

Abstract
Exposure of bone marrow–derived macrophages (BMDMs) to low concentrations of Bacillus anthracis lethal toxin (LT), whose catalytic subunit is lethal factor (LF), results in induction of a robust apoptotic response dependent on activation of Toll-like receptor (TLR)4. A similar TLR4-dependent apoptotic response is observed when BMDMs are infected with live B. anthracis (Sterne strain). However, TLR4 is considered to be a specific signaling receptor for lipopolysaccharide (LPS), a typical product of gram-negative bacteria, whereas B. anthracis is gram-positive. To understand how B. anthracis can activate TLR4, we analyzed its culture supernatants and found them to contain a potent TLR4-stimulating activity that can also induce apoptosis in macrophages in which the antiapoptotic p38 MAP kinase (whose activation is prevented by LF) was inhibited. Purification of this activity suggested it consists of anthrolysin O (ALO), a member of the cholesterol-dependent cytolysin (CDC) family. We show that recombinant ALO can activate TLR4 in a manner independent of LPS contamination and, together with LT, can induce macrophage apoptosis. We also provide genetic evidence that ALO is required for induction of macrophage apoptosis in response to infection with live B. anthracis and that other CDC family members share the ability to activate TLR4.
Keywords: macrophage, anthrax, innate immunity, apoptosis, infection
Introduction
Bacillus anthracis is a highly virulent gram-positive bacillus that is the causative agent of different forms of anthrax (1). At least part of the extreme virulence of B. anthracis is due to production of several exotoxins or virulence factors: lethal factor (LF), edema factor (EF), and protective antigen (PA; references 2, 3). Although PA binds to receptors expressed on the surface of host cells and allows cellular entry of LF and EF (4, 5), LF and EF possess essential enzymatic activities that alter host cell signaling (6–8). LF is metalloproteinase with unique specificity to mitogen-activated protein kinase (MAPK) kinases (MKKs), which severs the COOH-terminal MKK catalytic domain from the NH2-terminal regulatory domain (8, 9). This cleavage results in dismantling of MAPK activation cascades (8), whose normal function depends on interaction of the NH2-terminal MKK regulatory domain with upstream MKK kinases (MEKKs or MAP3Ks) and downstream MAPKs (10). We found that incubation with low amounts of B. anthracis lethal toxin (LT), a hetero-oligomer of PA and LF (2, 3), renders macrophages sensitive to LPS-induced apoptosis by preventing activation of the p38 MAPK pathway (11). This response, which depends on the proteolytic cleavage of MKK6 by LF, can be mimicked by the use of low molecular weight p38 inhibitors, such as SB202190 (11). More recently, we demonstrated that infection of BM-derived macrophages (BMDMs) with live B. anthracis (Sterne strain) also results in extensive apoptosis that depends on signaling from the LPS-responsive Toll-like receptor (TLR)4, which activates the proapoptotic double-stranded, RNA-dependent protein kinase PKR (12).
However, B. anthracis is a gram-positive bacterium that does not produce LPS and, thus, it remained to be identified which component of B. anthracis activates TLR4 and induces apoptosis of macrophages exposed to LF. In this work, we sought to identify a specific TLR4 agonist from B. anthracis. This investigation revealed that anthrolysin O (ALO), a cholesterol-dependent cytolysin (CDC) secreted by B. anthracis (13), specifically targets TLR4 and acts together with LT to induce macrophage apoptosis. Other members of the CDC family were also found capable of TLR4 activation.
Materials and Methods
Mice and Macrophages.
C57BL/6J, C3H/OuJ, C3H/HeJ, B6.MRL-Tnfrsf6 lpr/J (Faslpr/lpr), and C57BL/6-Tnfrsf1a tm1Imx (TNFR1−/−) mice were obtained from The Jackson Laboratory. IFNR1−/− mice in the 129/SvEv background were obtained from E. Raz (University of California, San Diego, CA). BMDMs were prepared as described previously (11).
Reagents.
B. anthracis cell walls were purchased from List Biological Laboratories, Inc. Other reagents used for treatment of BMDMs included the following: LPS (Escherichia coli; Sigma-Aldrich), peptidoglycan (Fluka), poly(I-C) (Amersham Biosciences), CpG oligodeoxynucleotide (TIB MOLBIOL), Pam3CSK4 (EMC Microcollections), and R-848 (GLS Synthesis). SB202190 was from Calbiochem.
Bacterial Strains, Culture, and Infection.
B. anthracis Sterne strain 7702 and its derivatives were described previously (13). Bacteria were grown in brain heart infusion broth (BHI; Difco), without added bicarbonate, with shaking (200 revolutions/min) at 37°C in an air shaker incubator or on BHI agar in a humidified incubator. Bacterial infection of macrophage cultures was described previously (12).
Purification of ALO from Bacterial Culture Supernatants.
All buffers used in dialysis and column chromatography were prepared with double-distilled and autoclaved water and contained protease inhibitors (10 μM phenylmethylsulfonyl fluoride, 20 nM pepstatin A, 6 nM leupeptin, and 20 μM bisbenzamidine). The buffers were confirmed to be endotoxin free by testing their ability to induce TNF-α expression in macrophages. To purify macrophage-stimulating activity from B. anthracis culture supernatants, bacteria were grown in BHI broth until OD595 reached 1.0. After removing bacteria by centrifugation, the supernatant (two liters) was filtered through 0.2-μm-pore Nylon filters (Nalgene), concentrated up to 80-fold on a Centricon Plus-20 Filter Device (Millipore), and dialyzed in buffer D100 (20 mM Tris-Cl, pH 7.0, 100 mM NaCl, and 0.1 mM EDTA). 84 mg of protein in the culture concentrate were applied to a DEAE-sepharose column (10 ml) equilibrated with buffer D100. The macrophage-stimulating activity (see Results) was found to pass through this column under this particular loading condition. Proteins in the flow-through fraction (61 mg) were equilibrated in buffer S50 (20 mM Hepes-KOH, pH 7.0, 50 mM NaCl, and 0.1 mM EDTA) by dialysis and applied to a Mono S column (1 ml per 20 mg of protein) equilibrated with buffer S50. After washing with buffer S50, bound proteins were eluted with a linear gradient of 50–1,000 mM NaCl. The major peak fractions of macrophage-stimulating activity (3.1 mg) were pooled and mixed with an equal volume of 100 mM Tris-Cl, pH 7.0, and 3 M (NH4)2SO4, and applied to a 0.5-ml phenyl-sepharose column equilibrated with buffer P1500 (50 mM Na2HPO4, pH 7.0, 1.5 M (NH4)2SO4, and 0.1 mM EDTA). After washing with buffer P1500, bound proteins were eluted with an inverse linear gradient of 1.5 to 0 M (NH4)2SO4. The phenyl-sepharose fractions active in macrophage stimulation were stored at −80°C.
Preparation of Recombinant Proteins.
Recombinant LF, PA, and CDCs were expressed in and purified from E. coli strain BL21 (DE3) bearing the appropriate plasmid construct as described previously (13–15). Purified listeriolysin O (LLO) and LLO expression vector were provided by D. Portnoy (University of California, Berkeley, CA), and perfringolysin O (PFO) and streptolysin O (SLO) expression vectors by R. Tweten (University of Oklahoma Health Sciences Center, Oklahoma City, OK). The analyses of macrophages' activation by CDCs (see Figs. 3, B and C, and 6 A) were performed with proteins synthesized in reticulocyte lysates using T7 expression plasmids harboring a CDC gene and the TNT T7 kit (Promega).
Figure 3.

ALO activates TLR4 independently of its cytolytic activity. (A) The levels of hemolysis, macrophage cytolysis (permeability to Hoechst 33258), and TNF-α mRNA induction were determined after treatment of either erythrocytes or macrophages with the indicated ALO concentrations. (B) Reticulocyte lysates programmed with either an empty backbone vector (−, pRSET-A), T7 expression vectors for full-length (FL) ALO, or the ΔD4 derivative, lacking amino acids 409–512, were analyzed by immunoblotting with an ALO-specific antibody. The hemolytic activity of the reticulocyte lysates was measured and is shown above the immunoblot. (C) BMDMs (C57BL/6J) were incubated with reticulocyte lysates prepared as in B. After 4 h, total RNA was isolated, and relative expression of TNF-α mRNA (black bars) was determined by real-time PCR. (D) BMDMs (C57BL/6J) were treated with 100 ng/ml ALO or 100 μg/ml melittin, or subjected to two cycles of freezing at −80°C and thawing at 37°C. After 4 h, the level of cell permeabilization was analyzed by staining with H33258 (left). The conditioned media (CM) collected after the first treatment were applied to fresh BMDMs and TNF-α mRNA induction was analyzed after 4 h by real-time PCR (right). (E) CM collected from ALO-treated cells was depleted with anti-ALO or preimmune serum (Mock). The level of ALO in the immunodepleted CM was analyzed by immunoblotting and the TNF-α–inducing activity of the immunodepleted CM was analyzed as in D.
Figure 6.

TLR4 activation by ALO and other CDCs of gram-positive bacteria. (A) BMDMs (C57BL/6J) were incubated with reticulocyte lysates programmed with either an empty backbone vector (Mock; pRSET-A), or T7 expression vectors for ALO, perfringolysin O (PFO), listeriolysin O (LLO), or streptolysin O (SLO). After 12 h, cell lysates were prepared and analyzed by immunoblotting for iNOS induction. (B) BMDMs (C57BL/6J) were preincubated with or without 25 μg/ml polymyxin B for 1 h and treated with100 ng/ml LPS or 50 ng/ml CDC proteins. After 12 h, culture supernatants were collected and examined for TNF-α secretion by ELISA. (C) BMDMs from C3H/OuJ (tlr4 OuJ/OuJ) or C3H/HeJ (tlr4 He HeJ/HeJ) mice were stimulated with the indicated amounts of recombinant ALO, PFO, LLO, and SLO, or sBLP (1 μg/ml). After 4 h, total RNA was isolated, and the levels of TNF-α and IL-6 mRNA were measured by real-time PCR.
Protein Analysis.
Whole-cell extracts for immunoblot analysis were prepared with lysis buffer (20 mM Hepes-KOH at 150 mM NaCl, pH 7.6, 10% glycerol, 1% Triton X-100, 25 mM β-glycerophosphate, 2 mM EDTA, and protease inhibitors) and subjected to SDS-PAGE. Proteins transferred to nitrocellulose membrane were probed with rabbit antiserum against recombinant ALO (13), and antibodies directed against actin (Sigma-Aldrich), iNOS, phospho-p38α, p38α, and IκBα (all from Santa Cruz Biotechnology, Inc.), and the immune complexes were visualized with the ECL Western blot reagent (Pierce Chemical Co.). TNF-α secretion was measured by an ELISA (R&D Systems).
RNA Analysis.
Total RNA was isolated using the RNAwiz reagent (Ambion). For real-time PCR analysis, cDNAs were synthesized with the Superscript II reverse transcriptase system (Invitrogen). An amount of cDNA equivalent to 0.2 μg of total RNA was subjected to 40 cycles of amplification consisting of a 15-s incubation at 95°C and a 1-min incubation at 60°C. Output was monitored using SYBR green core reagents and the ABI Prism 7700 System (PE Applied Biosystems). The results were normalized to the level of cyclophilin mRNA. Individual primer sequences are available upon request.
Measurement of Cell Viability and Hemolysis.
TUNEL assay and Hoechst staining were performed as described previously (11). MTT assay and annexin V staining were performed using an MTT kit and annexin V-Alexa 568, respectively, according to the manufacturer's instructions (Roche). The hemolysis assay was performed by incubating mouse erythrocytes (whole blood diluted 10-fold with phosphate-buffered saline) with CDCs for 30 min at 37°C, centrifuging the mixture at 8,000 g for 1 min, and measuring A350 of the supernatant. The relative optical density was compared with that of erythrocytes treated with 0.1% Triton X-100 and used to determine percentage of hemolysis.
Immunodepletion of ALO.
0.4 ml of culture supernatants was mixed with 5 μl of ALO-specific antiserum on a rotating wheel for 12 h at 4°C. 50 μl of protein A–agarose beads was added and incubated for 1 h. Samples were briefly centrifuged to precipitate the beads, and supernatants were collected for further analyses.
Results
Identification of Macrophage-stimulating Activity from B. anthracis Culture Supernatant.
To identify the TLR4 agonist from B. anthracis that is required for triggering the apoptosis of LT-treated macrophages, we tested B. anthracis cell wall preparations and culture supernatants for their ability to stimulate TNF-α gene expression and induce apoptosis of BMDMs in the presence of the p38 inhibitor SB202190. Treatment of BMDMs with a crude, commercially available B. anthracis cell wall preparation did not strongly induce TNF-α mRNA expression or apoptosis (Fig. 1 A, I and II). However, a B. anthracis culture supernatant induced both TNF-α mRNA and apoptosis under the same conditions (Fig. 1 A, III). The TNF-α– and apoptosis-inducing activity in the culture supernatant was sensitive to proteinase K digestion (Fig. 1 A, IV), suggesting that a proteinaceous component is responsible for both activities. As only TLR4 agonists, but not agonists for other TLRs, can strongly potentiate macrophage apoptosis in the presence of SB202190 (Fig. 2 A and references 11, 12), this protein is likely to act as a TLR4 agonist.
Figure 1.
Identification of a macrophage-stimulating activity in B. anthracis culture supernatant. (A) C57BL/6J BMDMs were left untreated (I), treated with a B. anthracis cell wall preparation (II), or with B. anthracis culture supernatant before (III) and after (IV) proteinase K pretreatment. After 4 h, total RNA was isolated, and relative expression of TNF-α mRNA (black bars) was determined by real-time PCR. In a separate experiment, BMDMs were preincubated with 10 μM SB202190 for 2 h, and treated as described before. After 24 h, cell viability (white bars) was measured by the MTT assay. (B) Partially purified B. anthracis culture supernatant was fractionated on a phenyl-sepharose column. Column fractions were tested for their ability to induce TNF-α expression (top) or macrophage apoptosis (middle) as described before. The protein composition and ALO-content of the column fractions were examined by SDS-PAGE and silver staining and immunoblotting with an anti-ALO antibody (bottom).
Figure 2.

Macrophage activation by ALO is TLR4 dependent. (A) BMDMs were left untreated (−), or treated with 50 ng/ml recombinant ALO, or other TLR agonists, including 10 μg/ml peptidoglycan (PGN), 1 μg/ml synthetic bacterial lipopeptide (sBLP; Pam3CSK), 10 μg/ml poly(I-C), 100 ng/ml LPS, 1 μM R-848, or 1 μM CpG oligodeoxynucleotide (CpG ODN). Total RNA was analyzed as in Fig. 1 A for expression of different cytokine mRNAs (black bars). BMDMs were also preincubated with 10 μM SB202190 for 2 h and treated as before. Cell viability was measured by the MTT assay (white bars). (B) BMDMs from C3H/OuJ (tlr4 OuJ/OuJ; Ou) or C3H/HeJ (tlr4 HeJ/HeJ; He) mice were incubated with 50 ng/ml ALO, 100 ng/ml LPS, or 1 μg/ml sBLP. At the indicated time points, cell lysates were prepared and analyzed by immunoblotting with the antibodies indicated on the left for IκBα degradation and p38 activation (appearance of phosphorylated p38α). (C) BMDMs (C57BL/6J) were preincubated with or without 25 μg/ml polymyxin B for 1 h and treated with 50 ng/ml ALO, 10 μM taxol, or 100 ng/ml LPS. After 15 min, cell lysates were prepared and analyzed as in B. An IκBα signal indicates no IKK activation, whereas a phospho-p38α signal indicates p38 activation. (D) BMDMs (C57BL/6J) were incubated with LPS or ALO in either serum-containing or serum-free media (SFM) before or after pretreatment with proteinase K (PK). After 15 min, cell lysates were prepared and analyzed as in B for p38 activation.
To identify the protein responsible for this activity, the B. anthracis culture supernatant was sequentially purified through DEAE-sepharose, Mono S, and phenyl-sepharose chromatography columns. On the phenyl-sepharose column, the TNF-α– and apoptosis-inducing activities cofractionated as a single peak centered at fraction 26 (Fig. 1 B). Analysis of the composition of the different column fractions revealed that a 63-kD polypeptide copurified with both activities (Fig. 1 B). Among the secreted proteins predicted by the B. anthracis genome sequence (16), we noted the presence of ALO, a CDC, encoded by the BA3355 gene (13). The ALO polypeptide consists of 512 amino acids with the NH2-terminal 35 residues coding for a signal peptide, a size consistent with the aforementioned 63-kD band. Of note, it was recently shown that pneumolysin, a closely related CDC of Streptococcus pneumoniae, is capable of activating TLR4 (17). This raised the possibility that the TNF-α and apoptosis-inducing activity of the B. anthracis culture supernatant may be attributable to ALO. Hence, we analyzed the phenyl-sepharose fractions by immunoblotting with anti-ALO antibody and found that ALO indeed copurified with the 63-kD protein, as well as the macrophage-stimulating and apoptosis-inducing activities (Fig. 1 B).
Activation of Macrophages by ALO via TLR4.
To directly test whether ALO can stimulate macrophages in a TLR4-dependent manner, we prepared pure recombinant ALO (rALO) by expression in E. coli. Treatment of BMDMs with rALO or other TLR agonists with different receptor specificities resulted in a strong induction of TNF-α mRNA (Fig. 2 A). To compare the gene induction specificity of ALO to those of other TLR agonists, we examined the mRNA levels of other cytokines including IL-1α, IL-1β, and IL-6. Although TNF-α was induced to similar extents by ALO and the different TLR agonists, the IL-1 and IL-6 genes were most strongly induced by ALO and LPS, but were less responsive to other TLR agonists (Fig. 2 A). Furthermore, treatment of BMDMs with either ALO or the different TLR agonists in conjunction with SB202190 revealed that only ALO and LPS were able to cause a robust apoptotic response (Fig. 2 A).
To specifically examine the role of TLR4 in the response to ALO, we prepared BMDMs from wild-type (C3H/OuJ) and TLR4 mutant (C3H/HeJ) mice and compared their responses with ALO treatment. ALO induced activation of p38 MAPK and degradation of IκBα in TLR4 wild type, but not in TLR4 mutant, BMDMs (Fig. 2 B). The TLR4 mutant BMDMs showed no defect in their response to the TLR2 agonist-synthetic bacterial lipopeptide (sBLP; Pam3CSK4; Fig. 2 B). ALO also failed to induce TNF-α and IL-6 gene expression in TLR4 mutant BMDMs (see Fig. 6 C and not depicted). These observations indicate that ALO activates macrophages via TLR4. To rule out the possibility that TLR4 activation by recombinant ALO is due to the presence of contaminating LPS in the preparation, we treated BMDMs with ALO, LPS, and taxol, another TLR4 agonist, in the presence of polymyxin B, which blocks LPS binding to TLR4. Activation of p38 MAPK by LPS, but not by ALO or taxol, was inhibited by polymyxin B (Fig. 2 C). Polymyxin B neutralized the activity of LPS at concentrations as high as 0.5 μg/ml (unpublished data). We also examined the requirement of serum for biological activity and the proteinase K sensitivity of ALO and LPS. TLR4 activation by LPS depends strictly on factors such as soluble CD14 and LPS-binding protein that are provided by inclusion of serum (18). We found that p38 MAPK activation in macrophages by rALO can occur in serum-free medium, in which LPS fails to activate p38 MAPK (Fig. 2 D). The converse was observed after proteinase K digestion. Although proteinase K treatment completely abolished the ability of ALO to activate p38 MAPK, the activity of LPS was largely unaffected (Fig. 2 D). Together, these experiments unequivocally demonstrate that TLR4 activation by ALO is not due to contamination with LPS, whose activity is proteinase K resistant and serum dependent.
TLR4 Activation by ALO Independent of the Cytolytic Activity.
It has been shown that certain proteins derived from damaged host cells can also activate TLR4 (19). This raised the possibility that ALO may activate TLR4 indirectly by lysis of macrophages leading to the release of endogenous TLR4 agonists.
To address this possibility, we first examined whether ALO by itself exerts a cytotoxic effect on BMDMs at a range of concentrations that support TNF-α induction. Although ALO manifested strong hemolytic activity on red blood cells at concentrations lower than those required for TNF-α mRNA induction, no significant cytotoxicity toward BMDMs was observed, even at the highest ALO concentration tested (Fig. 3 A). Hence, macrophage activation by ALO is not accompanied by host cell permeabilization. To obtain more direct evidence that TLR4 activation by ALO does not involve host cell permeabilization, we generated a derivative (designated ΔD4) of ALO that lacks a carboxy (COOH)-terminal fragment spanning amino acids 409–512. This COOH-terminal fragment is homologous to domain 4 of PFO that possesses a cholesterol-binding activity and plays an indispensable role in forming pores in cholesterol-containing membranes (20). Full-length (FL) ALO and ΔD4 were produced by in vitro translation in reticulocyte lysates and tested for hemolytic activity. FL ALO, but not ΔD4, exhibited hemolytic activity against mouse erythrocytes (Fig. 3 B), demonstrating the importance of the PFO domain 4–like region of ALO for pore formation. However, both of the in vitro translated proteins were capable of inducing TNF-α gene expression by BMDMs, whereas a control lysate programmed with an empty backbone plasmid was inactive (Fig. 3 C). This finding indicates that ALO activates macrophages independently of its cytolytic activity. In an alternative approach, we examined culture supernatant of ALO-treated as well as lysed macrophages for TLR4-stimulating activities. To permeabilize the membranes of BMDMs, the cells were treated with melittin, a bee venom toxin with potent cytolytic activity (21), or subjected to two cycles of freezing and thawing. Treatment with melittin or freezing–thawing induced significant levels of cell permeabilization as monitored by staining with the membrane-impermeable dye Hoechst 33258 (Fig. 3 D). However, as in Fig. 3 A, little or no comparable cytolysis was observed upon ALO treatment.
The conditioned media (CM) from these cultures were applied to fresh BMDMs, and TNF-α mRNA induction was analyzed. Although the CM of ALO-treated cells potently induced TNF-α mRNA, those of cells damaged by melittin treatment or freeze–thawing did not (Fig. 3 D). The macrophage-stimulating activity contained in the CM of ALO-treated cells may still originate from endogenous host factors that are specifically released from BMDMs in response to ALO treatment. To investigate this possibility, the CM from ALO-treated cells was depleted of ALO with an ALO-specific antiserum and applied to untreated BMDMs. The ALO-depleted CM lost its ability to activate TNF-α mRNA induction, but CM immunodepleted with preimmune serum was still capable of inducing TNF-α mRNA (Fig. 3 E). Therefore, macrophage activation by CM from ALO-treated cells is not due to any protein other than ALO itself. Together, these independent lines of evidence unequivocally demonstrate that TLR4 activation by ALO is not a secondary response to host cell damage caused by the cytolytic activity of ALO.
Macrophage Apoptosis Induced by Treatment with B. anthracis Proteins.
As we identified ALO as a TLR4 agonist that can lead to macrophage apoptosis under conditions of SB202190 pretreatment, we checked if it can also act together with B. anthracis LF to induce macrophage apoptosis. To this end, we used ALO, LF, and PA. The three B. anthracis proteins were produced in E. coli and purified to apparent homogeneity (Fig. 4 A). Addition of ALO, LF, and PA to BMDMs induced apoptosis of the latter as shown by either TUNEL assay or annexin V staining (Fig. 4 B). Each of the three proteins was indispensable for induction of macrophage apoptosis. In a separate experiment, different amounts of ALO were combined with fixed amounts of LF and PA and the mixture was added to BMDMs. Under these conditions, ALO induced macrophage apoptosis in a dose-dependent manner, but even at the highest concentration tested did not induce apoptosis on its own. This shows that the ability to induce macrophage apoptosis does not represent a nonspecific cytotoxic activity of ALO.
Figure 4.
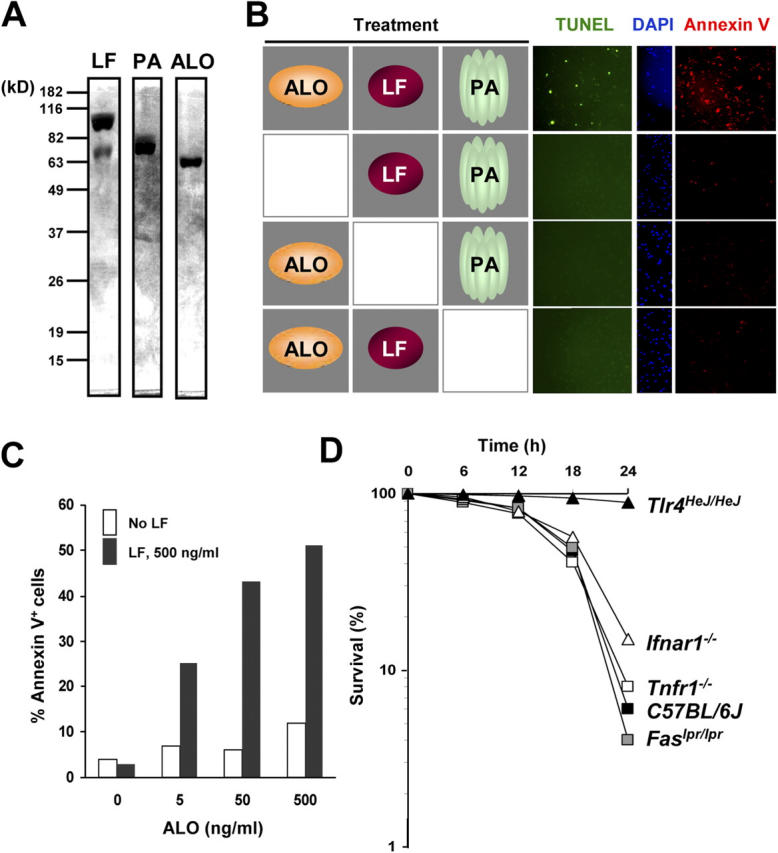
Reconstitution of macrophage apoptosis with recombinant LF, PA, and ALO proteins. (A) Recombinant LF, PA, and ALO proteins were analyzed by SDS-PAGE and Coomassie blue staining. (B) BMDMs (C57BL/6J) were treated with different combinations of 500 ng/ml recombinant LF, 1 μg/ml PA, and 50 ng/ml ALO as indicated. After 24 h, the cells were analyzed by TUNEL, DAPI, and annexin V staining. (C) BMDMs were treated with 1 μg/ml PA and the indicated amounts of ALO in the absence (white bars) or presence (black bars) of 500 ng/ml LF. The number of apoptotic cells was determined by annexin V staining. (D) BMDMs from wild type (C57BL/6J), TLR4 mutant (Tlr4 HeJ/HeJ), IFNR1 knockout (Ifnar1 −/−), TNFR1 knockout (Tnfr1 −/−), and Fas mutant (Fas lpr/lpr) mice were preincubated with 10 μM SB202190 and treated with 50 ng/ml ALO. At different time points, cell viability was determined by Hoechst 33258 staining. Cells that were not stained were counted as viable cells.
As ALO induces expression of proinflammatory genes by macrophages, the observed apoptotic response may represent a secondary response induced by one of these proinflammatory cytokines, such as TNF-α. To address this issue, we tested BMDMs harboring mutations in genes encoding different proapoptotic cytokine receptors for their response to ALO and SB202190. BMDMs from mice homozygous for deletions or inactivating mutations in the genes for TNF receptor 1, Fas, and the IFNR1 underwent apoptosis after treatment with ALO and SB202190, similar to wild-type BMDMs (Fig. 4 D). Only TLR4 mutant BMDMs were resistant to the apoptotic effect of ALO and SB202190.
Requirement for ALO in B. anthracis-induced Macrophage Apoptosis.
The aforementioned studies defined a minimal set of B. anthracis proteins, including ALO, that can trigger macrophage apoptosis in culture. Next, we examined the contribution of ALO to macrophage apoptosis caused by infection with live B. anthracis (12). BMDMs infected with the wild-type Sterne strain of B. anthracis underwent apoptosis detected by staining with Hoechst 33258 (Fig. 5 A) or by a TUNEL assay (Fig. 5 B). In contrast, macrophages infected with the same multiplicity of infection of an ALO-deficient mutant derived from the Sterne strain (13) exhibited a considerably reduced apoptotic response (Fig. 5, A and B). The inability of the Δalo mutant to induce macrophage apoptosis was rescued by transformation with a plasmid containing the alo gene (Fig. 5, A and B, and reference 13). These results indicate that ALO is necessary for the ability of B. anthracis to induce macrophage apoptosis, which was previously shown to be TLR4 dependent (12).
Figure 5.

Requirement for ALO in B. anthracis–induced macrophage apoptosis. (A) BMDMs (C57BL/6J) were infected with wild-type B. anthracis (7702), alo mutant (7702Δalo), or alo mutant complemented with the alo gene (7702Δalo; pUTE544[Alo]) at a multiplicity of infection of 0.1. After 6 h, cell death was measured by H33258 staining. (B) BMDMs were infected with B. anthracis as in A. After 12 h, cells were stained with DAPI (blue) or TUNEL (green).
TLR4-dependent Macrophage Activation by CDC from Different Gram-positive Bacteria.
ALO and pneumolysin are closely related to other CDCs produced by gram-positive pathogens. Our finding that ALO and pneumolysin (17) activate TLR4 prompted us to test whether this property is shared among other CDCs. Interestingly, it has been observed that CDCs, such as LLO from Listeria monocytogenes and SLO from Streptococcus pyogenes, can activate macrophages and mast cells to produce proinflammatory cytokines and chemokines (22, 23). We explored the possibility that CDCs may serve as gram-positive pathogen-associated molecular patterns specific for TLR4. LLO and SLO, as well as PFO from Clostridium perfringens and ALO, were produced by in vitro translation in reticulocyte lysates and applied to BMDMs. All of the in vitro–translated CDCs were found to induce iNOS expression by BMDMs, whereas a control lysate programmed with an empty backbone plasmid was inactive (Fig. 6 A). Hence, all four CDCs are capable of activating macrophages. Next, we tested recombinant CDCs produced in and purified from E. coli for their dependence on TLR4 for macrophage activation. First, we measured TNF-α produced from BMDMs after treatment with recombinant CDCs. All four CDCs induced TNF-α secretion into the medium to similar extents (Fig. 6 B). TNF-α production induced by all of the CDCs was refractory to polymyxin B treatment (Fig. 6 B), showing that LPS contamination is not responsible for macrophage activation by recombinant CDCs. Treatment of BMDMs with increasing amounts of ALO, PFO, LLO, and SLO also resulted in induction of TNF-α and IL-6 mRNAs (Fig. 6 C). In most cases, the gene induction response reached its maximum at 100 ng/ml of CDCs (or ∼1.7 nM for an ∼60-kD protein). In contrast, TLR4 mutant BMDMs did not respond with cytokine gene expression to the same concentration of any of the CDCs (Fig. 6 B). These results strongly suggest that the ability to activate TLR4 is a general property shared by CDCs from gram-positive bacteria.
Discussion
TLRs play pivotal roles in recognizing and resisting microbial infection (24, 25). Among the immediate outcomes of the TLR-dependent immune response is the production of cytokines by dedicated inflammatory cells such as macrophages. The production and release of cytokines and chemokines is responsible for the inflammatory response that accompanies bacterial infection and, in the case of TLR4 activation by LPS, can lead to septic shock (26). Several lines of circumstantial evidence have suggested that B. anthracis interacts with host immune cells through TLRs: B. anthracis infection elicits a cytokine response in both mice and cultured macrophages (27, 28) and causes macrophage apoptosis in a manner dependent on TLR4 signaling (12). Nonetheless, the identity of the B. anthracis component that is responsible for inducing cytokine and apoptotic responses in macrophages was not hitherto known. It was reported previously that macrophages treated with very low doses of LT produce TNF-α and IL-1β (29), suggesting a role for LT in cytokine response. However, unlike TLR agonists, LT induces IL-1β production by causing the release of mature IL-1β derived from a presynthesized pool of IL-1β precursor rather than activating IL-1β gene expression (30, 31). This effect is reminiscent of the Shigella IpaB and Salmonella SipB invasins, which cause IL-1β release through activation of caspase-1 (32, 33). It is noteworthy that the pathological features associated with LT-induced lethality in mice are also quite different from those seen in B. anthracis–infected mice. LT-injected mice manifest hypoxia-associated liver failure and pleural edema without mounting the massive cytokine response that usually accompanies either gram-negative or gram-positive septic shock (34). Hence, LT alone does not account for the inflammatory cytokine response that accompanies B. anthracis infections.
In this paper, we have identified the B. anthracis cytolysin ALO as a potent TLR4 agonist that is responsible for inducing cytokine gene expression and apoptotic responses in macrophages. TLR4-specific activation of immune cells appears to be a general property shared among gram-positive CDCs, including ALO, LLO, SLO, PFO, and pneumolysin (this paper and reference 17). It is well established that CDCs are major virulence factors in gram-positive infections (35–37). The mechanisms by which CDCs contribute to pathogenesis in gram-positive infections have been attributed thus far to their cytolytic activity and their role in regulating intracellular compartmentalization of pathogenic bacteria (38). In addition, SLO has been shown to mediate vectorial transport of streptococcal proteins into the cytoplasm of host cells (39, 40). Our results suggest that the ability of CDCs to induce inflammatory and apoptotic responses in host immune cells via TLR4 should be taken into account in understanding their virulence mechanism and can be a major contributor to gram-positive–induced septic shock. Notably, cultured macrophages exhibit maximal cytokine gene induction at considerably lower molar concentrations of CDCs (∼0.8 nM) than LPS (∼10 nM; based on an average molecular mass of 10 kD).
We have defined a minimal set of three anthrax proteins (ALO, LF, and PA) that can trigger macrophage apoptosis. Identifying the critical interplay between ALO and LT in modulating innate immune responses and inducing macrophage apoptosis provides new insights to the development of effective strategies for fighting inhalation anthrax, which may improve the current therapeutic scheme based on the use of antibiotics (41). Notably, LLO is a major target antigen of antilisterial immunity (42, 43) and protective immunity to L. monocytogenes can be induced by either an adoptive transfer of LLO-reactive cytotoxic T lymphocytes (44) or immunization with LLO-derived antigens (45, 46). Therefore, the use of an ALO-directed vaccination in the prophylaxis of inhalation anthrax should be considered. Overall, we suggest that ALO represents an important target for both the development of vaccines and the design of antitoxin therapies effective for preventing and treating anthrax diseases.
Acknowledgments
We thank J. Collier and R. Tweten for plasmids, D. Portnoy for plasmids and purified LLO, E. Raz for IFNR1−/− mice, and E. Mosser and D. Guiney for helpful discussions and intellectual contributions.
J.M. Park is the Bristol-Myers Squibb Foundation Research Fellow at the Irvington Institute. S. Maeda is supported by a postdoctoral fellowship from the Japan Society for the Promotion of Science. This work was supported by a National Institutes of Health (NIH) grant no. AI61712 (to M. Karin) and by NIH Research Centers of Excellence (RCE) in Biodefense and Emerging Infectious Diseases grant no. U54 AI57168 (to R.F. Rest). M. Karin is the Frank and Else Schilling American Cancer Society Research Professor.
The authors have no conflicting financial interests.
Abbreviations used in this paper: ALO, anthrolysin O; BHI, brain heart infusion; BMDM, BM-derived macrophage; CDC, cholesterol-dependent cytolysin; CM, conditioned media; EF, edema factor; LF, lethal factor; LT, lethal toxin; MAPK, mitogen-activated protein kinase; MKK, MAPK kinase; PA, protective antigen; PFO, perfringolysin O; SLO, streptolysin O; TLR, Toll-like receptor.
References
- 1.Dixon, T.C., M. Meselson, J. Guillemin, and P.C. Hanna. 1999. Anthrax. N. Engl. J. Med. 341:815–826. [DOI] [PubMed] [Google Scholar]
- 2.Collier, R.J., and J.A. Young. 2003. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 19:45–70. [DOI] [PubMed] [Google Scholar]
- 3.Moayeri, M., and S.H. Leppla. 2004. The roles of anthrax toxin in pathogenesis. Curr. Opin. Microbiol. 7:19–24. [DOI] [PubMed] [Google Scholar]
- 4.Bradley, K.A., J. Mogridge, M. Mourez, R.J. Collier, and J.A. Young. 2001. Identification of the cellular receptor for anthrax toxin. Nature. 414:225–229. [DOI] [PubMed] [Google Scholar]
- 5.Scobie, H.M., G.J. Rainey, K.A. Bradley, and J.A. Young. 2003. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA. 100:5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leppla, S.H. 1982. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA. 79:3162–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klimpel, K.R., N. Arora, and S.H. Leppla. 1994. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol. Microbiol. 13:1093–1100. [DOI] [PubMed] [Google Scholar]
- 8.Duesbery, N.S., C.P. Webb, S.H. Leppla, V.M. Gordon, K.R. Klimpel, T.D. Copeland, N.G. Ahn, M.K. Oskarsson, K. Fukasawa, K.D. Paull, and G.F. Vande Woude. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 280:734–737. [DOI] [PubMed] [Google Scholar]
- 9.Vitale, G., R. Pellizzari, C. Recchi, G. Napolitani, M. Mock, and C. Montecucco. 1998. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 248:706–711. [DOI] [PubMed] [Google Scholar]
- 10.Xia, Y., Z. Wu, B. Su, B. Murray, and M. Karin. 1998. JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev. 12:3369–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park, J.M., F.R. Greten, Z.W. Li, and M. Karin. 2002. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 297:2048–2051. [DOI] [PubMed] [Google Scholar]
- 12.Hsu, L.C., J.M. Park, K. Zhang, J.L. Luo, S. Maeda, R.J. Kaufman, L. Eckmann, D.G. Guiney, and M. Karin. 2004. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 428:341–345. [DOI] [PubMed] [Google Scholar]
- 13.Shannon, J.G., C.L. Ross, T.M. Koehler, and R.F. Rest. 2003. Characterization of anthrolysin O, the Bacillus anthracis cholesterol-dependent cytolysin. Infect. Immun. 71:3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham, K., D.B. Lacy, J. Mogridge, and R.J. Collier. 1998. Characterization of membrane translocation by anthrax protective antigen. Biochemistry. 37:15737–15746. [DOI] [PubMed] [Google Scholar]
- 15.Shepard, L.A., A.P. Heuck, B.D. Hamman, J. Rossjohn, M.W. Parker, K.R. Ryan, A.E. Johnson, and R.K. Tweten. 1998. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an alpha-helical to beta-sheet transition identified by fluorescence spectroscopy. Biochemistry. 37:14563–14574. [DOI] [PubMed] [Google Scholar]
- 16.Read, T.D., S.N. Peterson, N. Tourasse, L.W. Baillie, I.T. Paulsen, K.E. Nelson, H. Tettelin, D.E. Fouts, J.A. Eisen, S.R. Gill, et al. 2003. The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature. 423:81–86. [DOI] [PubMed] [Google Scholar]
- 17.Malley, R., P. Henneke, S.C. Morse, M.J. Cieslewicz, M. Lipsitch, C.M. Thompson, E. Kurt-Jones, J.C. Paton, M.R. Wessels, and D.T. Golenbock. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. USA. 100:1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulevitch, R.J., and P.S. Tobias. 1995. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu. Rev. Immunol. 13:437–457. [DOI] [PubMed] [Google Scholar]
- 19.Seong, S.Y., and P. Matzinger. 2004. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 4:469–478. [DOI] [PubMed] [Google Scholar]
- 20.Tweten, R.K., M.W. Parker, and A.E. Johnson. 2001. The cholesterol-dependent cytolysins. Curr. Top. Microbiol. Immunol. 257:15–33. [DOI] [PubMed] [Google Scholar]
- 21.Dempsey, C.E. 1990. The actions of melittin on membranes. Biochim. Biophys. Acta. 1031:143–161. [DOI] [PubMed] [Google Scholar]
- 22.Tsukada, H., I. Kawamura, T. Fujimura, K. Igarashi, M. Arakawa, and M. Mitsuyama. 1992. Induction of macrophage interleukin-1 production by Listeria monocytogenes hemolysin. Cell. Immunol. 140:21–30. [DOI] [PubMed] [Google Scholar]
- 23.Stassen, M., C. Muller, C. Richter, C. Neudorfl, L. Hultner, S. Bhakdi, I. Walev, and E. Schmitt. 2003. The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor α by p38 mitogen-activated protein kinase- and protein kinase C-dependent pathways. Infect. Immun. 71:6171–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopp, E., and R. Medzhitov. 2003. Recognition of microbial infection by Toll-like receptors. Curr. Opin. Immunol. 15:396–401. [DOI] [PubMed] [Google Scholar]
- 25.Beutler, B., and E.T. Rietschel. 2003. Innate immune sensing and its roots: the story of endotoxin. Nat. Rev. Immunol. 3:169–176. [DOI] [PubMed] [Google Scholar]
- 26.Cohen, J. 2002. The immunopathogenesis of sepsis. Nature. 420:885–891. [DOI] [PubMed] [Google Scholar]
- 27.Popov, S.G., T.G. Popova, E. Grene, F. Klotz, J. Cardwell, C. Bradburne, Y. Jama, M. Maland, J. Wells, A. Nalca, et al. 2004. Systemic cytokine response in murine anthrax. Cell. Microbiol. 6:225–233. [DOI] [PubMed] [Google Scholar]
- 28.Pickering, A.K., and T.J. Merkel. 2004. Macrophages release tumor necrosis factor alpha and interleukin-12 in response to intracellular Bacillus anthracis spores. Infect. Immun. 72:3069–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanna, P.C., D. Acosta, and R.J. Collier. 1993. On the role of macrophages in anthrax. Proc. Natl. Acad. Sci. USA. 90:10198–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erwin, J.L., L.M. DaSilva, S. Bavari, S.F. Little, A.M. Friedlander, and T.C. Chanh. 2001. Macrophage-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect. Immun. 69:1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cordoba-Rodriguez, R., H. Fang, C.S. Lankford, and D.M. Frucht. 2004. Anthrax lethal toxin rapidly activates caspase-1/ICE and induces extracellular release of interleukin (IL)-1beta and IL-18. J. Biol. Chem. 279:20563–20566. [DOI] [PubMed] [Google Scholar]
- 32.Chen, Y., M.R. Smith, K. Thirumalai, and A. Zychlinsky. 1996. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 15:3853–3860. [PMC free article] [PubMed] [Google Scholar]
- 33.Hersh, D., D.M. Monack, M.R. Smith, N. Ghori, S. Falkow, and A. Zychlinsky. 1999. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA. 96:2396–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moayeri, M., D. Haines, H.A. Young, and S.H. Leppla. 2003. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 112:670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Portnoy, D.A., T. Chakraborty, W. Goebel, and P. Cossart. 1992. Molecular determinants of Listeria monocytogenes pathogenesis. Infect. Immun. 60:1263–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paton, J.C. 1996. The contribution of pneumolysin to the pathogenicity of Streptococcus pneumoniae. Trends Microbiol. 4:103–106. [DOI] [PubMed] [Google Scholar]
- 37.Rood, J.I. 1998. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 52:333–360. [DOI] [PubMed] [Google Scholar]
- 38.Decatur, A.L., and D.A. Portnoy. 2000. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science. 290:992–995. [DOI] [PubMed] [Google Scholar]
- 39.Madden, J.C., N. Ruiz, and M. Caparon. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in Gram-positive bacteria. Cell. 104:143–152. [DOI] [PubMed] [Google Scholar]
- 40.Meehl, M.A., and M.G. Caparon. 2004. Specificity of streptolysin O in cytolysin-mediated translocation. Mol. Microbiol. 52:1665–1676. [DOI] [PubMed] [Google Scholar]
- 41.Gilligan, P.H. 2002. Therapeutic challenges posed by bacterial bioterrorism threats. Curr. Opin. Microbiol. 5:489–495. [DOI] [PubMed] [Google Scholar]
- 42.Pamer, E.G., J.T. Harty, and M.J. Bevan. 1991. Precise prediction of a dominant class I MHC-restricted epitope of Listeria monocytogenes. Nature. 353:852–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouwer, H.G., C.S. Nelson, B.L. Gibbins, D.A. Portnoy, and D.J. Hinrichs. 1992. Listeriolysin O is a target of the immune response to Listeria monocytogenes. J. Exp. Med. 175:1467–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harty, J.T., and M.J. Bevan. 1992. CD8+ T cells specific for a single nonamer epitope of Listeria monocytogenes are protective in vivo. J. Exp. Med. 175:1531–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sirard, J.C., C. Fayolle, C. de Chastellier, M. Mock, C. Leclerc, and P. Berche. 1997. Intracytoplasmic delivery of listeriolysin O by a vaccinal strain of Bacillus anthracis induces CD8-mediated protection against Listeria monocytogenes. J. Immunol. 159:4435–4443. [PubMed] [Google Scholar]
- 46.Cornell, K.A., H.G. Bouwer, D.J. Hinrichs, and R.A. Barry. 1999. Genetic immunization of mice against Listeria monocytogenes using plasmid DNA encoding listeriolysin O. J. Immunol. 163:322–329. [PubMed] [Google Scholar]