNA enzymes with specificity for target sequences in human and rat PAI-1 mRNA. Our results indicate that inhibition of PAI-1 expression in the heart after a myocardial infarction results in a striking augmentation of human angioblast–dependent neovascularization, cardiomyocyte regeneration, and functional cardiac recovery.
Materials and Methods
DNA Enzymes and RNA Substrates.
DNA enzymes with 3′-3′ inverted thymidine were synthesized by Integrated DNA Technologies and purified by RNase-free ion exchange HPLC or reverse phase HPLC. The short RNA substrates corresponding to target DNA enzyme sequences were chemically synthesized followed by RNase-free PAGE purification and also made by in vitro transcription from a DNA template.
In Vitro Transcription.
Human PAI-1 cDNA was amplified by RT-PCR from total RNA of cultured human umbilical vein endothelial cells (HUVECs) using the following primer pair: 5′-CCAAGAGCGCTGTCAAGAAGAC-3′ (forward primer) and 5′-TCACCGTCTGCTTTGGAGACCT-3′ (reverse primer; data are available from GenBank/EMBL/DDBJ under accession no. J03764, position 25–10600). Length of the PCR product was 1,598 bases. Rat PAI-1 cDNA was amplified using RT-PCR from total RNA of cultured rat arotic endothelial cells (provided by G. Ceballos-Reyes, Instituto Politecnico Nacional de Mexico, Mexico City, Mexico). The primer pair used was 5′-AGC ACA CAG CCA ACC ACA GCT-3′ (forward primer) and 5′-CTT CGA GAG TCT GAG GTC TG-3′ (reverse primer; data are available from GenBank/EMBL/DDBJ under accession no. M24067, position 48–1499) and the length of PCR product was 1,452 bases. Both PAI-1 cDNAs were cloned into pGEM-T vector (Promega) to obtain plasmid constructs pGEM-hPAI and pGEM-rPAI, and cDNA sequences were verified by automatic sequencing. A 32P-labeled nucleotide from the human or rat PAI-1 RNA transcript was prepared by in vitro transcription (SP6 polymerase; Promega) and cut before transcription with NcoI. 20 μl of transcription reaction consisted of 4 μl 5× buffer, 2 μl DTT, 1 μl RNasin inhibitor, 4 μl NTP mixture (1 μl A, G, C, and 1 μl H2O), 100 μM UTP, 2 μl template (0.3 μg/μl), α–32P-UTP (10 μci/μl), and 1 μl SP6 polymerase (20 U/μl). Reaction time was 1 h at 32°C. Unincorporated label and short nucleotides (<350 bases) were separated from radiolabeled species by centrifugation on Chromaspin-200 columns (CLONTECH Laboratories, Inc.).
Cleavage Reactions.
Synthetic RNA substrate was end-labeled with 32P using T4 polynucletiod kinase. The cleavage reaction system included 60 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 150 mM NaCl, 0.5 μM 32P-labeled RNA oligo, and 0.05–5 μM DNA enzyme. For cleavage of in vitro transcripts, the reaction system contained 1% of PAI-1 transcripts from transcription reaction system, 25 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 100 mM NaCl, and 0.2–20 μM DNA enzyme. Reactions were allowed to proceed at 37°C and were quenched by transfer of aliquots to tubes containing 90% formamide, 20 mM EDTA, and loading dye. Samples were separated by electrophoresis on TBE-urea–denaturing polyacrylamide gels (5% gel for in vitro transcripts and 15% for synthetic RNA). Densitometric analysis of band autoradiographs was performed using a Personal Densitometer and ImageQuant software (Molecular Dynamics). The percentage of the transcript mRNA cleaved by DNA enzyme was calculated as density of degraded mRNA bands/density of degraded plus undegraded bands.
DNA Enzyme Stability in Serum.
DNA enzymes were 32P-radiolabeled using T4 polynucleotide kinase. Labeling reaction (20 μl of volume) consisted of 1 μl DNA enzyme (20 uM), 2 μl 10× kinase buffer, 1 μl T4 PNK (10 U/μl), 10 μl H2O, and 6 μl 32P-ATP (3,000 μCi/mmol, 10 μCi/μl). Reaction time was 30 min at 37°C. 32P-labeled DNA enzymes were incubated at 37°C in media containing 2% FCS (EGM; Clonetics) or 20% FCS (DMEM; GIBCO BRL) at a final concentration of 100 nM. Aliquots of the mixture were removed at different time points of 0, 3, 6, 12, and 24 h, quenched with phenol/chloroform, and frozen until use. At the end of each experiment, all samples were phenol extracted and analyzed by 15% denaturing polyacrylamide gels and visualized by autoradiography.
Culture Conditions and DNA Enzyme Transfection.
Primary HUVECs were obtained from Clonetic and grown in EGM medium containing 2% FCS. Primary rat arotic endothelial cells were cultured in DMEM medium, pH 7.4 (Life Technologies), containing 10% FCS, 100 μg/ml streptomycin, and 100 IU/ml penicillin at 37°C in a humidified atmosphere of 5% CO2. For transfection of DNA enzyme, rat endothelial cells were plated into each well of six-well plates (∼105 cells/well). Subconfluent (∼70–80%) endothelial cells were washed twice with 1 ml Hepes buffer, pH 7.4, and transfected using 0.5 ml of serum-free DMEM containing 1 μM of test molecule (E2 or E0) and 7 μl/ml Superfect (QIAGEN). 3 h after incubation, 0.5 ml of 5% serum DMEM was added to each well. 3 h later, TGF-β was added to half of the wells at a final concentration of 1.8 ng/ml. Transfected cells continued to be incubated for 8 h and were lysed to isolate RNA (for RT-PCR) and protein (for Western blot) using Trizol reagent (Life Technologies).
RT-PCR.
Total RNA from each well of six-well plates was isolated using 1 ml Trizol reagent and dissolved in 20 μl DEPC-treated H2O. 2-μl samples were used to perform reverse transcription in 20 μl of reaction system, and 4 μl of products was then used as templates to amplify rat PAI-1 or GAPDH in 50 μl PCR system (5 μl 10× buffer, 1 μl dNTP, 0.25 μl Taq polymerase, 1 μl forward and reverse primer, 38 μl H2O, 2 μl 32P dCTP [3,000 ci/mmol, 10 μci/μl]). The reaction conditions were as follows: 95°C for 0.5 min, 58°C for 1 min, 72°C for 2 min, and 30 cycles at 72°C for 10 min. Rat GAPDH was used as internal control to quantify PAI-1 (PAI-1 primers as noted above). Rat GAPDH forward primer is 5′-CTCTACCCACGGCAAGTTCAA-3′ and the reverse primer is 5′-GGGATGACCTTGCCCACAGC-3′. PCR product was 515 bases long.
Western Blot.
Control and DNA enzyme–treated cells were lysed and protein pellets were dissolved in 1% SDS solution and then boiled for 4 min after an equal volume of 2× lysis buffer was added. Equal amounts of protein (10 μg/lane) were separated by SDS gel electrophoresis with 10% polyacrylamine separating gel using minigels (Bio-Rad Laboratories). After electrophoresis, proteins were transferred to nitrocellulose membrane (Amersham Biosciences) by electrotransfer and blocked for at least 1 h at room temperature with 5% (wt/vol) nonfat milk in TBS-T buffer (0.1 M Tris-base, pH 7.5, 0.15M NaCl, and 0.1% Tween-20). After this step, membranes were immunoblotted with goat IgG polyclonal anti-PAI antibodies (Santa Cruz Biotechnology, Inc.) and then visualized by the ECL system (Amersham Biosciences) using horseradish peroxidase–conjugated anti–goat IgG (Sigma-Aldrich).
Measurement of PAI-1 Activity.
At 70–80% confluence, HUVECs seeded in 24-well plates were transfected with 2 μM DNA enzymes (E1 and E3 as well as E0 in the presence or absence of 2 ng/ml TGF-β1 for 24 h. After washing twice with PBS, cell lysates were collected using 400 μl 0.5% Triton X-100 in PBS. 25 μl of cell lysate was incubated with plasmin substrate in a 96-well microtiter plate (final volume, 230 μl) for 134 min at room temperature. PAI-1 activity was measured by reading the difference of absorbances at 405 and 492 nm and was calculated against a plasmin standard regression line according to the protocol of American Diagnostica, Inc. (catalog no. 101201).
Transendothelial Migration Assay of Human Bone Marrow Stem Cells.
In brief, rat endothelial cell monolayers (∼2 × 105 cells/well) were grown to subconfluence (∼70–80%) on PET transwells in the presence or absence of rat vitronectin. To the top chamber of each well in triplicate experiments, 105 human CD34+ cells (use of human cells was approved by the Columbia University Institute for Animal Care and Use Committee) were added from a single donor together with 2% FCS, the presence or absence of 1.8 ng/ml TGF-β, and either E2 or scrambled DNA enzyme complexed with 20 μg/ml DOTAP. After 24 h in culture at 37°C, the cells in the top and bottom chambers were recovered and counted with a hemocytometer. The proportion of cells migrating across the endothelial monolayer was calculated as the number of cells in the bottom chamber divided by the total number of cells counted, and normalized to the proportion of human CD34+ cells migrating across the membrane in the absence of any endothelial monolayer for each condition tested. Additionally, after supplemental transmigration, an assay was performed to evaluate the effect of exogenous rat PAI-1 or antibody against rat PAI-1 on CD34+ cell transmigration. Rat endothelial cells (3.5 × 104 cells/100 μl) were seeded into the upper chamber of transwell plates (6.5-mm diameter, pore size of 5 μm; Costar, Inc.) and 300 μl DMEM (2% FCS) was placed into the lower chamber. After cultured cells reached full confluence and upper chambers were placed into fresh lower chambers, 2 μg rat PAI-1 (American Diagnostics) in 50 μl DMEM without FCS was added into some upper chambers, and 2 and 3 μg IgG against rat PAI-1 (American Diagnostics) was added into some upper and lower chambers, respectively. 30 min later, 105 CD34+ cells in 100 μl DMEM without FCS were added into each upper chamber. Cells migrating from the upper chamber into the lower chamber were harvested and counted using a hemocytometer 24 h after the addition of CD34+ cells.
Animals, Surgical Procedures, and Injection of DNA Enzyme and Human Cells.
Rowett (rnu/rnu) athymic nude rats (Harlan Sprague Dawley) were used in studies approved by the Columbia University Institute for Animal Care and Use Committee. After anesthesia, a left thoracotomy was performed, the pericardium was opened, and the left anterior descending (LAD) coronary artery was ligated. At the time of surgery, animals were randomized into three groups, each receiving three intracardiac injections at the peri-infarct region consisting, respectively, of E2 DNA enzyme, E0 scrambled control, or saline. 100 μl of injection solution included 30 μl DNA oligonucleotide (300 μg), 20 μl Superfect, and 50 μl saline. For studies on neovascular rization and effects on myocardial viability and function, 2.0 × 106 human cells obtained from a single donor after G-CSF mobilization were reconstituted with 2.0 × 105 immunopurified CD34+ CD117bright cells (1) and injected into the rat tail vein 48 h after LAD ligation. Each group consisted of 6–10 rats.
Immunohistochemistry and Quantitation of Capillary Density.
To quantitate and characterize PAI-1–expressing cells at 48 h after LAD ligation, sections from the hearts of control animals killed at this time point were freshly stained with goat IgG polyclonal anti-PAI antibodies (Santa Cruz Biotechnology, Inc.), mouse mAbs directed against rat CD68, factor VIII–related antigen, and cardiac troponin I (DakoCytomation), and then visualized by immunoperoxidase technique using an Avidin/Biotin Blocking Kit, a rat-absorbed biotinylated anti–goat IgG, and a peroxidase conjugate (all from Vector Laboratories). Capillary density and species origin of the capillaries were determined in sections from the hearts of animals killed at 2 wk. Sections were freshly stained with mAbs directed against rat CD31 (Serotec and Research Diagnostics, respectively), factor VIII–related antigen (DakoCytomation), and rat MHC class I (Accurate Chemicals). Arterioles were differentiated from large capillaries by the presence of a smooth muscle layer, identified by staining sections with an mAb against myocyte-specific desmin (DakoCytomation). Staining was performed by immunoperoxidase technique using an Avidin/Biotin Blocking Kit, a rat-absorbed biotinylated anti–mouse IgG, and a peroxidase conjugate (all from Vector Laboratories). Capillary density was determined at 2 wk after infarction from sections labeled with anti-CD31 mAb and confirmed with mAb against factor VIII–related antigen and compared with the capillary density of the unimpaired myocardium. Values are expressed as the number of CD31+ capillaries per high power field (a magnification of 400).
Quantitation of Cardiomyocyte Proliferation.
Cardiomyocyte DNA synthesis and cell cycling was determined by dual staining of rat myocardial tissue sections obtained from LAD-ligated rats 2 wk after injection of either saline or CD34+ human cells, and from healthy rats as negative controls, for cardiomyocyte-specific troponin I and human- or rat-specific Ki-67. In brief, paraffin-embedded sections were microwaved in 0.1 M EDTA buffer and stained with either a polyclonal rabbit antibody with specificity against rat Ki-67 at a 1:3,000 dilution (provided by G. Cattoretti, Columbia University, New York, NY), or a mouse mAb recognizing both human and rat Ki-67 and MIB-1 at a 1:300 dilution (DakoCytomation), and incubated overnight at 4°C. After washes, sections were incubated with a species-specific secondary antibody conjugated with alkaline phosphatase at a 1:200 dilution (Vector Laboratories) for 30 min, and positively staining nuclei were visualized as blue with a BCIP/NBT substrate kit (DakoCytomation). Sections were then incubated overnight at 4°C with an mAb against cardiomyocyte-specific troponin I (Accurate Chemicals), and positively staining cells were visualized as brown through the Avidin/Biotin system described above. Cardiomyocytes progressing through cell cycle in the infarct zone, peri-infarct region, and areas distal to the infarct were calculated as the proportion of troponin I+ cells per high power field coexpressing Ki-67. For confocal microscopy, FITC-conjugated rabbit anti–mouse IgG was used as secondary antibody to detect Ki67 in nuclei. A Cy5-conjugated mouse mAb against α-sarcomeric actin (clone 5C5; Sigma-Aldrich) was used to detect cardiomyocytes, and propidium iodide was used to identify all nuclei. In separate experiments, animals receiving saline or CD34+ cells after LAD ligation were fed BrdU ad libitum daily in drinking water. After death, paraffin-embedded tissue was incubated with a mouse anti-BrdUrd antibody (Roche Molecular Biochemicals) followed by a biotinylated rabbit anti–mouse IgG antibody (Jackson ImmunoResearch Laboratories), diluted at 1:3,000 with D-PBS. The biotin is detected by using the Avidin/Biotin Complex Kit (Vector Laboratories) as described above.
Analyses of Myocardial Function.
Echocardiographic studies were performed using a high frequency liner array transducer (SONOS 5500; Hewlett Packard). Two-dimensional images were obtained at mid-papillary and apical levels. End-diastolic (EDV) and end-systolic (ESV) left ventricular volumes were obtained by bi-plane area length method, and the percent of left ventricular ejection fraction was calculated as [(EDV-ESV)/EDV] × 100. All echocardiographic studies were performed by a blinded investigator.
Statistical Analysis.
Data are presented as mean ± SD. Comparisons between groups were made by Student's t test. Values of P < 0.05 were considered significant.
Results
Construction of DNA Enzymes Targeting Human and Rat PAI-1 mRNA.
We designed three DNA enzymes to specifically target pyrimidine–purine junctions at or near the translational start site AUG of human and rat PAI-1 messenger RNA, a region that is conserved between species and has low relative free energy (31). As shown in Fig. 1 A, the three DNA enzymes, termed E1, E2, and E3, contained identical 15-nucleotide catalytic domains that were flanked by two arms of eight nucleotides (E1 and E2) or nine nucleotides (E3) with complementarity to human (E1 and E3) or rat (E2) PAI-1 mRNA. To produce the control DNA enzyme E0, the nucleotide sequence in the two flanking arms of E2 was scrambled without altering the catalytic domain (Fig. 1 A). The 3′ terminus of each molecule was capped with an inverted 3′-3′–linked thymidine for resistance to 3′ to 5′ exonuclease digestion (see below).
Figure 1.
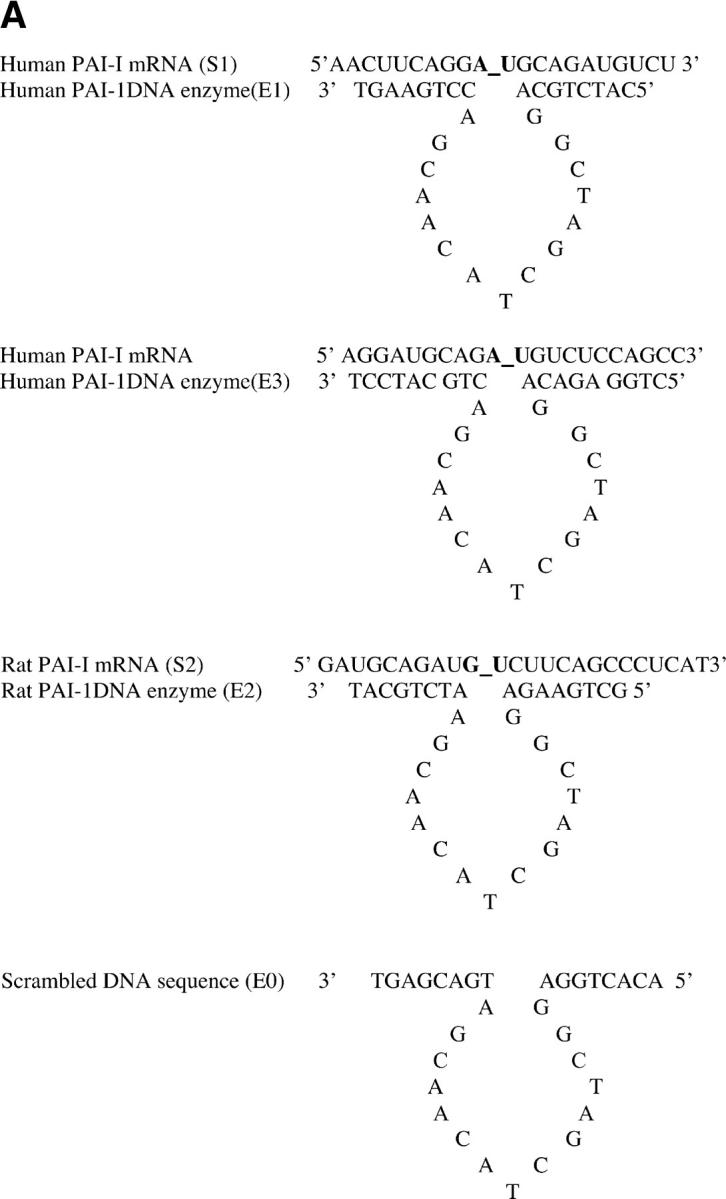


Construction of DNA enzymes and specific cleavage of human PAI-1 mRNA by E1 and E3. (A) Three DNA enzymes were constructed, termed E1, E2, and E3, containing identical 15-nucleotide catalytic domains that were flanked by two arms of eight nucleotides (E1 and E2) or nine nucleotides (E3) with complementarity to human (E1 and E3) or rat (E2) PAI-1 mRNA. To produce the control DNA enzyme E0, the nucleotide sequence in the two flanking arms of E2 was scrambled without altering the catalytic domain. The 3′ terminus of each molecule was capped with an inverted 3′-3′–linked thymidine for resistance to 3′ to 5′ exonuclease digestion. (B) 32P-labeled 21-base oligomer S1, synthesized from human PAI-1 mRNA, was cleaved in a time-dependent manner by E1 at a 10:1 substrate/enzyme excess. (C) Sequence-specific cleavage of human PAI-1 mRNA in vitro generated transcript by DNA enzyme E1 compared with control DNA enzyme E0. In lanes 2 and 4, incubation with E1, but not E0, after preheating transcript to 72°C for 10 min demonstrates further increase in cleavage. (D) Concentration-dependent cleavage of 32P-labeled human PAI-1 mRNA in vitro generated transcripts by E3. The 520 nucleotide transcript was cleaved to products of 320 and 200 nucleotides. (E) Quantitation of representative transcript cleavage by PAI-1 DNA enzymes from two separate experiments, showing time and dose dependence of the cleavage by DNA enzyme.
Specific Cleavage of Human PAI-1 mRNA by E1 and E3 DNA Enzymes.
As shown in Fig. 1 B, the 21-base oligonucleotide S1, synthesized from human PAI-1 mRNA and labeled at the 5′ end with 32P, was cleaved within 2 min when cultured with E1 at a 10:1 substrate/enzyme excess, with maximal cleavage occurring by 2 h. The 10-nucleotide cleavage product is consistent with the size of the 32P-labeled fragment at the 5′ end. E1 also cleaved larger 32P-labeled fragments of human PAI-1 mRNA prepared by in vitro transcription (Fig. 1 C). The 520-nucleotide transcript was cleaved by 4 h to expected cleavage products of 320 and 200 nucleotides. The sequence-specific nature of the DNA enzymatic cleavage is also shown in Fig. 1 C, where the control DNA enzyme E0, containing an identical catalytic domain to E1 and E3, but scrambled sequences in the flanking arms, caused no cleavage of human PAI-1 mRNA transcripts. Preheating the transcript to 72°C for 10 min (Fig. 1 C, lane 4) before incubation with E1, but not E0, further increased cleavage (percent of cleaved mRNA increased from 65 to 76%). Fig. 1 D shows similar cleavage of human PAI-1 mRNA transcripts by DNA enzyme E3 for 2 h. As shown in Fig. 1 E, the cleavage of mRNA transcripts by DNA enzymes occurred in a time- and concentration-dependent manner, with 65% cleavage by E1 for 4 h and 74% cleavage by E3 for 2 h, both at a concentration of 20 μM.
DNA Enzymes Inhibit Induction of Endogenous PAI-1 mRNA and Protein.
To determine the effect of DNA enzymes on endogenous PAI-1 production, endothelial monolayers of human and rat origin were grown to confluence and transfected with species-specific DNA enzymes or scrambled control. Transfected cells were then activated with TGF-β for 8 h to maximally induce expression of PAI-1. Densitometric analysis of RT-PCR products after reverse transcription of cellular mRNA showed that E2 inhibited TGF-β–inducible steady-state mRNA levels in cultured rat endothelium by 52%, relative to the E0 scrambled DNA enzyme (Fig. 2 A). Basal level of PAI-1 mRNA revealed a small decrease after transfection with the DNA enzymes (ratio of PAI1 mRNA to GAPDH, 27 ± 1% for E0 vs. 19 ± 2% for E2). Fig. 2 B shows the effect of endothelial cell transfection with E2 DNA enzyme on TGF-β–mediated induction of PAI-1 protein. Endothelial cells transfected with scrambled DNA enzyme demonstrated an ∼50% increase in cytoplasmic PAI-1 protein as detected by Western blot. In contrast, this effect was almost completely abrogated by transfection with the PAI-1 DNA enzyme E2.
Figure 2.

DNA enzymes inhibit induction of endogenous PAI-1 mRNA and protein, demonstrate serum resistance, and reduce PAI-1 activity. (A and B) Results of RT-PCR and immunoblots demonstrating that transfection of rat endothelial monolayers with DNA enzyme E2 results in significant inhibition of TGF-β–induced expression of PAI-1 mRNA and protein, relative to the E0 scrambled DNA (P < 0.01 for both). (C) 32P-labeled PAI-1 DNA enzyme with inverted thymidine at the 3′ end demonstrates resistance to degradation in culture with 2%, but not 20%, serum. (D) Transfection of HUVEC monolayers with the human PAI-1–specific DNA enzymes E1 or E3 reduces functional PAI-1 activity, as measured by chromogenic reaction of plasmin substrate (P < 0.01). (A, B, and D) Results are expressed as the mean ± SEM of three separate experiments.
PAI-1 DNA Enzyme Is Resistant to Serum-dependent Degradation and Inhibits PAI-1 Activity in Serum-cultured Endothelial Monolayers.
Because DNA enzymes with a thymidine in the correct orientation at the 3′ end are significantly degraded by factors in serum (1–5% concentration) within 24 h (25), we investigated the protective effect of an inverted thymidine at the 3′ end on serum-dependent nucleolytic degradation of PAI-1 DNA enzymes. The DNA enzyme E2 was labeled with 32P and incubated at 37°C for 3–24 h in medium containing 2 or 20% serum concentrations. Although the DNA enzyme was significantly degraded within 6 h in medium containing 20% serum, it remained intact during the entire 24-h incubation with medium containing 2% serum (Fig. 2 C). To evaluate whether PAI-1 was functionally inhibited by DNA enzymes after prolonged serum exposure, the conversion of plasminogen to plasmin corresponding to residual tissue type plasminogen activator 1 was measured in endothelial monolayers transfected with PAI-1–specific or –scrambled DNA enzymes and cultured for 12 h in 2% serum. Under these conditions, the E1 and E3 DNA enzyme inhibited TGF-β–dependent PAI-1 activity in endothelial monolayers by a mean of 25% (P < 0.01; Fig. 2 D). Because the residual tissue type plasminogen activator activity in the sample catalyzed the conversion of plasminogen to plasmin, which in turn hydrolyzed the chromogenic substrate, reduction in PAI-1 activity by DNA enzymes meanwhile indicated increase in plasmin activity. Together, these results indicate that an inverted thymidine at the 3′ end can protect the PAI-1 DNA enzyme against nucleolytic degradation at 2% serum concentrations likely to be encountered in vivo, enabling inhibition of cellular PAI-1 activity.
PAI-1 DNA Enzymes Increase Transendothelial Migration of Human Bone Marrow Stem Cells.
To investigate the physiologic significance of inhibiting PAI-1 activity in endothelial cells, we examined migration of human CD34+ cells across rat endothelial monolayers transfected with either E2 or the scrambled DNA enzyme E0. As shown in Fig. 3 A, treatment of rat endothelial monolayers with E2 increased human CD34+ migration by approximately threefold relative to medium alone in the presence of vitronectin, but not in the absence of vitronectin (P < 0.05). In contrast, treatment with the scrambled DNA enzyme did not significantly increase transendothelial migration relative to medium alone. The addition of exogenous recombinant rat PAI-1 or antibody against rat PAI-1 caused, respectively, either a marked reduction or an increase in transmigrating CD34+ cells compared with untreated controls (53.1 ± 18.7% or 168.7 ± 33% vs. 100 ± 20.4%, P < 0.05 for both; Fig. 3 B). These results demonstrate that inhibition of PAI-1 mRNA and protein expression augments the ability of human CD34+ cells to migrate across endothelial monolayers via interactions involving plasmin generation and vitronectin binding.
Figure 3.



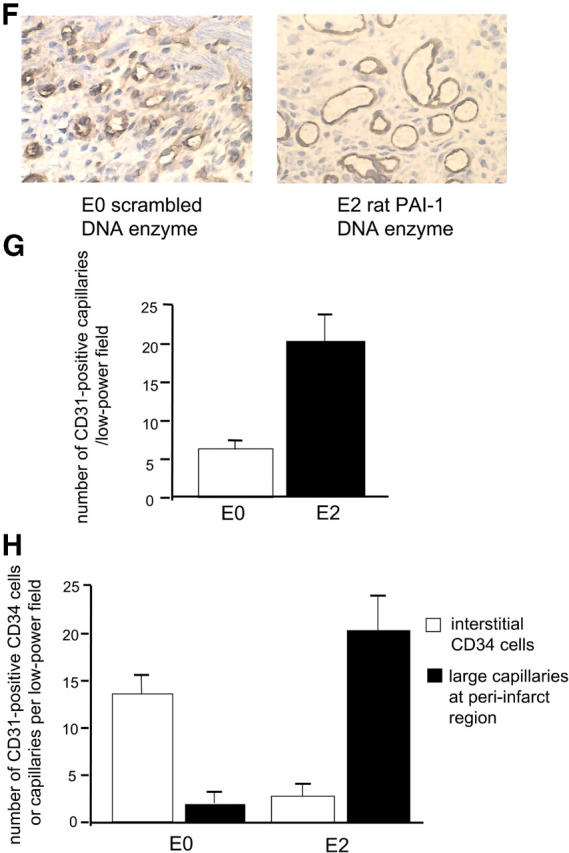
Effects of intramyocardial PAI-1 DNA enzyme injection on myocardial neovascularization. (A) Treatment of rat endothelial cell monolayers with the rat PAI-1 DNA enzyme E2, but not with the scrambled DNA enzyme E0, increased transendothelial migration of human CD34+ cells by approximately threefold relative to medium alone in the presence, but not in the absence, of vitronectin (P < 0.05). (B) The addition of rat recombinant PAI-1 or antibody against rat PAI-1 caused, respectively, either a significant reduction or an increase in CD34+ cells transmigrating across endothelial cell monolayers (P < 0.05 for both). (C) Immunohistochemical analyses using polyclonal PAI antiserum, showing reactivity in endothelial cells lining capillaries at the peri-infarct region (arrows). (D) Results of RT-PCR showing that intramyocardial injection with E2 results in reduced myocardial PAI-1 mRNA levels at 48 h and 2 wk after LAD ligation (P < 0.01 for both, relative to E0- and saline-treated controls). (E) Results of quantitative immunohistochemical analyses, showing reduced numbers of PAI-1–expressing cells at the peri-infarct region after E2 injection relative to the E0-treated controls (P < 0.01). (F) Immunohistochemical studies using anti-CD31 mAb demonstrating thin-walled capillaries with very large lumens (more than six nuclei), diameters in excess of 30 μm, and no smooth muscle outer layers, at the peri-infarct region after coadministration of E2 and human CD34+ cells (right). (G) Coadministration of E2 DNA enzyme intramyocardially induced significantly greater neovascularization at the peri-infarct region in comparison to E0 when human CD34+ cells were injected intravenously (P < 0.01). (H) Coadministration of E2 intramyocardially with human CD34+ cells intravenously resulted in reduced numbers of unincorporated, discrete, interstitial human CD34+ cells and increased numbers of large lumen capillaries (more than six nuclei) at the peri-infarct region, as determined by staining with a human anti-CD31 mAb, compared with E0 scrambled control enzyme (P < 0.01). (A, B, D, E, G, and H) Results are expressed as the mean ± SEM of three or four separate experiments.
Intracardiac Injection of E2 Reduces PAI-1 mRNA and Protein Expression after Acute Infarction.
Next, we investigated the in vivo biological activities of the PAI-1 DNA enzyme. As shown in Fig. 3 C, at 48 h after ligation of the LAD artery and induction of myocardial infarction in rats, immunohistochemical studies showed that PAI-1 expression occurred predominantly in vascular endothelial cells at the peri-infarct region and in infiltrating mononuclear cells. PAI-1 expression was not detected in cardiomyocytes either within or distal to the infarct zone. As shown in Fig. 3 D, direct intramyocardial injection of E2 at the peri-infarct region at the time of LAD ligation resulted in 60% lower levels of PAI-1 mRNA in the heart 48 h later, whereas the scrambled E0 control did not significantly reduce PAI-1 levels as measured by RT-PCR (P < 0.01). This effect was maintained for at least 2 wk, with cardiac PAI-1 mRNA levels remaining 39% lower in LAD-ligated animals treated with E2 compared with saline-treated animals at this time point (P < 0.01). As shown in Fig. 3 E, quantitative immunohistochemical analyses showed that at 48 h after LAD ligation and DNA enzyme injection, there was an over threefold reduction in the number of PA-1+ cells at the peri-infarct region of PAI-1 DNA enzyme-treated animals compared with scrambled DNA enzyme-treated controls (17 ± 5 low power field vs. 5 ± 1 low power field, P < 0.01).
Intracardiac Injection of E2 Together with Angioblasts Results in Enhanced Neovascularization at the Peri-Infarct Region.
When CD34+ cells containing angioblasts were administered intravenously with intramyocardial E2 DNA enzyme, as shown in Fig. 3 (F and G), a further 3.5-fold increase in neovascularization was seen over that induced by the CD34+ cells alone (P < 0.01). Moreover, thin-walled capillaries with very large lumens (more than six nuclei) and diameters in excess of 30 μm were frequently seen at the peri-infarct region when E2 and CD34+ cells were coadministered, but not when either was injected alone (Fig. 3 F). Notably, the large lumen capillaries overlapped in size with arterioles, but differed in that they only contained a thin endothelial layer, whereas the arterioles could be distinguished by an outer layer containing two to three smooth muscle cells of rat origin, as determined by positive staining with desmin and rat MHC class I mAbs. Although many of the newly formed vessels were of rat origin (angiogenesis), at least 25% of the vessels at the peri-infarct region were of human origin (vasculogenesis), as defined by a human anti-CD31 mAb. Fig. 3 H shows the effect of E2 on coalescence and vascular incorporation of human CD34+ cells in infarcted rat myocardium. In the presence of E2, the number of unincorporated, discrete, interstitial human CD34+ cells in the infarct zone and peri-infarct region, as determined by staining with a human anti-CD31 mAb, was reduced by 5.2-fold compared with E0 control enzyme (P < 0.01). Conversely, the number of large lumen capillaries (more than six nuclei) at the peri-infarct region was increased by 14-fold after E2 injection as compared with E0. We conclude that by inhibiting PAI-1 expression at the peri-infarct region, E2 enables human CD34+ cells to more effectively migrate through cardiac tissue, coalesce, and participate in new vessel formation.
Angioblast-dependent Neovascularization Induces Cardiomyocyte Regeneration.
Although myocyte hypertrophy and increase in nuclear ploidy have generally been considered the primary mammalian cardiac responses to ischemia, damage, and overload (32, 33), recent observations have suggested that human cardiomyocytes have the capacity to proliferate and regenerate in response to injury (34, 35). Because organogenesis in the prenatal period is preceded by signals derived from neovasculature (36), and fetal cardiomyocytes have the capacity to enter the cell cycle, we next examined whether neovascularization of the adult heart by human angioblasts might induce proliferation/regeneration of endogenous cardiomyocytes. As shown in Fig. 4 A, at 2 wk after LAD ligation, rats receiving human CD34+ cells demonstrated numerous “fingers” of cardiomyocytes of rat origin, as determined by expression of rat MHC class I molecules, extending from the peri-infarct region into the infarct zone. Similar extensions were seen very rarely in animals receiving saline. The islands of cardiomyocytes at the peri-infarct rim in animals receiving human CD34+ cells contained a high frequency of rat myocytes with DNA activity, as determined by dual staining with an mAb reactive against cardiomyocyte-specific troponin I and a polyclonal rabbit antiserum with reactivity against rat, but not human, Ki67. Triple immunofluorescence using confocal microscopy confirmed the presence of cycling rat cardiomyocytes and demonstrated a speckled pattern of Ki67 reactivity within cycling nuclei (Fig. 4 B). In contrast, in animals receiving saline, there was a high frequency of cells with fibroblast morphology and reactivity with rat Ki67, but not troponin I, within the infarct zone. The number of cardiomyocytes progressing through cell cycle at the peri-infarct region of rats receiving human CD34+ cells was 40-fold higher than that at sites distal to the infarct, where myocyte DNA activity was no different than in sham-operated rats.
Figure 4.



Effects of PAI-1 inhibition on regeneration of endogenous cardiomyocytes and on functional cardiac recovery. (A) Section from infarct of representative animal receiving CD34+ cells intravenously showing “finger” of cardiomyocytes of rat origin, as determined by expression of rat MHC class I molecules, extending from the peri-infarct region into the infarct zone is shown. These cellular islands contain a high frequency of myocytes staining positively for both cardiac-specific troponin I (brown) and rat-specific Ki-67 (dark blue; arrows). Sections from infarcts of representative animals receiving saline do not show same frequency of dual staining myocytes. (B) Confocal microscopy of peri-infarct tissue from representative animal receiving human CD34+ cells demonstrates cardiomyocytes whose nuclei (labeled blue by Cy5) concomitantly expressed Ki67 antigen (labeled green by FITC-conjugated secondary antibody reacting with polyclonal rabbit anti–rat primary antibody) and whose cytoplasm concomitantly stained for troponin I (labeled red by propidium iodide–conjugated anti-troponin mAb). (C) Animals receiving human CD34+ cells intravenously had a fourfold higher number of cycling cardiomyocytes at the peri-infarct region than that found in LAD-ligated controls receiving saline (P < 0.01). Combining intramyocardial E2 injection with intravenously delivered human CD34+ cells increased cardiomyocyte regeneration to levels 7.5-fold higher than in saline controls (P < 0.01). The scrambled DNA enzyme E0 had no such effect. (D) Combining intramyocardial injection of E2 with intravenous human CD34+ cells increased functional recovery of left ventricular ejection fraction at 2 wk from a mean of 22–50% (P < 0.01). The scrambled DNA enzyme E0 had no such effect. (C and D) Results are expressed as the mean ± SEM of three separate experiments.
E2 Augments Human CD34+ Cell–dependent Cardiomyocyte Regeneration and Improvement in Cardiac Function.
As shown in Fig. 4 C, animals receiving human CD34+ cells intravenously had a fourfold higher number of cell-cycling cardiomyocytes at the peri-infarct region than those found in LAD-ligated controls receiving saline (P < 0.01). Combining intramyocardial E2 injection with intravenously delivered human CD34+ cells resulted in a striking increase in the degree of cardiomyocyte regeneration, to levels 7.5-fold higher than in saline controls (P < 0.01). Moreover, as shown in Fig. 4 D, combining intramyocardial injection of E2 with human CD34+ cells delivered intravenously resulted in an almost doubling of the positive effect of CD34+ cells alone on cardiac function, as determined by recovery in left ventricular ejection fraction at 2 wk (P < 0.01). The scrambled DNA enzyme E0 had no such effect on either cardiomyocyte regeneration or cardiac functional recovery.
Discussion
Numerous clinical studies have documented direct correlations between elevated PAI-1 levels and poor cardiovascular outcomes, including myocardial infarction, atherosclerosis, and restenosis (13–16). Moreover, PAI-1 mRNA and protein expression are elevated in atherosclerotic human arteries and failed vein grafts (17–19), as well as in arterial walls and neointima formation in various animal models of arterial injury (20, 21). However, despite these associations, no study has to date established a causal link between elevated PAI-1 levels and cardiovascular disease. The results in this study suggest that elevated PAI-1 levels in patients with myocardial infarction might be causally related to poor outcome through mechanisms that result in direct inhibition of bone marrow–dependent neovascularization. Indeed, we demonstrated that reducing PAI-1 mRNA expression in situations of endogenous PAI-1 excess resulted in augmented transendothelial migration of human CD34+ cells in a vitronectin-dependent manner, increased fusogenicity and vascular formation by discrete CD34+ cells within ischemic myocardial tissue, and improved cardiac functional recovery.
Transendothelial egress, extracellular matrix degradation, and neovascularization are all related processes thought to require intravascular activation of latent metalloproteinases by plasmin, which is derived from plasminogen through activation by u-PA on the surface of the infiltrating bone marrow–derived cells and inhibited by PAI-1 (6–8). Recent studies have suggested that u-PA also binds to PAI-1 in tissues, where it is complexed to vitronectin (9, 10). The u-PA–PAI-1 interaction results in a rapid conformational change in PAI-1 that causes it to dissociate from vitronectin and increase its affinity for the low density lipoprotein receptor (11), leading to its clearance and degradation. Removal of PAI-1 from vitronectin exposes a cryptic epitope necessary for binding to the cell surface integrin alphavbeta3, which regulates cellular attachment and migration (9, 12). Together, these data indicate that angioblast-dependent neovascularization of the adult heart is inhibited in situations of excess circulating and tissue PAI-1 levels by overlapping mechanisms, including interference with u-PA–dependent plasmin generation and integrin binding to tissue vitronectin (8, 9, 22, 23).
In addition to augmenting cardiac neovascularization, inhibition of myocardial PAI-1 mRNA expression resulted in striking induction of cardiomyocyte regeneration and functional cardiac recovery in the presence of intravenously administered angioblasts. Although myocyte hypertrophy and increase in nuclear ploidy have generally been considered to be the primary mammalian cardiac responses to ischemia, damage, and overload (32, 33), recent observations have suggested that human cardiomyocytes have the capacity to proliferate and regenerate in response to injury (34, 35), although the signals required to induce such regeneration in meaningful numbers have not been identified. Common precursors giving rise to both cells of smooth muscle and cardiomyocyte lineage have been identified in the adult murine bone marrow (37), and it is possible that these cells might be mobilized coincident with acute ischemia or may seed the developing heart early in ontogeny and be locally resident in the adult. Whether the regenerative process described herein indeed involves bone marrow–derived or resident cardiomyocyte progenitors remains to be determined; however, it bears noting the striking similarity to the spontaneous myocardial regeneration seen to accompany prominent neovascularization after myocardial injury in MRL mice, a strain with abnormal stem cell development (38).
To develop an approach to inhibit PAI-1 expression that might have clinical applicability, various potential strategies for inhibiting specific PAI-1 mRNA activity were examined. Antisense oligonucleotides hybridize with their complementary target site in mRNA, blocking translation to protein by sterically inhibiting ribosome movement or by triggering cleavage by endogenous RNase H (24). Although current constructs are made more resistant to degradation by serum through phosphorothioate linkages, nonspecific biological effects due to “irrelevant cleavage” of nontargeted mRNA remains a major concern (25). Ribozymes are naturally occurring RNA molecules that contain catalytic sites, making them more potent agents than antisense oligonucleotides. However, wider use of ribozymes has been hampered by their susceptibility to chemical and enzymatic degradation and restricted target site specificity (26). Consequently, we focused on the use of a new generation of catalytic nucleic acids containing DNA molecules with catalytic activity for specific RNA sequences (27–30). These DNA enzymes exhibit greater catalytic efficiency than hammerhead ribozymes, producing a rate enhancement of ∼10 million-fold over the spontaneous rate of RNA cleavage, offer greater substrate specificity, are more resistant to chemical and enzymatic degradation, and are far cheaper to synthesize. Our results demonstrate that inhibition of myocardial PAI-1 mRNA by a sequence-specific catalytic DNA enzyme is a feasible approach for enhancing cardiac neovascularization, regeneration, and functional recovery after an acute ischemic insult.
Acknowledgments
This research was supported in part by National Institutes of Health grants RFA-HL-02-017 and RFA-AG-01-006.
The authors have no conflicting financial interests.
Abbreviations used in this paper: HUVEC, human umbilical vein endothelial cell; LAD, left anterior descending; PAI, plasminogen activator inhibitor; u-PA, urokinase-type plasminogen activator.
References
- 1.Kocher, A.A., M.D. Schuster, M.J. Szabolcs, S. Takuma, D. Burkhoff, J. Wang, S. Homma, N.M. Edwards, and S. Itescu. 2001. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 7:430–436. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy, M., M. Firpo, K. Choi, C. Wall, S. Robertson, N. Kabrun, and G. Keller. 1997. A common precursor for primitive erythropoiesis and definitive haematopoiesis. Nature. 386:488–493. [DOI] [PubMed] [Google Scholar]
- 3.Choi, K., M. Kennedy, A. Kazarov, and G. Keller. 1998. A common precursor for hematopoietic and endothelial cells. Development. 125:725–732. [DOI] [PubMed] [Google Scholar]
- 4.Elefanty, A.G., L. Robb, R. Birner, and C.G. Begley. 1997. Hematopoietic-specific genes are not induced during in vitro differentiation of scl-null embryonic stem cells. Blood. 90:1435–1447. [PubMed] [Google Scholar]
- 5.Labastie, M.C., F. Cortes, P.H. Romeo, C. Dulac, and B. Peault. 1998. Molecular identity of hematopoietic precursor cells emerging in the human embryo. Blood. 92:3624–3635. [PubMed] [Google Scholar]
- 6.Heymans, S., A. Luttun, D. Nuyens, G. Theilmeier, E. Creemers, L. Moons, M.J. Daemen, and P. Carmeliet. 1999. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 5:1135–1142. [DOI] [PubMed] [Google Scholar]
- 7.Johnsen, M., L.R. Lund, J. Romer, K. Almholt, and K. Dano. 1998. Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr. Opin. Cell Biol. 10:667–671. [DOI] [PubMed] [Google Scholar]
- 8.Lijnen, H.R., B. Van Hoef, F. Lupu, L. Moons, P. Carmeliet, and D. Collen. 1998. Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolysis. Arterioscler. Thromb. Vasc. Biol. 18:1035–1045. [DOI] [PubMed] [Google Scholar]
- 9.Stefansson, S., and D.A. Lawrence. 1996. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature. 383:441–443. [DOI] [PubMed] [Google Scholar]
- 10.Deng, G., G. Royle, S. Wang, K. Crain, and D.J. Loskutoff. 1996. Structural and functional analysis of the plasminogen activator inhibitor-1 binding motif in the somatomedin B domain of vitronectin. J. Biol. Chem. 271:12716–12723. [DOI] [PubMed] [Google Scholar]
- 11.Stefansson, S., S. Muhammad, X.F. Cheng, F.D. Battey, D.K. Strickland, and D.A. Lawrence. 1998. Plasminogen activator inhibitor-1 contains a cryptic high affinity binding site for the low density lipoprotein receptor-related protein. J. Biol. Chem. 273:6358–6366. [DOI] [PubMed] [Google Scholar]
- 12.Stefansson, S., E. Petitclerc, M.K. Wong, G.A. McMahon, P.C. Brooks, and D.A. Lawrence. 2001. Inhibition of angiogenesis in vivo by plasminogen activator inhibitor-1. J. Biol. Chem. 276:8135–8141. [DOI] [PubMed] [Google Scholar]
- 13.Hamsten, A., U. de Faire, G. Walldius, G. Dahlen, A. Szamosi, C. Landou, M. Blomback, and B. Wiman. 1987. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. 2:3–9. [DOI] [PubMed] [Google Scholar]
- 14.Juhan-Vague, I., S.D. Pyke, M.C. Alessi, J. Jespersen, F. Haverkate, and S.G. Thompson. 1996. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. Circulation. 94:2057–2063. [DOI] [PubMed] [Google Scholar]
- 15.Huber, K., M. Jorg, P. Probst, E. Schuster, I. Lang, F. Kaindl, and B.R. Binder. 1992. A decrease in plasminogen activator inhibitor-1 activity after successful percutaneous transluminal coronary angioplasty is associated with a significantly reduced risk for coronary restenosis. Thromb. Haemost. 67:209–213. [PubMed] [Google Scholar]
- 16.Sakata, K., F. Miura, H. Sugino, M. Shinobe, M. Shirotani, H. Yoshida, N. Mori, T. Hoshino, and A. Takada. 1996. Impaired fibrinolysis early after percutaneous transluminal coronary angioplasty is associated with restenosis. Am. Heart J. 131:1–6. [DOI] [PubMed] [Google Scholar]
- 17.Schneiderman, J., M.S. Sawdey, M.R. Keeton, G.M. Bordin, E.F. Bernstein, R.B. Dilley, and D.J. Loskutoff. 1992. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc. Natl. Acad. Sci. USA. 89:6998–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kauhanen, P., V. Siren, O. Carpen, A. Vaheri, M. Lepantalo, and R. Lassila. 1997. Plasminogen activator inhibitor-1 in neointima of vein grafts: its role in reduced fibrinolytic potential and graft failure. Circulation. 96:1783–1789. [DOI] [PubMed] [Google Scholar]
- 19.Padro, T., J.J. Emeis, M. Steins, K.W. Schmid, and J. Kienast. 1995. Quantification of plasminogen activators and their inhibitors in the aortic vessel wall in relation to the presence and severity of atherosclerotic disease. Arterioscler. Thromb. Vasc. Biol. 15:893–902. [DOI] [PubMed] [Google Scholar]
- 20.Hasenstab, D., R. Forough, and A.W. Clowes. 1997. Plasminogen activator inhibitor type 1 and tissue inhibitor of metalloproteinases-2 increase after arterial injury in rats. Circ. Res. 80:490–496. [DOI] [PubMed] [Google Scholar]
- 21.DeYoung, M.B., C. Zamarron, A.P. Lin, C. Qiu, R.M. Driscoll, and D.A. Dichek. 1999. Optimizing vascular gene transfer of plasminogen activator inhibitor 1. Hum. Gene Ther. 10:1469–1478. [DOI] [PubMed] [Google Scholar]
- 22.Brooks, P.C., A.M. Montgomery, M. Rosenfeld, R.A. Reisfeld, T. Hu, G. Klier, and D.A. Cheresh. 1994. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 79:1157–1164. [DOI] [PubMed] [Google Scholar]
- 23.Brooks, P.C., R.A.F. Clark, and D.A. Cheresh. 1994. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 264:569–571. [DOI] [PubMed] [Google Scholar]
- 24.Bennett, M.R., and S.M. Schwartz. 1995. Antisense therapy for angioplasty restenosis: some critical considerations. Circulation. 92:1981–1993. [DOI] [PubMed] [Google Scholar]
- 25.Stein, C.A. 2000. Is irrelevant cleavage the price of antisense efficacy? Pharmacol. Ther. 85:231–236. [DOI] [PubMed] [Google Scholar]
- 26.Simayama, T., F. Nishikawa, S. Nishikawa, and K. Taira. 1993. Nuclease-resistant chimeric ribozymes containing deoxyribonucleotides and phosphorothioate linkages. Nucleic Acids Res. 21:2605–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santoro, S.W., and G.F. Joyce. 1997. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA. 94:4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santiago, F.S., H.C. Lowe, M.M. Kavurma, C.N. Chesterman, A. Baker, D.G. Atkins, and L.M. Khachigian. 1999. New DNA enzyme targeting Egr-1 mRNA inhibits vascular smooth muscle proliferation and regrowth after injury. Nat. Med. 5:1264–1269. [DOI] [PubMed] [Google Scholar]
- 29.Breaker, R.R. 2000. Making catalytic DNAs. Science. 290:2095–2096. [DOI] [PubMed] [Google Scholar]
- 30.Khachigian, L.M. 2000. Catalytic DNAs as potential therapeutic agents and sequence-specific molecular tools to dissect biological function. J. Clin. Invest. 106:1189–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuker, M. 1989. On finding all suboptimal foldings of an RNA molecule. Science. 244:48–52. [DOI] [PubMed] [Google Scholar]
- 32.Soonpaa, M.H., and L.J. Field. 1997. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 272:H220–H226. [DOI] [PubMed] [Google Scholar]
- 33.Kellerman, S., J.A. Moore, W. Zierhut, H.G. Zimmer, J. Campbell, and A.M. Gerdes. 1992. Nuclear DNA content and nucleation patterns in rat cardiac myocytes from different models of cardiac hypertrophy. J. Mol. Cell. Cardiol. 24:497–505. [DOI] [PubMed] [Google Scholar]
- 34.Kajstura, J., A. Leri, N. Finato, N. di Loreto, C.A. Beltramo, and P. Anversa. 1998. Myocyte proliferation in end-stage cardiac failure in humans. Proc. Natl. Acad. Sci. USA. 95:8801–8805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beltrami, A.P., K. Urbanek, J. Kajstura, S.M. Yan, N. Finato, R. Bussani, B. Nadal-Ginard, F. Silvestri, A. Leri, C.A. Beltrami, and P. Anversa. 2001. Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 344:1750–1757. [DOI] [PubMed] [Google Scholar]
- 36.Lammert, E., O. Cleaver, and D. Melton. 2001. Induction of pancreatic differentiation by signals from blood vessels. Science. 294:564–567. [DOI] [PubMed] [Google Scholar]
- 37.Orlic, D., J. Kajstura, S. Chimenti, I. Jakoniuk, S.M. Anderson, B. Li, J. Pickel, R. McKay, B. Nadal-Ginard, D.M. Bodine, et al. 2001. Bone marrow cells regenerate infarcted myocardium. Nature. 410:701–705. [DOI] [PubMed] [Google Scholar]
- 38.Leferovich, J.M., K. Bedelbaeva, S. Samulewicz, X.M. Zhang, D. Zwas, E.B. Lankford, and E. Heber-Katz. 2001. Heart regeneration in adult MRL mice. Proc. Natl. Acad. Sci. USA. 98:9830–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]