

Abstract
CD1c-mediated T cells are activated by a mycobacterial phospholipid antigen whose carbohydrate structure precisely corresponds to mammalian mannosyl β-1-phosphodolichol (MPD), but contains an unusual lipid moiety. Here, we show that this T cell antigen is a member of a family of branched, alkane lipids that vary in length (C30-34) and are produced by medically important mycobacteria such as M. tuberculosis and M. bovis Bacille-Calmette-Guerin. The alkane moiety distinguished these mycobacterial lipid antigens from mammalian MPDs and was necessary for activation of CD1c-restricted T cells, but could not be accounted for by any known lipid biosynthetic pathway. Metabolic labeling and mass spectrometric analyses suggested a mechanism for elongating lipids using alternating C2 and C3 units, rather than C5 isopentenyl pyrophosphate. Inspection of the M. tuberculosis genome identified one candidate gene, pks12, which was predicted to encode the largest protein in M. tuberculosis, consisting of 12 catalytic domains that correspond to key steps in the proposed pathway. Genetic deletion and complementation showed that Pks12 was necessary for antigen production, but did not affect synthesis of true isoprenols. These studies establish the genetic and enzymatic basis for a previously unknown type of polyketide, designated mycoketide, which contains a lipidic pathogen-associated molecular pattern.
Keywords: tuberculosis, CD1 antigens, polyketide synthase, polyisoprenyl phosphate monosaccharides, lipids
Introduction
Mycobacterium tuberculosis remains one of the world's most prevalent and harmful pathogens, having infected 1.7 billion humans, one third of the world's population. M. tuberculosis causes tuberculosis based on its ability to infect and persist in lysosomes of host phagocytes and persist for years or decades in human hosts.
The interface with the host is an unusually thick, lipid-laden cell wall composed of the plasma membrane, peptidoglycan, arabinogalactan complexed to an outer membrane of long chain fatty acids known as mycolic acids, and a layer of surface exposed glycolipids. Synthesis of the outer layers of this structure requires the export of carbohydrate and lipids through plasma membrane using membrane channels such as MmpLs (1) and lipid carbohydrate transporters such as phosphopolyprenols, which transport sugars by flipping from the inner to the outer leaflet of membranes (2). Compared with eukaryotic cells and other bacteria, mycobacteria have an unusually diverse repertoire of lipids, with ∼6% of the M. tuberculosis genome encoding lipid-modifying enzymes, including those that lead synthesis of phthiocerols, mycolic acids, sulfolipids, and other lipids that are not found in eubacteria or mammalian cells. Because many of these mycobacteria-specific lipids play specialized, nonredundant roles in cell wall function, small molecule inhibitors of mycobacterial lipid synthesis, such as isoniazid, are among the most effective and least toxic antimicrobial drugs in clinical use.
The mycobacterial cell wall functions as an immunoprotective barrier against host mechanisms of recognition and killing, but also sheds lipids that activate immune responses via Toll-like receptors and antigen presentation by group 1 CD1 proteins: CD1a, CD1b, and CD1c. CD1 proteins are expressed in complex with β2-microglobulin on B cells, Langerhans cells, and myeloid DCs, including those found at the sites of mycobacterial infection (3, 4). Human CD1a, CD1b, and CD1c proteins bind several types of mycobacterial lipids, including mycolates (5–7), lipoarabinomannan (8), trehalose sulfolipids (9), dideoxymycobactin (10), and mannosyl-β-1-phosphoisoprenoids (MPIs; reference 11). The resulting CD1–lipid complexes traffic to the cell surface, allowing direct contact of intracellularly derived mycobacterial lipids with T cell antigen receptors of lipid-reactive T cells. CD1-restricted T cells express antimicrobial proteins such as γ-interferon and granulysin, and they have the ability to lyse mycobacteria-infected target cells in vitro (12, 13). In addition, human CD1-restricted T cells recognizing mannosyl-β1-phospholipids (11), mycolyl glycolipids (14), and sulfotrehalose (9) can be detected in the bloodstream of human patients acutely infected with M. tuberculosis, but not healthy control subjects, indicating that T cell recognition of mycobacterial cell wall lipids occurs during the natural history of tuberculosis infections.
To date, the only known microbial lipid presented by CD1c is an MPI antigen with an unusual structure that was isolated from Mycobacterium avium (11). This lipid is of interest in understanding the molecular basis of T cell discrimination between self and foreign lipid structures because it contains a mannosyl-β-1-phosphate moiety that is identical to those found in mannosyl-β-1-phosphodolichols (MPDs) from mammalian cells. However, the lipid portion of the MPI antigen is composed of an unusual 4, 8, 12, 16, 20-pentamethylpentacosyl unit, which has a fully saturated backbone and short length (C30) compared with polyunsaturated C95 polyisoprenols found in mammalian cells. These observations suggested that the unusual lipid moiety might be the key chemical element that allows CD1c-restricted T cells to distinguish foreign MPIs from self-MPDs, and thereby detect cellular infection (15). However, it was unknown whether this or similar phosphoglycolipids are produced by mycobacteria or other pathogens of medical significance. Furthermore, the branched, fully saturated structure of the MPI lipid moiety did not conform to the expected products of any known fatty acyl, polyprenol, or polyketide biosynthetic pathway, which led us to study the structural diversity of naturally occurring antigens, as well as the genes and biosynthetic mechanisms involved in their production.
We found that antigenic MPIs were made by mycobacterial strains, which are capable of growth in human cells, but were not detected in rapidly growing saprophytic mycobacteria or nonmycobacterial pathogens. Mass spectrometric analysis of MPIs revealed that they are a chemically diverse family of structurally related mannosyl-β-1-phospholipids with C30–34 alkane chains. Although a repeating motif of C5 unit initially suggested that the lipid might be made from isopentenyl pyrophosphate (IPP) using a modified polyisoprenol mechanism, variations in the expected length and methyl branching patterns could not be explained by this model. Structural variations among natural lipids and labeling studies suggested a hypothetical mechanism for production of the isoprene-like alkyl backbone via malonyl (C2) and methylmalonyl (C3) units and polyketide synthases (PKSs). This led us to target pks12 for knockout in M. tuberculosis and Mycobacterium bovis Bacille-Calmette-Guerin (BCG), which was found to be necessary for MPI production and the CD1c-mediated T cell response. These studies establish the genetic and enzymatic basis for a previously unknown lipid biosynthetic pathway leading to the production of an antigenic mycobacterial polyketide, designated mycoketide.
Materials and Methods
Bacteria.
M. tuberculosis H37Rv and H37Ra were provided by P. Brennan (Colorado State University, Fort Collins, CO). M. avium serovar 4 was provided by H. Remold (Harvard Medical School, Boston, MA). Escherichia coli was provided by H. Band (Northwestern University, Evanston, IL). M. tuberculosis CDC1551 and Mycobacterium smegmatis 607 were obtained from Center for Disease Control and Prevention. M. bovis BCG Pasteur, Leishmania niger, Leishmania tropicana, Mycobacterium phlei, Mycobacterium fallax, Rhodocccus equi, Nocardia farcinica, Saccharomyces pombe, Candida albicans, and Aspergillus niger were purchased from American Type Culture Collection. M. tuberculosis H37Rv and CDC1551 strains and M. bovis BCG were maintained on Middlebrook 7H10 plates supplemented with oleic acid–albumin–dextrose complex. For liquid culture, a single colony was inoculated in Middlebrook 7H9 media with 0.2% glycerol supplemented with 10% albumin–dextrose and grown to mid-log phase. For labeling, bacteria were grown to an optical density of 0.4 at 600 nm (OD600), changed to Proskauer-Beck medium for 1 d of growth, treated with 1[13C]-sodium propionate or propionate (2 mM), and further cultured for 3 d (OD600 ∼ 0.9) before harvesting.
Cellular Assays.
Monocyte-derived DCs were prepared from human PBMCs by centrifugation over Ficoll-Hypaque, adherence to plastic, and treatment with 300 U/ml granulocyte/monocyte-colony stimulating factor and 400 U/ml IL-4 for 72 h, followed by γ-irradiation (5,000 rad) as described previously (16). CD1c-restricted, MPI-reactive T cells (CD8-1; references 11, 17) were tested for IL-2 release using the HT-2 bioassay as described previously (7). In brief, 5 × 104 CD8-1 T cells and 5 × 104 γ-irradiated monocyte-derived DCs were incubated in 200 μL of T cell media for 24 h after which 50 μl of culture supernatant was transferred to wells containing 100 μl of the media and 104 IL-2–dependent HT-2 cells, which were cultured for 24 h before adding 1 μCi [3H]thymidine for an additional 6 h of culture, followed by harvesting and counting β emissions. Assays were done in triplicate and reported as the mean ± standard deviation. For cytolysis, 106 C1R lymphoblastoid cells transfected with the vector pSRα-NEO containing the cDNAs encoding human CD1a, CD1b, CD1c, and CD1d were labeled with 51Cr in media (1 mCi/ml) with or without M. avium MPI overnight, washed, and added at 2,000 cells per well to a 96-well plate. CD8-1 T cells were added at an effector:target ratio of 20:1 for 4 h before measuring the 51Cr release in supernatants by scintillation counting and expressed as percent lysis compared with that found in wells treated with detergent (2% NP-40).
Deletion and Complementation of pks12 Gene in M. tuberculosis and BCG.
The pks12 gene was deleted by specialized transduction as described previously (18). For genetic manipulations of mycobacteria, 75 μg/ml hygromycin or 20 μg/ml kanamycin was used for selection of transformants or transductants, whereas 150 μg/ml hygromycin or 40 μg/ml kanamycin was used for selecting E. coli HB101 transformants. To generate an allelic exchange substrate for gene replacement, chromosomal sequences flanking M. tuberculosis H37Rv pks12 (Rv2048c) were PCR amplified from M. tuberculosis H37Rv genomic DNA. Primers PKS12-1, 5′-ATCACTAGTGTTGCGGTCGGTCTCATAG-3′, and PKS12-2, 5′-ATCAGATCTGCGCTCCAGCAACGCACG-3′, which contained a SpeI and a BglII site, respectively, were used to amplify a 1,016-bp fragment containing upstream flanking sequences. Primers PKS12-3, 5′-CAATCTAGACAGGACCCGGCGTTGGCG-3′, and PKS12-4, 5′-TATCTTAAGTCTGGGCCGGCCAATCTG-3′, which contained XbaI and AflII sites, respectively, were used to amplify a 1,019-bp fragment containing downstream flanking sequences. After cloning in pCR2.1-TOPO (Invitrogen and Life Technologies) and sequencing, the cloned fragments were excised using the introduced restriction sites and cloned into the allelic exchange plasmid vector pJSC347. The resultant plasmid pΔpks12 was cloned into a conditionally replicating shuttle plasmid phAE159 (increased cloning capacity, but similar to phAE87; unpublished data) as described before (18), to generate the specialized transduction phage phAEΔpks12. M. tuberculosis H37Rv, M. tuberculosis CDC1551, and M. bovis BCG were transduced with phAEΔpks12 as described previously (18) and hygromycin-resistant (HygR) colonies were screened by Southern blot, using flanking sequences as probes to detect replacement of the pks12 ORF by the hyg cassette. One positive clone of each strain was chosen for further work, and these were designated as mc24701 for H37Rv Δpks12, mc24702 for CDC1551 Δpks12, and mc24703 for BCG Δpks12.
For complementation, a genomic DNA fragment (bp 2277324–2322018) containing M. tuberculosis pks12 was excised from cosmid pYUB2212 (HygR) by PacI digestion, and ligated into PacI-digested, single copy integrative cosmid vector pJK93 (kanamycin resistant; unpublished data), packaged into empty λ phage particles and transduced into E. coli HB101, to generate the kanamycin-resistant (KanR) complementing cosmid pYUB2410. Cosmid DNA obtained from KanR HB101 colonies was electroporated (19) into H37Rv Δpks12, and HygR KanR transformants were analyzed by Southern blot to confirm chromosomal integration of pYUB2410.
Lipids and Lipid Antigens.
Bacteria harvested at mid-log phase or cells were extracted at 5 mg/ml in chloroform:methanol (C/M) (2:1, vol/vol) for 1 h at room temperature followed by centrifugation (2,000 g) and removal of supernatants. The pellet was reextracted in C/M (1:1, vol/vol) and C/M (1:2, vol/vol), and all three supernatants were pooled to give “total lipids.” For purification of MPI compounds, total lipids (∼50 mg) were applied to a silica gel column (23 ml of bed volume; Alltech), equilibrated with chloroform, and washed with 40 ml of chloroform, 40 ml of acetone, and 50 ml of methanol. MPI antigens were recovered solely in the methanol eluates, which were further fractionated by TLC using a silica gel G plate (Analtech Inc.) with a solvent system of chloroform:methanol:water:ammonium hydroxide (60:35:7.2:0.8, vol/vol/vol/vol).
The lipid fraction with T cell activation activity was extracted with C/M (1:1, vol/vol) from the gel and subjected to further separation by HPLC with a Monochrome Diol column (46 mm × 250 mm, 3 μm; Varian Inc.) and coupled on-line to a LCQ Advantage ion-trap mass spectrometer (ThermoElectron) equipped with an electrospray ionization (ESI) source. Mobile phase A was hexane:isopropanol (60:40, vol/vol) containing 0.1% (vol/vol) formic acid, 0.05% (vol/vol) ammonium hydroxide, and 0.05% (vol/vol) triethylamine. Mobile phase B was methanol containing 0.1% (vol/vol) formic acid, 0.05% (vol/vol) ammonium hydroxide, and 0.05% (vol/vol) triethylamine. Using a flow rate of 0.7 ml/min and the column temperature of 25°C, separation was obtained by using a binary gradient starting at 5% mobile phase B, linearly increasing to 15% B in 6 min and held at 15% B for 10 min, linearly increasing to 95% B in 8 min, held at 95% mobile phase B for 6 min and finally back to 5% B in 2 min. The electrospray source was modified by using narrow stainless steel tubing (127 μm OD, 51 μm ID; Small Parts Inc.) and was operated at 1.9 kV. The heated capillary inlet was operated at 220°C. The HPLC effluent was split using fused silica tubing and a micro-Tee union (Upchurch Scientific) to allow fraction collection. This method allowed removal of phosphatidylinositol and yielded MPI compounds judged as pure based on total ion current profiles and ESI-mass spectrometry (MS) using nanospray. To separate MPI homologues with differing alkyl backbones, reverse phase HPLC with a Vydac C8 reverse phase column (46 mm × 250 mm, 3 μm; Grace Vydac) was used. Compounds were eluted with isopropanol:methanol:acetonitrile:hexane:water (37:30:18:3:12, vol/vol/vol/vol/vol) containing 6 mM ammonium acetate.
The MPI compounds collected during the aforementioned LC-MS separation were dried under nitrogen gas, dissolved in chloroform:isopropanol:water (5:75:20, vol/vol/vol) containing 20 mM sodium acetate, and analyzed by MS/MS using both low and high energy collisionally induced dissociation (CID) to produce product ions. Low-energy CID-MS/MS was performed on the ion-trap mass spectrometer equipped with a nanoelectrospray ion source (JA Hill, Instrument Services, Inc.). The spray voltage and capillary temperature were set to 0.8 kV and 215°C, respectively. Collision energy was 30–40% of maximum and the trapping of product ions were performed with a q value of 0.25.
High-energy CID was performed using a double focusing magnetic sector mass spectrometer (MStation; JEOL) which was operated at 5 kV in the linked scan mode using helium leaked into the collision cell (first field free region) at a sufficient pressure to effect a 50% decrease in the parent beam.
Semi-synthetic mannosyl-1-β-phosphodolichol with 95 carbon alkyl chain (C95 MPD) was synthesized from human hepatic dolichol as described previously (11).
Online Supplemental Material.
Two-dimensional TLC analyses of 14C-labeled lipids from M. tuberculosis H37Rv and M. tuberculosis CDC1551 are shown in Fig. S1. The lipids were developed with petroleum ether:ethyl acetate (98:2, vol/vol) in the first dimension and with petroleum ether:acetone (98:2, vol/vol) in the second dimension to clearly separate phthiocerol dimycocerosate (DIM) species. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20041429/DC1.
Results
Infectious Mycobacteria Produce a Family of Structurally Related MPIs.
A prior paper described an antigenic MPI from a facultative human pathogen, M. avium (11). To determine whether such unusual lipids are made by mycobacteria or other pathogens that infect humans, we used a CD1c-restricted, MPI-reactive T cell line named CD8-1 as a reporter line to screen total lipid preparations from several species of mycobacteria, nonmycobacterial Actinomycetales, Gram-positive bacteria, Gram-negative bacteria, protozoal parasites, fungi, and human cells (Table I and reference 20) (20). Initial studies using B lymphoblastoid target cells transfected with various human CD1 isoforms confirmed that the MPI-mediated recognition of target cells by CD8-1 was absolutely dependent on expression of CD1c by antigen-presenting cells and could not be mediated by other CD1 proteins (Fig. 1 A). T cells can detect ∼0.2 ng of purified MPI, which under the conditions of this assay, represented ∼1/100 of the level produced by BCG (Fig. 1 B). Among the many types of bacterial and cellular organisms tested, T cell activation was only seen in response to mycobacteria (Table I). Among the mycobacterial strains tested, rapid growing saprophytes such as M. phlei, M. fallax, and M. smegmatis did not produce antigenic compounds at levels detected by this assay. In contrast, all tested species or strains that are capable of infecting human cells stimulated the response at high titers, including M. avium, M. bovis BCG, and several strains of M. tuberculosis. These results indicated that antigenic MPIs are produced by mycobacteria including a clinically important vaccine strain (BCG Pasteur) and a natural isolate of M. tuberculosis from human patients (M. tuberculosis CDC1551).
Table I.
MPI Compounds from Bacteria and Eukaryotic Cells
| T cell stimulationb |
Fragment ions (MS/MS)e
|
||||||
|---|---|---|---|---|---|---|---|
| Speciesa | [M-H]−
c(normalized intensity)d |
Lipid length |
[M-H2O-H]− | [M-C4H8O4-H]− | Alkyl phosphate ion |
Hexosyl phosphate ion |
|
| M. tuberculosis | + | 679 (10.7) | C30 | 661 | 559 | 517 | 241 |
| H37Rv | 693 (3.9) | C31 | 675 | 573 | 531 | ||
| 707 (100) | C32 | 689 | 587 | 545 | 241 | ||
| 721 (2.4) | C33 | 703 | 601 | 559 | |||
| 735 (5.9) | C34 | 717 | 615 | 573 | 241 | ||
| M. tuberculosis | + | 679 (100) | C30 | 661 | 559 | 517 | 241 |
| CDC1551 | 707 (5.7) | C32 | 689 | 587 | 545 | 241 | |
| M. bovis BCG | + | 679 (10.6) | C30 | ||||
| Pasteur | 693 (6.1) | C31 | |||||
| 707 (100) | C32 | 689 | 587 | 545 | 241 | ||
| 721 (5.6) | C33 | ||||||
| 735 (5.0) | C34 | ||||||
| M. avium | + | 679 (100) | C30 | 661 | 559 | 517 | |
| 693 (0.2) | C31 | ||||||
| 707 (0.8) | C32 | 689 | 587 | 545 | |||
| 721 (0.8) | C33 | ||||||
| M. tuberculosis | + | 707f | C32 | ||||
| H37Ra | |||||||
| Human mononuclear cells |
− | ||||||
Total lipid extracts from M. smegmatis, M. phlei, M. fallax, R. equi, N. farcinica, S. aureus, E. coli, S. pombe, C. albicans, A. niger, L. donovani, and L. tropicana did not stimulate T cells when tested as in Fig. 1 B.
The CDIc-mediated T cell activity in the crude lipid extracts were considered as (+) when greater than twofold proliferation of T cells was seen in response to total lipid fractions tested as in Fig. 1 B.
[M-H]− ions of MPI compound detected by LC-MS analyses.
Intensity of each MPI compound when the most abundant MS signal from each preparation is 100.
Detected fragment ions by low energy CDI-MS analysis for a listed parent ion (left).
See reference 11.
Figure 1.

CD1c-restricted T cell responses for mycobacterial phospholipids. (A) Human C1R B lymphoblastoid cells transfected with the indicated CD1 genes were pulsed with 10 μg/ml MPI, labeled with 51Cr, and lysed by the CD8-1 T cell line with an effecter target ratio of 20:1. (B) CD1c-mediated IL-2 production in response to DCs treated with total lipid extracts from mycobacteria was measured using the HT-2 bioassay. (C) CD1c-mediated IL-2 release was measured in response to C30 MPI compounds purified from M. avium and C95 MPDs made from human dolichol.
In the MPI made by M. avium, the mannosyl-β-1-phosphate group, which is predicted to protrude from the CD1c antigen binding groove and function as part of the epitope for responding T cells, is structurally identical to phosphoglycan in MPDs found in all eukaryotic cells. Therefore, we sought to determine whether MPI-reactive T cells would cross-react with MPDs or other phospholipids from human cells. CD8-1 was activated by total lipid extracts from mycobacteria but not human cells, suggesting that T cells might preferentially recognize mannosyl-β-1-phsopholipids with the unusual lipid moiety found in mycobacterial MPIs (Table I). Alternatively, the preferential response to mycobacterial lipid extracts might simply have resulted from lower absolute levels of mammalian MPDs in cellular lipid extracts. To clarify this issue, we synthesized MPD from human C95 dolichol and compared its potency to the smaller, fully saturated C30 MPI from M. avium. Human C95 MPD showed faint or absent CD1c-mediated T cell activation, in contrast with the potent activity of purified C30 MPI, and thus provided direct evidence that the unusual lipid moiety found in mycobacterial MPI was responsible for its recognition by CD8-1 T cells (Fig. 1 C). This prompted us to consider possible biosynthetic mechanisms for producing this saturated, branched alkane lipid that characterizes mycobacterial MPIs.
Structures of MPIs Are Not Consistent with Known Lipid Biosynthetic Pathways.
These lipid antigens were initially described as being polyisoprene-like because, like true polyisoprenols made from serial condensation of IPP, they contained a discernable repeating C5 structure with a methyl branch at every fourth carbon on the alkyl chain. This suggested a candidate mechanism whereby serial condensation of IPP units might be followed by enzymatically catalyzed saturation of the double bonds, as seen in biosynthesis of phytol (21), a branched, saturated lipid found in plants (Fig. 2 A, model 1). This simple model was attractive because it used known enzymatic mechanisms and precursors and would be viewed as an alternate form of polyisoprenol biosynthesis.
Figure 2.

MPI species from M. tuberculosis H37Rv and selective labeling of the alkyl backbone of C32 MPI. (A) Model 1 is based on sequential condensation of IPP moieties to form a polyisoprenol, which is subsequently acted upon an enzyme with saturase activity. In Model 2, the alkyl backbone is primed by a fatty acyl group and extended by alternating condensation of malonate and methylmalonate in repeated cycles to yield a carboxylic acid product. (B and C) An MPI antigen purified from M. tuberculosis H37Rv was subjected to ESI-MS in the negative mode. The major product, C32 MPI (m/z 707.6), differed in size from the major product from M. avium (m/z 707.6; Table I), and lower abundance ions at m/z 679.6, 693.7, 721.6, and 735.6 were also seen, providing evidence for structural diversity of these natural compounds. Low energy CID-MS/MS for compounds of m/z 679.6, 707.6, and 735.6 generated fragments ions corresponding to a hexosyl phosphate ion (m/z 241) as well as saturated alkyl phosphates corresponding to C30 (m/z 517), C32 (m/z 545), and C34 (m/z 573), respectively, demonstrating that the size variations are accounted for by differences in sizes of the alkyl moiety. (D) High energy CID-MS/MS (double-focusing magnetic sector mass spectrometer) for C32 MPI shows a series of ions at 14-u or 28-u intervals. The 28-u intervals (arrows) correspond to methyl branches at C4, C8, C12, C16, and C20. (E) M. tuberculosis H37Rv was grown in the presence of 13C-labeled propionate or nonlabeled propionate and subjected to lipid extraction and analysis using a quadruple ion trap mass spectrometer. Compared with unlabeled MPI, the mass shift of the main product from m/z 707.6 to 713.6 corresponded to the incorporation of six C3 units in each molecule. MS/MS spectra of m/z 713.6 shows that the alkyl phosphate fragment (m/z 551.5) was shifted by six mass units compared with the corresponding fragment from unlabeled C32 MPI (m/z 545.5; panel C), indicating that as many as six C3 units are incorporated into the alkyl chain.
To evaluate this model, we first isolated highly purified MPIs from M. tuberculosis and BCG and M. avium and used collisional MS to identify naturally occurring variations in chain length and branching patterns of the alkyl portion of antigenic compounds. This was accomplished by development of normal and reversed phase HPLC methods, which generated purified preparations of MPIs and in most cases achieved separation of individual molecular species of MPIs based on C2H4 differences in chain length (Table I).
The MPI produced by M. avium predominantly yielded an [M-H]− ion at m/z 679.6, corresponding to a mannosyl phosphate with a C30 lipid tail (Table I). The predominant MPI from M. tuberculosis H37Rv generated an [M-H]− ion at m/z 707.6, which corresponded to an MPI with a C32 alkyl chain (Table I and Fig. 2 B). Analysis of phospholipids from M. tuberculosis H37Rv, M. tuberculosis strain CDC1551, and M. bovis BCG showed multiple molecular species of homologous MPIs that varied from one another in increments of 14 u (m/z 679, m/z 693, m/z 707, m/z 721, and m/z 735; Table I), suggesting that natural MPIs with C31, C33, and C34 lipids also exist. This was confirmed by low energy MS/MS analyses of M. tuberculosis H37Rv-derived MPIs (Fig. 2 C), each of which yielded hexosyl phosphate product ions at m/z 241.1, but differed in the size of the alkyl phosphate moieties, which were between C30 and C34 in length. The same alkyl chain length distribution was seen for BCG MPIs, and MPIs from M. tuberculosis CDC1551 showed diversity and did not differ in C5 increments (Table I). Thus, naturally occurring MPIs represented an unexpectedly diverse family of structurally related lipids that differed from one another in alkyl chain length by single CH2 increments, providing strong evidence against model 1 in which condensation of IPP units would be produce lipid tails that varied in length by C5 increments (Fig. 2 A).
High energy collisional dissociation MS using charge remote fragmentation showed that the methyl branching patterns on the alkane backbone of MPI did not correspond to those predicted by model 1. Although both true isoprenols and the MPI structures incorporate repeating C5 units with a methyl branch at every fourth carbon, IPP is methylated at the third (γ) carbon and would be expected to yield γ-methylated lipid moieties. However, the high energy collisional fragmentation of the MPI from M. tuberculosis (Fig. 2 D) yielded fragment ions at m/z 327, 397, 467, 537, and 607, which correspond to cleavage products distal to each methyl branch, and fragment ions were not seen at m/z 313, 383, 453, 523, and 593 as expected for methyl branches at the δ-C and every fourth carbon thereafter (Fig. 2 D, arrows), similar to the pattern previously seen with M. avium MPI (11). Therefore, the observed methyl branches were out of register compared with those expected from condensation of C5 IPP units.
An Alternate Model Using PKSs.
This prompted us to consider model 2, in which the repeating C5 units were made by alternating addition of C2 and C3 units, which would be supplied from malonate and methylmalonate, respectively (Fig. 2 A). Although this mechanism had no direct precedent, elongation in C5 increments could be performed by a plausible mixed fatty acyl-polyketide mechanism in which a carboxylate primer of varying length is coupled to the repeating C5 motif, which actually represents repeating cycles of C2 and C3 unit condensation. The final carboxylate moiety would be reduced to an alcohol and phosphorylated to yield a substrate for glycosylation. This biosynthetic mechanism was considered plausible because mycobacteria were known to use PKSs to produce polymethylated alkane lipids of various sorts, including phthiocerol and mycocerosic acid (22). Importantly, this model could account for the unusual structures of natural MPIs because, unlike IPP, all precursors are saturated. Furthermore, the C2 and C3 condensation product would contain a δ-methyl branch, and the C1 increments in chain length could come from varying fatty acyl primers, so that this model could account for all of the observed features of the natural alkane lipids isolated from M. avium, BCG, and M. tuberculosis.
Although isoprenoid biosynthesis involves assembly of IPP units made from acetate or glyceraldehyde phosphate and pyruvate, a prediction of model 2 is that the alkyl backbone is made by assembly of up to five methylmalonate units. To test this model, we biosynthetically labeled M. tuberculosis lipids by growth in media formulated with 13C-propionate, which is a precursor of methylmalonate in bacteria. Labeled and unlabeled MPIs were analyzed using ESI-MS (Fig. 2 E). Negative mode mass spectra of C32 MPI from bacteria grown in control media showed a predominant ion [M-H]− at 707.6 with adjacent isotope peaks whose abundance corresponded to the expected ratios of naturally occurring 13C and 2H. In contrast, MPI from M. tuberculosis grown with 13C-labeled propionate showed two predominant ion clusters near m/z 707.6 and 713.6. Isotopically labeled ions were shifted by ∼6 u, consistent with incorporation of labeled carbon at the site of one C3 primer from propionate and five C3 elongation units from methylmalonate per molecule.
To confirm that the ion at m/z 713.6 was indeed MPI, we performed MS/MS analysis of this labeled molecule (Fig. 2 E) in comparison with unlabeled MPI from the same species (Fig. 2 B). As expected, decomposition of the ion at 713.6 detected fragments corresponding to mannose phosphate (m/z 241.1), alkyl phosphate (m/z 551.5), and a through-ring cleavage product, [M-C4H8O4-H]− (m/z 593.6). Compared with the corresponding fragments from unlabeled MPI, the 6-u mass shift of all products containing the alkyl unit provided direct evidence that six 13C labels were incorporated into the branched lipid. Conversely, the lack of mass shift in the mannosyl phosphate product (m/z 241.1) provided evidence that the pulse labeling method used here did not diffusely label distantly downstream products through multiple biochemical conversions. Thus, the unusual lipid unit was produced from propionate precursors and was incorporated as predicted by model 2.
PKS12 Is Necessary for Production of Antigenic MPIs.
The natural MPI structures suggested a polyketide rather than polyisoprenoid mechanism of biosynthesis, so we sought to identify the mycobacterial genes, which performed MPI synthesis. We considered whether the detailed biosynthetic predictions of model 2 might support a targeted, reverse genetics approach in which the responsible genes might be deduced based on the necessary enzymatic activities that would be required to carry out model 2. By searching the TubercuList database (http://genolist.pasteur.fr/TubercuList; reference 23), we found 23 genes that were annotated as likely having PKS activities in the M. tuberculosis genome, though their functions remain largely undetermined. Because the MPI backbone is completely saturated and lacks hydroxyl or keto groups, a complete set of fatty acid synthase (FAS)-like catalytic domains would be necessary to catalyze acyltransfer, ketosynthesis, ketoreduction, dehydration, and enoyl reduction. Furthermore, because the C2 and C3 units were incorporated strictly in an alternating manner (rather than at random), we reasoned that two-sets of FAS would likely be required, one for C2 units and one for C3 units. Among the 23 putative PKSs made by M. tuberculosis, pks12, the largest open reading frame in the genome, was a good candidate because it was predicted to encode an extremely large polypeptide with 12 enzymatic domains, including two complete sets of FAS-like catalytic domains. Moreover, a recent paper on substrate specificity of the acyltransferase (AT) domains of pks12 predicted that one of them is specific for malonyl-CoA and the other is for methylmalonyl-CoA (24), consistent with model 2 (Fig. 2 A). In addition, there was evidence for retention of pks12 genes in the M. tuberculosis CDC1551 strain and BCG, two species that were known to make antigenic MPIs (Table I).
To directly test this hypothesis, we used homologous recombination to disrupt pks12 genes in M. tuberculosis H37Rv, BCG, and M. tuberculosis CDC1551 and measured the ability of mutants to activate CD1c-restricted T cells. Allelic exchange of pks12 with a hygromycin resistance gene (hyg) was performed using specialized transduction (18) with a conditionally replicating phage carrying the homologous recombination substrate. Hygromycin-resistant colonies obtained after transduction were analyzed by Southern blot to detect replacement of pks12 by hyg (Fig. 3 A). The left and right flanking DNA, used as probes, hybridized with 6.083 kb and 8-kb StuI-digested bands in all wild-type M. tuberculosis strains, and 6.083-kb and 12.445-kb StuI bands in BCG, and with an 8.09-kb band in all mutant strains, consistent with what was expected from a bonafide gene replacement. Expected band patterns were also observed in Southern blots of KpnI-digested genomic DNA (unpublished data), confirming the results. Allelic exchange resulted in the replacement of bp 97–12,360 of pks12 by hyg, thus removing sequences corresponding to all the twelve domains of the PKS. The three pks12-deleted mutant strains of H37Rv, CDC1551, and BCG (mc24701, mc24702, and mc24703, respectively) are referred to as H37Rv Δpks12, CDC1551 Δpks12, and BCG Δpks12. As shown in Fig. 3 (B–D), deletion of pks12 genes in each of these three strains completely abolished the ability of all three strains to activate CD1c-restricted T cells. Conversely, introduction of pks12 into H37Rv Δpks12 restored the T cell response to levels seen in wild-type bacteria (Fig. 3 B). This restoration of MPI production was observed in two independent pks12-complemented H37Rv clones (unpublished data).
Figure 3.

Deletion and complementation of pks12. (A) Southern blot of StuI-digested genomic DNA from M. tuberculosis H37Rv, M. tuberculosis CDC1551, and M. bovis BCG, and the respective pks12 deletion mutant strains H37Rv Δpks12, CDC1551 Δpks12, and BCG Δpks12. The adjoining map shows the chromosomal regions of BCG, M. tuberculosis H37Rv, and H37Rv Δpks12. res, γδ resolvase site; hyg, hygromycin resistance gene. The regions used as probes are indicated (♦-♦). (B–D) Total lipid extracts from parental, pks12-deficient strains, and strains complemented with the pYUB2410 vector containing pks12 were tested for their ability to stimulate IL-2 production of the CD1c-restricted T cell line, CD8-1, using DCs as antigen-presenting cells.
In parallel, LC-MS methods were used to analyze MPIs and related lipids in phospholipid extracts of M. tuberculosis H37Rv and H37Rv Δpks12. Under conditions in which parental and mutant strains gave similar overall phospholipid profiles as determined by total ion currents in LC-MS detection, mass chromatograms targeting the molecular ions (m/z 707–708) of the MPI gave a strong signal in the parental M. tuberculosis strain at the expected retention time (10 min) but no signal in H37Rv Δpks12 (Fig. 4 A). Furthermore, the mass spectra at the retention time of 10 min from the parental and the mutant strains clearly show that MPIs are only found in the parental strain (Fig. 4 B). A mass spectral analysis for lipids from the pks12-complemented strain confirmed restoration of MPI production (unpublished data). Together, these data provide strong biochemical and immunological evidence that the PKSs encoded by pks12 is involved in MPI biosynthesis. In contrast with an earlier paper that suggested that an H37Rv pks12 mutant did not synthesize DIMs (25), TLC analysis of total lipids (labeled with 14C-acetate or propionate) extracted from H37Rv Δpks12 and CDC1551 Δpks12 revealed presence of DIMs (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041429/DC1).
Figure 4.
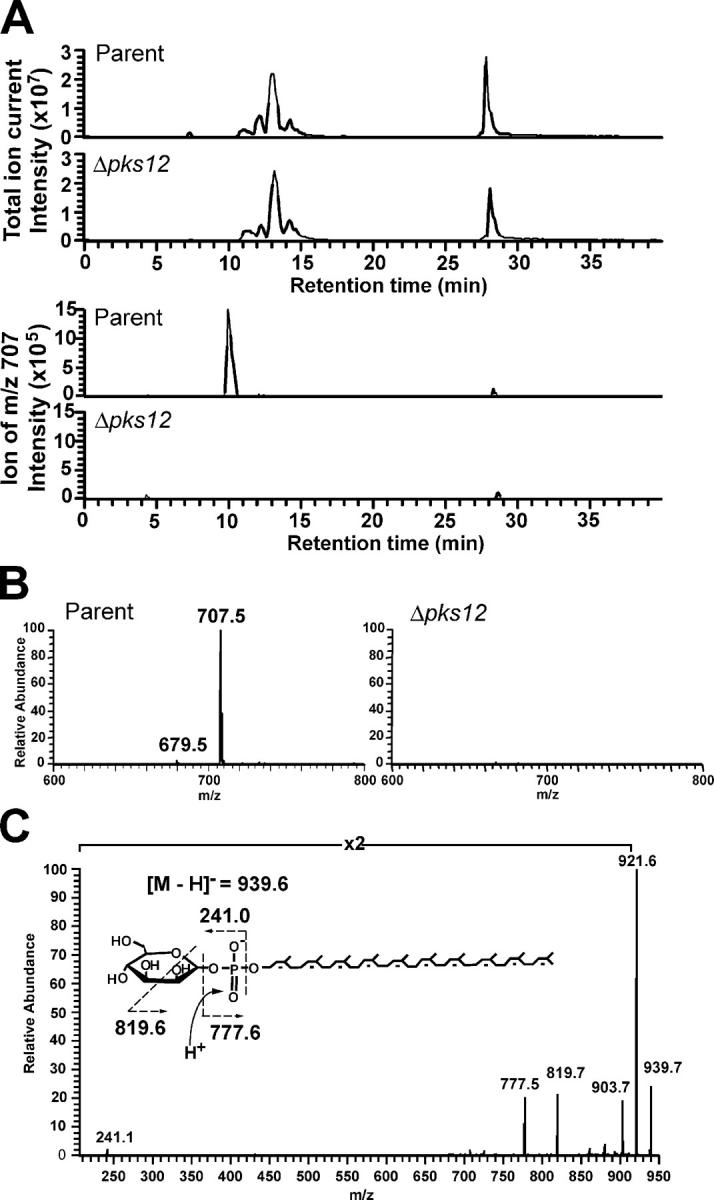
MPI and polyprenol biosynthesis pathways function independently. (A) Mass chromatograms of measured ions in the mass range corresponding to MPI (m/z 707.0–708.0; bottom) were compared with total ion chromatograms (top) in HPLC separation of phospholipids from M. tuberculosis H37Rv parental strain and the Δpks12 mutant. The total ion current trace shows that total phospholipid pattern was not substantially altered and confirms that similar amounts of lipids were analyzed for the comparison of m/z 707–708 ions. (B) Mass spectra at the retention time of 10 min in the chromatograms shown in A. The 100% relative abundance of the y axis was adjusted to the same intensity in each mass spectra from the parental and the mutant strains. (C) CDI-MS spectra of phospholipids from the Δpks12 mutant showed an ion of m/z 939.5, which corresponded to the expected mass and collision products of a true polyisoprenol phosphoglycolipid, mannosyl-β-1-phosphopolyisoprenol (C50 MPP). The ions of m/z 921.6 and m/z 903.7 corresponded to ones, which lost one or two water molecules from the parent ion and m/z 241.1 corresponded to a hexosyl phosphate ion.
Because Pks12 has the catalytic domains predicted to make the alkyl backbone of MPIs, we favored the possibility that Pks12 actually synthesizes this lipid, but there was some possibility that this Pks12 functioned in some indirect way to alter the synthesis of polyisoprenols. To test whether Pks12 affects polyprenol biosynthesis, we sought to measure a known product of polyisoprenol synthases, C50 mannosyl-β-1-phosphopolyisoprenol (C50 MPP) in the M. tuberculosis H37Rv Δpks12 mutant (26). ESI-MS analysis revealed a negative ion corresponding to the predicted [M-H]− of C50 MPP (m/z 939.5) in both the parental strain and mutant strains, and MS/MS analysis confirmed that the m/z values of its products matched the expected masses for hexose phosphates and a polyunsaturated phosphodecaprenol (Fig. 4 C). Thus, deletion of the pks12 gene did not disrupt biosynthesis of a polyisoprenol compound, providing clear evidence that polyketide and polyisoprenoid phospholipids are made by independent mechanisms. Furthermore, the failure of T cells to recognize phospholipids from this mutant (Fig. 3 B) indicated that mannosyl-β-1-phospholipids with true polyisoprenol lipid moieties do not substitute as potent antigens for the T cell response.
Discussion
MPIs were originally identified as scarce, but immunologically potent, phospholipid components of the mycobacterial cell wall that trigger CD1c-mediated human T cell responses (11). The mannosyl-β-1-phosphate head group, along with the repeating C5 structure, provided an obvious structural homology of these antigenic phospholipids with evolutionarily conserved mannosyl phosphopolyprenols that function as carbohydrate carriers (15). Here, we provide structural, biosynthetic, and genetic evidence that the repeating C5 units found in these lipids are made by Pks12 from methylmalonate and malonate precursors. This represents a new type of lipid synthesis mechanism that leads to the production of alkane lipids that structurally mimic polyisoprenols but are made by a polyketide mechanism. Based on their mode of synthesis and their conserved expression among infectious mycobacterial species, the 4-, 8-, 12-, 16-, and 20-branched alkane lipids can be simply referred to as mycoketides and the antigenic phosphoglycolipid forms as mannosyl-β-1-phosphomycoketides (MPMs).
Although fatty acid synthesis involves chain elongation by repeated acyltransfer, ketosynthesis, ketoreduction, dehydration, and enoyl reduction of C2 units, polyketide biosynthesis can involve all, some, or none of these functional group modifications. The AT modules of PKSs control whether malonyl (C2), methylmalonyl (C3), or ethylmalonyl (C4) enter into the elongation cycles, so that varied precursors and partial cycles allow PKSs to produce structurally diverse lipid products. The pks12 gene is predicted to encode a 430-kD protein, which is a typical type I PKS with 12 contiguous, but separately functioning, enzymatic domains. Although the function of the Pks12 has not been extensively analyzed, a detailed comparison of the sequence of pks12 to PKSs of known function had predicted that Pks12 had two complete sets of FAS-like domains in which substrate specificities of two AT domains were predicted to be specific for malonate (C2) and methylmalonate (C3), respectively (24). Thus, the overall domain organization of this enzyme, with duplicate FAS modules and two differing AT activities, is consistent with a mechanism of lipid elongation involving the alternating insertion of one C2 and one C3 units per cycle, as predicted by model 2 (Fig. 2 A).
The structures of mycoketides provide detailed information that independently support this model and rule out competing models based on polyisoprenol pathways involving IPP. Instead of a γ-methyl branch and an unsaturated structure typical of IPP, the sequential condensation of malonate and methylmalonate yields a C5 unit with fully saturated, δ-methyl units. Synthesis of mycoketides from this basic unit is consistent with the detection of fully saturated lipid moieties in all MPMs studied (Table I), the δ-methyl branches seen in M. avium (11) and M. tuberculosis (Fig. 2 D), the incorporation of the expected number of methylmalonyl units into the final MPM product (Fig. 2 E), and the precise positioning of 5 methyl branches at every fourth carbon in MPMs analyzed from M. avium and M. tuberculosis (Fig. 2 D). The incremental C1 differences in length seen among different MPMs produced by the same organism (Fig. 2 B) and species-specific differences in the C30 (M. avium) or C32 (M. tuberculosis H37Rv and BCG) can be explained by differences in the length of the carboxylate primer. However, it is noted that primer selection is not entirely understood and is somehow unusual because fatty acids with an even number of carbons usually predominate, but the most abundant species of MPM appear to use C5 (M. tuberculosis CDC1551 and M. avium) or C7 primers (M. tuberculosis H37Rv and H37Ra and BCG). The production and linkage of carboxylate primers to polyketides have not been directly established, but combinational biosynthesis by FAS and PKS systems were recently reported in mycobacteria, providing a precedent for this type of a mixed FAS-PKS mechanism (27).
This new pathway for production of mycoketides and the predicted steps by which mycoketides are converted to phosphomycoketides are outlined in Fig. 5. The one to one correspondence between the predicted enzymatic functions of the 12 subunits of Pks12 with the biochemical reactions need to produce mycoketide suggests that Pks12 may be sufficient for production of the mycoketide unit. This lipid would have to undergo offloading from Pks12, reduction, phosphorylation, and glycosylation by as yet unknown mechanisms to produce the final MPM phosphoglycolipid antigen. An earlier paper demonstrated that pks12-deleted mycobacteria lack DIM, a polyketide that might be plausibly produced by this mechanism. However, our data, which include analysis of pks12-complemented strains, demonstrate that DIM is produced by pks12-deficient M. tuberculosis. Because previous studies have reported that loss of DIM leads to attenuation (28, 29), it is difficult to determine whether the previously reported effects of pks12 on mycobacterial growth in mice were due to DIM loss, MPM loss, or both (25).
Figure 5.

Proposed biosynthetic pathway of MPM. R is the enzyme. In the case of C30 MPM, C2 and C3 unit elongation occurs in five cycles using a straight chain heptanoyl primer. Subsequently, the carboxyl group of the resulting product is predicted to be reduced to alcohol, followed by phosphorylation and mannosylation.
This complex biosynthesis pathway is energetically expensive, and is lacking in saprophytic mycobacteria, but is conserved in all tested infectious mycobacteria, suggesting that mycoketides play a functional role in intracellular growth. In general, polyketides are not abundant structural lipids with basic housekeeping functions, but instead are secondary metabolites that function as antimicrobial compounds, signaling molecules or other highly specialized roles. Consistent with this, we found that MPMs are present at low absolute levels, as they comprise ∼1 ppm of the total weight of the cell wall (unpublished data). The structural homology of MPM with MPDs that function in transmembrane carbohydrate transport raises that possibility that MPMs also function as lipid intermediates in mannose transmembrane transport or transfer.
One function of MPMs is quite clear, as these lipids are presented by CD1c proteins to human T cells and provide a signal for the detection of intracellular mycobacteria during tuberculosis infection (11). These results support this idea by showing that MPMs are produced by mycobacteria of medical importance, including the M. tuberculosis and vaccine strains of BCG. The mycoketide portion of the antigen is produced by a lipid biosynthetic pathway present in mycobacterial pathogens, but is lacking in mammalian species that express CD1c on their antigen-presenting cells. Furthermore, the shorter chain length and saturated structure of mycoketides, in contrast with the homologous mammalian polyisoprenols, represent two chemical elements that increase the potency of a CD1c-mediated T cell response (Fig. 1 C).
T cell recognition of lipid antigens occurs after the lipid portion of the antigen is inserted into an antigen-binding groove on the distal surface of the CD1 protein. The grooves of CD1d (30), CD1a (31), and CD1b (32) are optimized to accommodate lipid antigens of approximately C40, C32, and C60 in length, respectively. Unlike CD1b, which has small amino acid side chains at conserved positions on the floor of the groove, the CD1c sequence encodes amino acids with larger side chains like those seen in CD1a and CD1d, so that the CD1c groove size may be similar to that of CD1a and CD1d (32, 33). Thus, C95 mammalian MPDs are larger than any known CD1 groove, and the smaller C30-34 mycoketide may be of the correct size to fit within the CD1c groove. The discovery of the mycoketide biosynthetic pathway in mycobacteria provides insight into how the shorter, saturated lipid portion of mycoketide represents a pathogen-associated molecular pattern, which allows mycobacteria to be recognized as foreign by the human immune system (34).
Acknowledgments
The authors gratefully acknowledge Drs. P. Brennan, J. Belisle, H. Remold, and H. Band for providing mycobacteria or other organisms and Dr. J. Kriakov for providing the cosmid vectors pJK93 and phAE159 used in this work. We thank C.A. Debono for technical assistance.
This work was in part supported by grants from the National Institutes of Health (AI 49313 and AR 48632), the American College of Rheumatology Research and Education Foundation, the Mizutani Foundation for Glycoscience and Pew Scholars Program in the Biomedical Sciences (to D.B. Moody), National Institutes of Health grant nos. AI26170 (to W.R. Jacobs Jr.) and RR10888 (to C.E. Costello), and grants from The Medical Research Council (G0000895, G9901077) and The Wellcome Trust (060750) to G.S. Besra. G.S. Besra is a Lister-Jenner Research fellow.
The authors have no conflicting financial interests.
Abbreviations used in this paper: AT, acyltransferase; BCG, Bacille-Calmette-Guerin; CID, collisionally induced dissociation; DIM, phthiocerol dimycocerosate; ESI, electrospray ionization; FAS, fatty acid synthase; IPP, isopentenyl pyrophosphate; MPD, mannosyl-β-1-phosphodolichol; MPI, mannosyl-β-1-phosphoisoprenoid; MPM, mannosyl-β-1-phoshomycoketide; MPP, mannosyl-β-1-phosphopolyisoprenol; MS, mass spectrometry; PKS, polyketide synthase.
References
- 1.Converse, S.E., J.D. Mougous, M.D. Leavell, J.A. Leary, C.R. Bertozzi, and J.S. Cox. 2003. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc. Natl. Acad. Sci. USA. 100:6121–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baulard, A.R., S.S. Gurcha, J. Engohang-Ndong, K. Gouffi, C. Locht, and G.S. Besra. 2003. In vivo interaction between the polyprenol phosphate mannose synthase Ppm1 and the integral membrane protein Ppm2 from Mycobacterium smegmatis revealed by a bacterial two-hybrid system. J. Biol. Chem. 278:2242–2248. [DOI] [PubMed] [Google Scholar]
- 3.Sieling, P.A., D. Jullien, M. Dahlem, T.F. Tedder, T.H. Rea, R.L. Modlin, and S.A. Porcelli. 1999. CD1 expression by dendritic cells in human leprosy lesions: correlation with effective host immunity. J. Immunol. 162:1851–1858. [PubMed] [Google Scholar]
- 4.Uehira, K., R. Amakawa, T. Ito, K. Tajima, S. Naitoh, Y. Ozaki, T. Shimizu, K. Yamaguchi, Y. Uemura, H. Kitajima, et al. 2002. Dendritic cells are decreased in blood and accumulated in granuloma in tuberculosis. Clin. Immunol. 105:296–303. [DOI] [PubMed] [Google Scholar]
- 5.Beckman, E.M., S.A. Porcelli, C.T. Morita, S.M. Behar, S.T. Furlong, and M.B. Brenner. 1994. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature. 372:691–694. [DOI] [PubMed] [Google Scholar]
- 6.Moody, D.B., B.B. Reinhold, M.R. Guy, E.M. Beckman, D.E. Frederique, S.T. Furlong, S. Ye, V.N. Reinhold, P.A. Sieling, R.L. Modlin, et al. 1997. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science. 278:283–286. [DOI] [PubMed] [Google Scholar]
- 7.Moody, D.B., M.R. Guy, E. Grant, T.Y. Cheng, M.B. Brenner, G.S. Besra, and S.A. Porcelli. 2000. CD1b-mediated T cell recognition of a glycolipid antigen generated from mycobacterial lipid and host carbohydrate during infection. J. Exp. Med. 192:965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sieling, P.A., D. Chatterjee, S.A. Porcelli, T.I. Prigozy, R.J. Mazzaccaro, T. Soriano, B.R. Bloom, M.B. Brenner, M. Kronenberg, P.J. Brennan, et al. 1995. CD1-restricted T cell recognition of microbial lipoglycan antigens. Science. 269:227–230. [DOI] [PubMed] [Google Scholar]
- 9.Gilleron, M., S. Stenger, Z. Mazorra, F. Wittke, S. Mariotti, G. Bohmer, J. Prandi, L. Mori, G. Puzo, and G. De Libero. 2004. Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J. Exp. Med. 199:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moody, D.B., D.C. Young, T.Y. Cheng, J.P. Rosat, C. Roura-Mir, P.B. O'Connor, D.M. Zajonc, A. Walz, M.J. Miller, S.B. Levery, et al. 2004. T cell activation by lipopeptide antigens. Science. 303:527–531. [DOI] [PubMed] [Google Scholar]
- 11.Moody, D.B., T. Ulrichs, W. Muhlecker, D.C. Young, S.S. Gurcha, E. Grant, J.P. Rosat, M.B. Brenner, C.E. Costello, G.S. Besra, and S.A. Porcelli. 2000. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature. 404:884–888. [DOI] [PubMed] [Google Scholar]
- 12.Stenger, S., D.A. Hanson, R. Teitelbaum, P. Dewan, K.R. Niazi, C.J. Froelich, T. Ganz, S. Thoma-Uszynski, A. Melian, C. Bogdan, et al. 1998. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 282:121–125. [DOI] [PubMed] [Google Scholar]
- 13.Stenger, S., R.J. Mazzaccaro, K. Uyemura, S. Cho, P.F. Barnes, J.P. Rosat, A. Sette, M.B. Brenner, S.A. Porcelli, B.R. Bloom, and R.L. Modlin. 1997. Differential effects of cytolytic T cell subsets on intracellular infection. Science. 276:1684–1687. [DOI] [PubMed] [Google Scholar]
- 14.Ulrichs, T., D.B. Moody, E. Grant, S.H. Kaufmann, and S.A. Porcelli. 2003. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect. Immun. 71:3076–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moody, D.B. 2001. Polyisoprenyl glycolipids as targets of CD1-mediated T cell responses. Cell. Mol. Life Sci. 58:1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant, E.P., M. Degano, J.P. Rosat, S. Stenger, R.L. Modlin, I.A. Wilson, S.A. Porcelli, and M.B. Brenner. 1999. Molecular recognition of lipid antigens by T cell receptors. J. Exp. Med. 189:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beckman, E.M., A. Melian, S.M. Behar, P.A. Sieling, D. Chatterjee, S.T. Furlong, R. Matsumoto, J.P. Rosat, R.L. Modlin, and S.A. Porcelli. 1996. CD1c restricts responses of mycobacteria-specific T cells. Evidence for antigen presentation by a second member of the human CD1 family. J. Immunol. 157:2795–2803. [PubMed] [Google Scholar]
- 18.Bardarov, S., S. Bardarov Jr., M.S. Pavelka Jr., V. Sambandamurthy, M. Larsen, J. Tufariello, J. Chan, G. Hatfull, and W.R. Jacobs Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis Microbiol. 148:3007–3017. [DOI] [PubMed] [Google Scholar]
- 19.Snapper, S.B., L. Lugosi, A. Jekkel, R.E. Melton, T. Kieser, B.R. Bloom, and W.R. Jacobs Jr. 1988. Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc. Natl. Acad. Sci. USA. 85:6987–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosat, J.P., E.P. Grant, E.M. Beckman, C.C. Dascher, P.A. Sieling, D. Frederique, R.L. Modlin, S.A. Porcelli, S.T. Furlong, and M.B. Brenner. 1999. CD1-restricted microbial lipid antigen-specific recognition found in the CD8+ alpha beta T cell pool. J. Immunol. 162:366–371. [PubMed] [Google Scholar]
- 21.Addlesee, H.A., and C.N. Hunter. 2002. Rhodospirillum rubrum possesses a variant of the bchP gene, encoding geranylgeranyl-bacteriopheophytin reductase. J. Bacteriol. 184:1578–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolattukudy, P.E., N.D. Fernandes, A.K. Azad, A.M. Fitzmaurice, and T.D. Sirakova. 1997. Biochemistry and molecular genetics of cell-wall lipid biosynthesis in mycobacteria. Mol. Microbiol. 24:263–270. [DOI] [PubMed] [Google Scholar]
- 23.Cole, S.T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S.V. Gordon, K. Eiglmeier, S. Gas, C.E. Barry III, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 393:537–544. [DOI] [PubMed] [Google Scholar]
- 24.Yadav, G., R.S. Gokhale, and D. Mohanty. 2003. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J. Mol. Biol. 328:335–363. [DOI] [PubMed] [Google Scholar]
- 25.Sirakova, T.D., V.S. Dubey, H.J. Kim, M.H. Cynamon, and P.E. Kolattukudy. 2003. The largest open reading frame (pks12) in the Mycobacterium tuberculosis genome is involved in pathogenesis and dimycocerosyl phthiocerol synthesis. Infect. Immun. 71:3794–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurcha, S.S., A.R. Baulard, L. Kremer, C. Locht, D.B. Moody, W. Muhlecker, C.E. Costello, D.C. Crick, P.J. Brennan, and G.S. Besra. 2002. Ppm1, a novel polyprenol monophosphomannose synthase from Mycobacterium tuberculosis. Biochem. J. 365:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trivedi, O.A., P. Arora, V. Sridharan, R. Tickoo, D. Mohanty, and R.S. Gokhale. 2004. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 428:441–445. [DOI] [PubMed] [Google Scholar]
- 28.Cox, J.S., B. Chen, M. McNeil, and W.R. Jacobs Jr. 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 402:79–83. [DOI] [PubMed] [Google Scholar]
- 29.Camacho, L.R., D. Ensergueix, E. Perez, B. Gicquel, and C. Guilhot. 1999. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol. Microbiol. 34:257–267. [DOI] [PubMed] [Google Scholar]
- 30.Zeng, Z., A.R. Castano, B.W. Segelke, E.A. Stura, P.A. Peterson, and I.A. Wilson. 1997. Crystal structure of mouse CD1: an MHC-like fold with a large hydrophobic binding groove. Science. 277:339–345. [DOI] [PubMed] [Google Scholar]
- 31.Zajonc, D.M., M.A. Elsliger, L. Teyton, and I.A. Wilson. 2003. Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 A. Nat. Immunol. 4:808–815. [DOI] [PubMed] [Google Scholar]
- 32.Gadola, S.D., N.R. Zaccai, K. Harlos, D. Shepherd, J.C. Castro-Palomino, G. Ritter, R.R. Schmidt, E.Y. Jones, and V. Cerundolo. 2002. Structure of human CD1b with bound ligands at 2.3 A, a maze for alkyl chains. Nat. Immunol. 3:721–726. [DOI] [PubMed] [Google Scholar]
- 33.Roura-Mir, C., and D.B. Moody. 2003. Sorting out self and microbial lipid antigens for CD1. Microbes Infect. 5:1137–1148. [DOI] [PubMed] [Google Scholar]
- 34.Medzhitov, R., and C.A. Janeway Jr. 1997. Innate immunity: the virtues of a nonclonal system of recognition. Cell. 91:295–298. [DOI] [PubMed] [Google Scholar]