

Abstract
Chronic myelogenous leukemia (CML) is characterized by the presence of the chimeric p210bcr/abl oncoprotein that shows elevated and constitutive protein tyrosine kinase activity relative to the normal c-abl tyrosine kinase. Although several p210bcr/abl substrates have been identified, their relevance in the pathogenesis of the disease is unclear. We have identified a family of proteins, Dok (downstream of tyrosine kinase), coexpressed in hematopoietic progenitor cells. Members of this family such as p62dok(Dok-1) and p56dok-2(Dok-2) associate with the p120 rasGTPase-activating protein (rasGAP) upon phosphorylation by p210bcr/abl as well as receptor and nonreceptor tyrosine kinases. Here, we report the generation and characterization of single and double Dok-1 or Dok-2 knockout (KO) mutants. Single KO mice displayed normal steady-state hematopoiesis. By contrast, concomitant Dok-1 and Dok-2 inactivation resulted in aberrant hemopoiesis and Ras/MAP kinase activation. Strikingly, all Dok-1/Dok-2 double KO mutants spontaneously developed transplantable CML-like myeloproliferative disease due to increased cellular proliferation and reduced apoptosis. Furthermore, Dok-1 or Dok-2 inactivation markedly accelerated leukemia and blastic crisis onset in Tec-p210 bcr/abl transgenic mice known to develop, after long latency, a myeloproliferative disorder resembling human CML. These findings unravel the critical and unexpected role of Dok-1 and Dok-2 in tumor suppression and control of the hematopoietic compartment homeostasis.
Keywords: cell proliferation, apoptosis, knockout, CML leukemogenesis, signal transduction
Introduction
Chronic myelogenous leukemia (CML) is a clonal disorder of the hematopoietic cells characterized by the presence of the Philadelphia chromosome (Ph+), which is the result of a chromosomal translocation between the BCR gene on chromosome 22 and the ABL gene on chromosome 9 (1, 2). A bcr-abl chimeric protein originates from this translocation. Its p210 form, which is the causative mutation found in 95% of cases of CML, has elevated tyrosine kinase activity and exists exclusively in cytoplasm compared with endogenous c-ABL (1, 2). Two phases of the disease have been characterized: (a) a chronic phase with an average span of 3–5 yr during which the Ph+ cells populate the entire intermediate and late hematopoietic maturational compartments, and (b) an acute malignant and fatal stage known as blast crisis when the leukemic cells acquire additional genetic changes, lose their ability to differentiate and mature, and acquire the ability to infiltrate and colonize other organs (1, 2). Inhibition of p210bcr/abl activity by selective drugs such as STI571 leads to disease remission, making CML a paradigmatic example of targeted cancer therapy (3). However, patients do relapse upon STI571 treatment, underscoring the need to identify critical downstream events in the p210bcr/abl signaling cascade. Furthermore, the genetics of blastic crisis transformation is poorly understood. p62 dok (Dok-1) was cloned as a major phosphorylation substrate of the p210bcr/abl oncoprotein in Ph+ CML blasts, as well as a major substrate of many tyrosine kinases (4, 5). Soon after, additional members of the family (6-9) have been identified. These proteins resemble docking proteins in their structure because they contain PH and PTB domains as well as multiple binding motifs for SH2 and SH3 domains. Three of them (Dok-1–3) are differentially expressed in the hemopoietic compartment, coexpressed in the hemopoietic progenitors, and aberrantly phosphorylated by p210bcr/abl (4, 6, 7, 9). Several recent reports implicate Dok proteins in the negative regulation of signaling pathways activated by tyrosine kinases (9-14). Unlike Dok-3–5, Dok-1 and Dok-2 are able to associate with rasGAP when phosphorylated, suggesting that they may serve critical, but possibly redundant, functions (6, 9). Therefore, to determine the role of DOK as p210bcr/abl substrates in hematopoiesis and CML pathogenesis, we studied in vivo in the mouse the effect of combined inactivation of DOK family members. Here, we unravel the key tumor suppressive role of Dok-1 and Dok-2 in the hemopoietic compartment and their importance in CML pathogenesis.
Materials and Methods
Targeting Vector and Generation of Dok-2+/− Embryonic Stem Cells.
A 129/Sv mouse genomic library (Stratagene) was screened with a probe containing murine Dok-2 exon 1. Exon/intron boundaries of the isolated Dok-2 genomic clones were determined by restriction enzyme mapping, DNA sequencing, and PCR. To generate the targeting construct, a 2.7-Kb EcoRI–SacI Dok-2 genomic fragment (5′ arm) and a 4.5-Kb XhoI–HindIII Dok-2 genomic fragment (3′ arm) were cloned into the pPNT (13). The targeting construct was linearized with NotI and electroporated into CJ7 embryonic stem cells. Transfectants were selected in 350 μg/ml G418 and 2 μM gancyclovir and expanded for Southern blot analysis using a 5′ probe (see Fig. S1 A below).
Generation of Dok-2−/−, Double KO (DKO), and Tec-p210bcr/abl/Dok-1/Dok-2 Compound Mutants.
Chimeric mice and F1 offspring were produced as described previously (13). Chimeric males were then mated with 129/Sv females (The Jackson Laboratory) to obtain Dok-2 mutants in a 129/Sv background. Dok-1 −/− mice (13) and Tec-p210 bcr/abl transgenic mice (TM; references 15 and 16) have been described. Dok-1 −/−/Dok-2 −/− mice were obtained by interbreeding Dok-1 −/− mice with Dok-2 −/− mice both in 129/Sv background. To obtain Tec-p210 bcr/abl/Dok-1/Dok-2 compound mice, Tec-p210 bcr/abl TM were at first crossed with Dok-1 −/− mice (129/Sv) or Dok-2 −/− mice (129/Sv). F1 offspring were then mated with each other to get Tec-p210 bcr/abl/Dok null mice and to balance the genetic background. All mice studies were approved by The Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center.
Follow-up Design and Leukemia Diagnosis.
Mice were monitored monthly by peripheral blood (PB) counts and smears (biweekly in the case of Tec-p210 bcr/abl/Dok-1/Dok-2 compound mutants and BM transplantation). Diagnosis of leukemia was made on the criteria that two consecutive white blood cell counts are >20 × 103/μl. Autopsies were performed on dead or moribund animals as described previously (17). For B220 or CD3 detection, immunohistochemistry was performed on representative sections using anti–mouse B220 monoclonal antibody (RA3-6B2; BD Biosciences) or a rabbit anti-CD3 polyclonal antibody (DakoCytomation) according to the manufacturer's instructions.
BM Transplantation.
2 × 106 BM cells from WT or Dok-1 −/−/Dok-2 −/− mutant mice were injected via tail vein into lethally irradiated (920 rads) 129/Sv WT mice (6-wk-old female). Recipient mice were monitored and scored positive for disease according to criteria mentioned in the legend to Fig. S2 (see below).
Western Blot and Flow Cytometric Analysis.
These analyses were performed as described previously (13, 18). For Western blot analysis, we used a rabbit polyclonal anti–Dok-R/Dok2 antibody (Upstate Cell Signaling) to detect Dok-2 protein. To detect Erk 2 protein, we used a polyclonal anti–Erk 2 antibody (Santa Cruz Biotechnology, Inc.). For flow cytometry, we used the following conjugated antibodies: anti–c-Kit, anti–Sca-1, anti–Mac-1, anti–Gr-1, anti-F4/80, anti-CD3 complex, anti-B220, anti-CD4, and anti-CD8a. Anti-F4/80 was obtained from Caltag. All other antibodies were from BD Biosciences. Flow cytometry was performed using a FACScan (Becton Dickinson). The data were analyzed using FlowJo software (Tree Star).
Ras GTPase Activation Assay.
Activation of Ras was measured using GST-RBD (Ras-binding domain of Raf [RBD]) pull down assays (19). The underlying premise of this assay is that the RBD binds only to GTP-bound Ras proteins. Mac-1+ cells were isolated from freshly isolated BM cells using CD11b Microbeads (Miltenyi Biotec). Purity (>90%) was confirmed by flow cytometry. Mac-1+ cells were incubated in RPMI/0.1% FCS for 3 h, and then stimulated with 10 ng/ml GM-CSF for 10 min at 37°C. Cells were then lysed in 25 mM Hepes, pH 7.5, 150 mM NaCl, 1% NP-40, 10% glycerol, 25 mM NaF, 10 mM MgCl2, 1 mM EDTA, 2.5 mM sodium deoxycholic, and 1 mM Na3VO4 plus protease inhibitors. An equal amount of cell lysates was incubated with GST-RBD coupled to glutathione beads. Bead-associated Ras (GTP-bound Ras) and total Ras in cell lysates were detected by Western blotting with an anti–pan-Ras antibody (Transduction Laboratories). GM-CSF–induced Ras activation is measured by normalizing the amount of GTP-bound Ras to the total amount of Ras in cell lysates.
Proliferation, Apoptosis, and In Vitro Colony-forming Assay.
BM cells were flushed from murine femurs and tibiae. 2 × 104 cells were plated in MethoCult M3434 (StemCell Technologies Inc.). Colonies were counted on days 2 (CFU-E), 7 (CFU-GM and BFU-E), and 13 (CFU-GEMM). For proliferation assay of collected cells from in vitro colony-forming assay, 2 × 104 BM cells were plated in MethoCult M3234 with 20 ng/ml IL-3, 50 ng/ml G-CSF, and 20 ng/ml GM-CSF. At day 7, cells were collected from methylcellulose and washed by RPMI/10% FCS and counted. 2.5 × 104 cells were cultured without growth factor for 20 h. [3H]thymidine was then added for 4 h. For proliferation assays of BM cells, contaminating erythrocytes were removed by hypotonic lysis. Mac-1+ cells were isolated using CD11b Microbeads (Miltenyi Biotec). Purity (>90%) was confirmed by flow cytometry. Cells were treated with 10 ng/ml IL-3, 10 ng/ml stem cell factor (SCF), or 10 ng/ml GM-CSF for 42 h. [3H]thymidine was then added for 6 h. For apoptosis analysis, BM cells were cultured as described above. After 48 h, cells were harvested and incubated with anti-CD16/32 to block nonspecific binding. Cells were then stained with anti–Mac-1–APC and annexin V (BD Biosciences) according to the manufacturer's instructions. The percentage of apoptotic cells was determined by FACS analysis (FACSCalibur; Becton Dickinson).
Spectral Karyotyping Analysis.
BM cells from DKO mice were cultured in RPMI 1640/10% FCS with 6 ng/ml IL-3, 10 ng/ml IL-6, 100 ng/ml SCF, and 10 ng/ml BrdU for 18 h. The cells were then prepared for cytogenetic analysis performed using a mouse SkyPaint Kit (Applied Spectral Imaging) according to the manufacturer's instructions.
Online Supplemental Material.
Fig. S1 shows targeted disruption of the Dok-2 gene. Fig. S2 shows results of adoptive transfer of BM cells from DKO mice with myeloproliferative disease (MPD). Fig. S3 shows FACS analysis of BM and spleen cells from leukemic Tec-p210 bcr/abl, Tec-p210bcr/abl/Dok-1−/−, and Tec-p210 bcr/abl/Dok-2−/− mice and survival curves of these compound mutants. Table S1 shows the data of PB cell count. Figs. S1–S3 and Table S1 are available at http://www.jem.org/cgi/content/full/jem.20041306/DC1.
Results and Discussion
We inactivated the Dok-2 gene in the mouse and crossed Dok-2 mutants with Dok-1 −/− mice that we described previously (13). As in the case of Dok-1–targeted disruption, Dok-2 −/− mutants were born following mendelian frequencies. Postmortem pathological analysis and monthly flow cytometric and morphological assessments of the various hematopoietic organs from Dok-2 −/− mutants revealed normal steady-state hematopoiesis and organogenesis (unpublished data). Analysis of Dok-1 and Dok-2 protein expression in the various hematopoietic organs from Dok-1 −/− and Dok-2 −/− mutants, respectively, did not reveal compensatory up-regulation of the remaining Dok protein (Fig. S1 E, available at http://www.jem.org/cgi/content/full/jem.20041306/DC1). Furthermore, in standard colony-forming assays in methylcellulose, BM progenitors from Dok-2 −/− mutants yielded numbers of erythroid and myeloid colonies comparable to WT sex- and age-matched littermates (Fig. S1 F). Although myeloid differentiation was overall normal in Dok-2 −/− mice, we did observe a consistent defect in the activation of Dok-2 −/− mature granulocyte upon TPA treatment (unpublished data). Nevertheless, Dok-2 −/− mice did not display an increase incidence of spontaneous infections. The fact that both Dok-1 and Dok-2 KO mutants displayed normal steady-state hemopoiesis suggested that they may exert redundant function.
Therefore, we generated DKO mutants. These mutants were also developmentally normal (as revealed by pathological analysis of all organs; unpublished data) and fertile. However, monthly postmortem pathological, flow cytometric, and morphological assessments of the various hematopoietic organs from DKO mutants unraveled striking differences with respect to the Dok-1 and Dok-2 single KO mutants. In fact, DKO mutants developed at complete penetrance a CML-like MPD at 10–12 mo of age (Fig. 1 A; refer to Materials and Methods). DKO displayed a progressive increase in white blood cell counts in the PB after 4 mo of age (Fig. 1 B and Table S1, which is available at http://www.jem.org/cgi/content/full/jem.20041306/DC1; at 4 mo: WT [n = 6] 9,800 ± 2,078, DKO [n = 6] 9,725 ± 3,007; at 8 mo: WT [n = 6] 10,033 ± 1,286, DKO [n = 7] 15,575 ± 2,326; at 12 mo: WT [n = 6] 9,200 ± 3,268, DKO [n = 8] 24,180 ± 4,540). At leukemia onset, DKO mice invariably displayed a marked splenomegaly (Fig. 1 C; WT [n = 3] 0.051 ± 0.005 g, DKO [n = 3] 0.081 ± 0.02 g) as well as PB and BM hypercellularity (Fig. 1 D and Table S1). BM and spleen were both predominantly infiltrated by myeloid cells that retained the ability to terminally differentiate (Fig. 1 D). Automated and differential counts in the PB revealed a marked leukocytosis caused by an increase in the number of neutrophils and monocytes (Table S1). Interestingly, erythrocytes and platelets number counts remained relatively normal at this stage (Table S1). The increase in PB cellularity was accompanied by the appearance of undifferentiated blasts in the PB (Fig. 1 E and Table S1). Flow cytometric analysis of PB, BM, and spleen confirmed the expansion of the differentiated myeloid compartment (increase in the percentage of Mac-1+, Gr-1+, and Mac-1+ F4/80+ cells with a concomitant decrease in the percentage of B220+ [B cell] and CD3+ [T cell] cells) as well as the presence of undifferentiated cells in the PB (Sca-1+ cells: WT [n = 4] 6.5 ± 4.3%, DKO [n = 4] 13.5 ± 2.9%; Fig. 1, F–H). Colony-forming assays in methylcellulose from BM cells of DKO diseased mice revealed the expansion of myeloid progenitors (Fig. 1 I). A higher number of progenitors (CFU-GM) from BM cells of DKO mice was already observed in the preleukemic phase (4 mo of age; Fig. 2 A). Next, we assessed whether the MPD was transplantable (refer to Materials and Methods). To this end, BM cells from disease DKO mice or WT controls were transplanted into lethally irradiated recipient mice. Five out of the seven recipient mice that were transplanted with cells from DKO mice developed overt disease (Fig. S2 A) with splenomegaly (Fig. S2 B) and a marked increase in the percentage of Mac-1+ Gr-1+ cells in the spleen (Fig. S2 C). Thus, at onset, the MPD is fully transplantable, hence demonstrating the cell autonomy of the disorder. We also studied whether the MPD was the result of a clonal evolution by performing spectral karyotyping analysis (refer to Materials and Methods) on BM cells from diseased DKO mice. At least 12 metaphase cells per mouse (n = 4) were analyzed and no karyological abnormalities were found.
Figure 1.
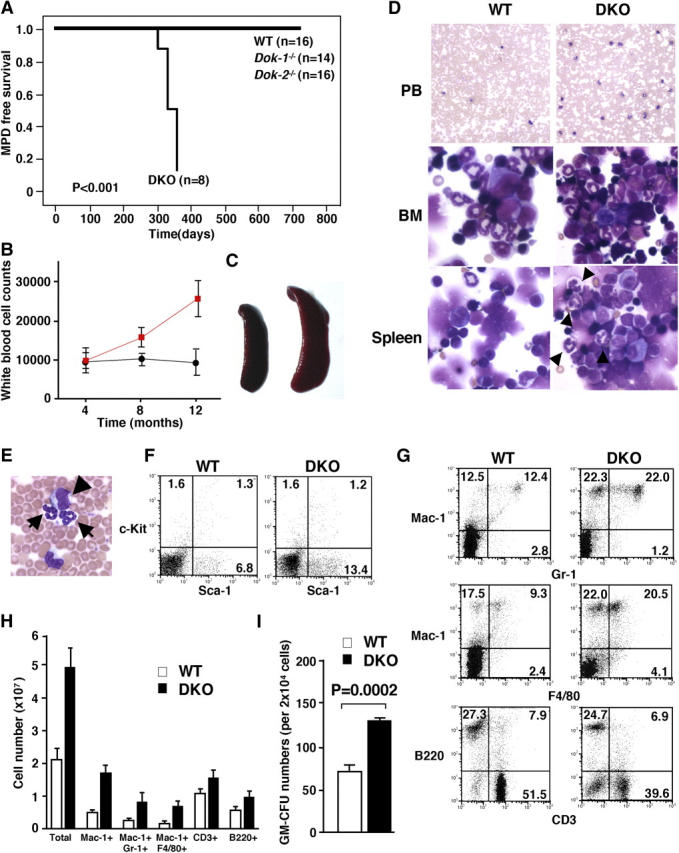
MPD in Dok-1 −/−/Dok-2 −/− mutants. (A) MPD-free survival curve of WT, Dok-1 −/−, Dok-2 −/− single mutants, and Dok-1 −/−/Dok-2 −/− mutant (DKO) mice. (B) Time course of white blood cell counts of WT (black line) and Dok-1 −/−/Dok-2 −/− mutants (red line). (C) Splenomegaly in Dok-1 −/−/Dok-2 −/− diseased mutants. Spleen from 1-yr-old WT (left) and Dok-1 −/−/Dok-2 −/− mice (right). (D) Morphology of PB, BM, and spleen cells from 1-yr-old WT and Dok-1 −/−/Dok-2 −/− mutants (DKO). The smears of PB and cytospins of BM and spleen were stained with Wright-Giemsa stain. Original magnifications: 100 for PB and 400 for BM and spleen. Differentiated granulocytes (arrowheads) are seen in spleen cytospin from Dok-1 −/−/Dok-2 −/− mutant (DKO). (E) High magnification (400) of PB from Dok-1 −/−/Dok-2 −/− mutant. Arrows indicate differentiated granulocytes and arrowhead indicates blast cell. (F) Flow cytometric analysis of PB from a Dok-1 −/−/Dok-2 −/− mutant (DKO) and WT littermate. PB cells were double stained with anti–c-Kit–PE and anti–Sca-1–FITC. Percentages of positive cells are shown in each quadrant. (G) Flow cytometric analysis of splenocytes from a Dok-1 −/−/Dok-2 −/− (DKO) mutant and WT littermate. (H) Absolute numbers of total, Mac-1+, Mac-1+/Gr-1+, Mac-1+/F4/80+, CD3+, and B220+ cells from 1-yr-old WT (white bars) and DKO mutant (black bars). Spleens from three WT and three DKO mutants were analyzed by flow cytometry. The number of positive cells for each marker was calculated. Means and SD are indicated. (I) In vitro colony-forming assay performed with BM cells isolated from a 1-yr-old WT (white bar) and diseased DOK mutant (black bar). P-value is also shown.
Figure 2.

Analysis of preleukemic Dok-1 −/−/Dok-2 −/− mutants. (A) In vitro colony-forming assay performed with BM cells isolated from a WT (white bar) and a Dok-1 −/−/Dok-2 −/− mutant (DKO; black bar) at 4 mo of age. P-value is also shown. (B) [3H]thymidine incorporation analysis of proliferative response of cells collected from in vitro colony assay of BM cells from 2-mo-old WT, Dok-1 −/−, Dok-2 −/−, and DKO mice. (C) Proliferative response determined by [3H]thymidine incorporation of Mac-1+ BM cells upon IL-3, GM-CSF, and SCF stimulation. (D) Apoptotic response to growth factor deprivation in BM cells. Cell death was analyzed by annexin V staining of WT (white bar) and Dok-1 −/−/Dok-2 −/− (DKO; black bar) Mac-1+ BM cells. (E) GM-CSF–induced Ras activation of Mac-1+ BM cells isolated from 4-mo-old WT, Dok-1 −/−, Dok-2 −/−, and DKO mice. A representative Western blot from three experiments is shown. Very low amounts of Ras-GTP were detected in serum-starved cells before GM-CSF treatment. (F) Relative Ras activity in the above cells treated with GM-CSF is quantified by the ratio of GTP-bound Ras over total Ras. The value of WT BM cells was set as 1 and the ratios of other types of BM cells were calculated correspondingly. (G) Levels of ERK 1/2 phosphorylation upon (lanes 3 and 4) or in the absence (lanes 1 and 2) of GM-CSF stimulation in BM cells from WT (lanes 1 and 3) and Dok-1 −/−/Dok-2 −/− (lanes 2 and 4) mice.
Next, we investigated the molecular and biological consequences of concomitant Dok-1 and Dok-2 inactivation in cells from DKO mutants before leukemia onset (2–4-mo-old mice). Myeloid BM cells from DKO mice displayed increased proliferative potential upon IL-3, GM-CSF, and SCF stimulation (Fig. 2 C). Furthermore, DKO myeloid cells collected from in vitro colony-forming assays displayed increased proliferative potential even in the absence of growth factor (Fig. 2 B). Furthermore, Dok-1 and Dok-2 inactivation protected myeloid BM cells from growth factor deprivation–induced cell death (Fig. 2 D).
As Dok-1 and Dok-2 can act as negative Ras regulator (12–14), we tested the level of Ras activation upon GM-CSF stimulation in Mac-1+ BM cells, which are known to normally coexpress these two proteins, from single and DKO mice and WT mice. DKO cells indeed displayed elevated levels of Ras activation (Fig. 2, E and F). Furthermore, we observed a marked elevation of MAP kinase (p44 and p42 Erk) activation upon GM-CSF stimulation in the BM from DKO mutants (Fig. 2 G). Thus, the concomitant inactivation of Dok-1 and Dok-2 causes profound biological and molecular outcomes, which result in overt disease at full penetrance.
On the basis of what we observed in DKO mutants, Dok-1 and Dok-2 may therefore oppose the leukemogenic potential of p210bcr/abl. To test this hypothesis genetically in vivo, we made use of a Tec-p210 bcr/abl transgenic model. Tec-p210 bcr/abl TM are faithful animal models of CML as they develop a chronic leukemic phase after a long latency, followed by an acute terminal phase that is reminiscent of a CML blastic crisis with appearance of blasts in the PB and organ infiltration (15, 16). Therefore, we crossed Tec-p210 bcr/abl TM with Dok-1 − / − and Dok-2 − / − mutants and assessed whether their inactivation would impact on the biology of the disease. Inactivation of either Dok-1 or Dok-2 accelerated chronic phase onset in compound mutants (Fig. 3, A and B). By contrast, the distinctive features of the chronic phase in Tec-p210 bcr/abl TM were not perturbed by Dok-1 or Dok-2 inactivation as revealed by comparable flow cytometric and morphological profiles of the major hematopoietic organs (Fig. S3 A and unpublished data). Furthermore, and importantly, Dok-1 or Dok-2 inactivation accelerated the onset of the fatal blastic phase of the disease resulting in a marked reduction in overall survival in the compound mutants (Fig. 3, C–F, and Fig. S2, B and C; mean survival of Dok-1 crosses: Tec-p210 bcr/abl/WT: 324.7 ± 50.0 d; Tec-p210 bcr/abl/Dok-1 +/−: 307.4 ± 67.1 d; Tec-p210 bcr/abl/Dok-1 −/−: 284.9 ± 65.4 d; mean survival of Dok-2 crosses: Tec-p210 bcr/abl/WT: 320.9 ± 54.8 d; Tec-p210 bcr/abl/Dok-2 +/−: 282.7 ± 96.0 d; p210 bcr/abl/Dok-2 −/−: 270.5 ± 48.7 d) as well as in shortening of the chronic phase (Dok-1 crosses: Tec-p210 bcr/abl/WT [n = 10] 84.6 ± 46.3 d; Tec-p210 bcr/abl/Dok-1 +/− [n = 21] 78.2 ± 47.5 d; Tec-p210 bcr/abl/Dok-1 −/− [n = 9] 78.2 ± 44.1 d; Dok-2 crosses: Tec-p210 bcr/abl/WT [n = 8] 83.8 ± 31.1 d; Tec-p210 bcr/abl/Dok-2 +/− [n = 13] 76.9 ± 39.6 d; Tec-p210 bcr/abl/Dok-2 −/− [n = 9] 70.0 ± 27.7 d). Postmortem analysis of compound mutants in various genotypes did not reveal qualitative differences in the biology and cellularity of the blast crisis. In mice from all genotypes, blasts and myeloid differentiating cells (e.g., neutrophils) were found to infiltrate solid organs such as the lung (Fig. 3, C–F). By contrast, a major difference was observed in analyzing the intestinal tract of the various compound mutants. Approximately 25% of the Tec-p210 bcr/abl/Dok-2 −/− mutants analyzed were found to develop an aggressive and infiltrating lymphoma of the small intestine of B cell origin (Fig. 3, G–J). By contrast, this malignancy was not observed in the Tec-p210 bcr/abl/Dok-1 −/− mutants analyzed (Fig. 3 J).
Figure 3.
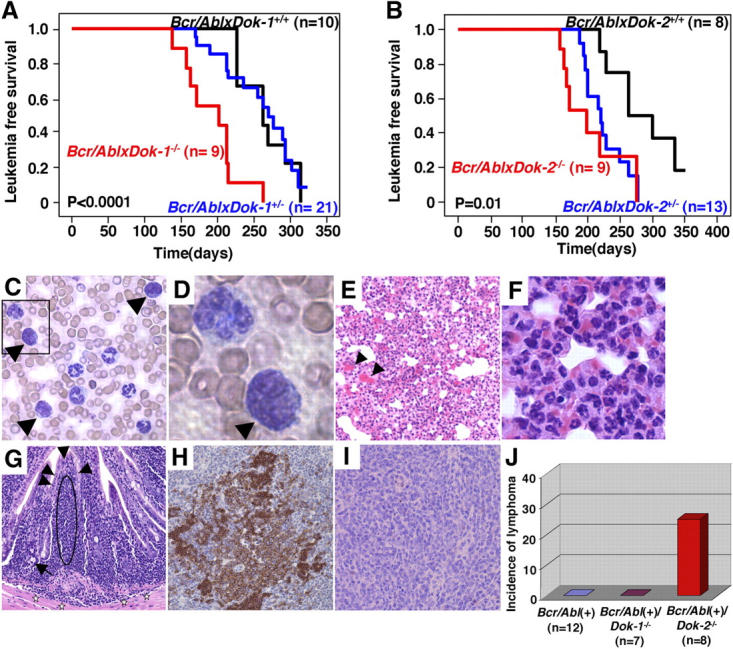
Leukemia onset and phenotype of Tec-p210 bcr/abl/Dok-1/Dok-2 compound mutants. (A) Leukemia-free survival curves of Tec-p210 bcr/abl(+)/Dok-1 mutants. P-value is also indicated. (B) Leukemia-free survival curves of Tec-p210 bcr/abl(+)/Dok-2 mutants. P-value is also indicated. (C) Morphology of PB cells from a Tec-p210 bcr/abl(+)/Dok-2 −/− mouse during the blast crisis. Blasts (arrowheads) are indicated. Original magnification of 200. (D) Magnification (1,000) of boxed region in C. Blast cell is indicated by arrowhead. (E) Hematoxylin and eosin staining of lung from a Tec-p210 bcr/abl(+)/Dok-2 −/− mouse killed during the blast crisis. Alveolar interstitial spaces are infiltrated by accumulation of erythrocytes (arrowheads) and invasion of both myeloid blasts and differentiated cells. Original magnification of 100. (F) High magnification of E. Many differentiated granulocytes are seen. Original magnification of 400. (G) Hematoxylin and eosin staining of an invasive lymphoma in the ileum in a Tec-p210 bcr/abl(+)/Dok-2 −/− mouse. A monomorphic infiltrate of lymphocytic cells (indicated by the dashed oval) with hyperchromatic and dysplastic nuclei is identified under the mucosa (arrowheads). These cells are invading beyond the muscularis mucosa into the submucosa (stars). Cobblet cells (arrow) are seen. Original magnification of 200. (H and I) Immunohistochemical staining of the lymphoma shown in G using anti-B220 antibody (H; for B cell identification) and anti-CD3 antibody (I; for T cell identification). Original magnification of 200. (J) Incidence of invasive lymphoma in Tec-p210 bcr/abl(+), Tec-p210 bcr/abl(+)/Dok-1 −/−, and Tec-p210 bcr/abl(+)/Dok-2 −/− mice.
Our analysis leads to three major conclusions: (a) Dok-1 and Dok-2 play a pivotal cooperative role in the control of the homeostasis of the hematopoietic compartment and in tumor suppression, as their combined loss triggers a CML-like MPD at complete penetrance; (b) Dok-1 and Dok-2 negatively regulate Ras and MAP kinase activation, and their loss leads to the hematopoietic cells' proliferative and survival advantage in the presence or the absence of growth factors, respectively; and (c) Dok-1 and Dok-2 oppose p210bcr/abl-driven leukemogenesis and lymphomagenesis. Therefore, Dok-1 and Dok-2 functional loss may exacerbate and accelerate the natural history of the human disease.
Acknowledgments
In memory of Hisamaru Hirai.
We thank B. Clarkson, D. Wisniewski, A. Strife, H. Matsushita, P. Scaglioni, T. Maeda, L-F. Cai, L. DiSantis, J. Lauchle, K. Shannon, and Y. Yamanashi for advice and discussions, and V. Sahi, M. Jiao, and M. Leversha for technical assistance.
Supported by a grant (CA-64593) from the National Cancer Institute to P.P. Pandolfi.
The authors have no conflicting financial interests.
A. Di Cristofano's present address is Human Genetics Program, Fox Chase Cancer Center, Philadelphia, PA 19111.
References
- 1.Wong, S., and O.N. Witte. 2001. Modeling Philadelphia chromosome positive leukemias. Oncogene. 20:5644–5659. [DOI] [PubMed] [Google Scholar]
- 2.Van Etten, R.A. 2002. Studying the pathogenesis of BCR-ABL+ leukemia in mice. Oncogene. 21:8643–8651. [DOI] [PubMed] [Google Scholar]
- 3.Druker, B.J., C.L. Sawyers, R. Capdeville, J.M. Ford, M. Baccarani, and J.M. Goldman. 2001. Chronic myelogenous leukemia. Hematology (Am Soc Hematol Educ Program). 1:87–112. [DOI] [PubMed] [Google Scholar]
- 4.Carpino, N., D. Wisniewski, A. Strife, D. Marshak, R. Kobayashi, B. Stillman, and B. Clarkson. 1997. p62dok: a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 88:197–204. [DOI] [PubMed] [Google Scholar]
- 5.Yamanashi, Y., and D. Baltimore. 1997. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 88:205–211. [DOI] [PubMed] [Google Scholar]
- 6.Cong, F., B. Yuan, and S.P. Goff. 1999. Characterization of a novel member of the DOK family that binds and modulates Abl signaling. Mol. Cell. Biol. 19:8314–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Cristofano, A., N. Carpino, N. Dunant, G. Friedland, R. Kobayashi, A. Strife, D. Wisniewski, B. Clarkson, P.P. Pandolfi, and M.D. Resh. 1998. Molecular cloning and characterization of p56dok-2defines a new family of RasGAP-binding proteins. J. Biol. Chem. 273:4827–4830. [DOI] [PubMed] [Google Scholar]
- 8.Grimm, J., M. Sachs, S. Britsch, S. Di Cesare, T. Schwarz-Romond, K. Alitalo, and W. Birchmeier. 2001. Novel p62dok family members, dok-4 and dok-5, are substrates of the c-Ret receptor tyrosine kinase and mediate neuronal differentiation. J. Cell Biol. 154:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemay, S., D. Davidson, S. Latour, and A. Veillette. 2000. Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell. Biol. 20:2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelms, K., A.L. Snow, J. Hu-Li, and W.E. Paul. 1998. FRIP, a hematopoietic cell-specific rasGAP-interacting protein phosphorylated in response to cytokine stimulation. Immunity. 9:13–24. [DOI] [PubMed] [Google Scholar]
- 11.Suzu, S., M. Tanaka-Douzono, K. Nomaguchi, M. Yamada, H. Hayasawa, F. Kimura, and K. Motoyoshi. 2000. p56dok-2as a cytokine-inducible inhibitor of cell proliferation and signal transduction. EMBO J. 19:5114–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamanashi, Y., T. Tamura, T. Kanamori, H. Yamane, H. Nariuchi, T. Yamamoto, and D. Baltimore. 2000. Role of the rasGAP-associated docking protein p62dokin negative regulation of B cell receptor-mediated signaling. Genes Dev. 14:11–16. [PMC free article] [PubMed] [Google Scholar]
- 13.Di Cristofano, A., M. Niki, M. Zhao, F.G. Karnell, B. Clarkson, W.S. Pear, L. Van Aelst, and P.P. Pandolfi. 2001. p62dok, a negative regulator of Ras and mitogen-activated protein kinase (MAPK) activity, opposes leukemogenesis by p210bcr-abl. J. Exp. Med. 194:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao, M., A.A. Schmitz, Y. Qin, A. Di Cristofano, P.P. Pandolfi, and L. Van Aelst. 2001. Phosphoinositide 3-kinase–dependent membrane recruitment of p62dok is essential for its negative effect on mitogen-activated protein (MAP) kinase activation. J. Exp. Med. 194:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honda, H., H. Oda, T. Suzuki, T. Takahashi, O.N. Witte, K. Ozawa, T. Ishikawa, Y. Yazaki, and H. Hirai. 1998. Development of acute lymphoblastic leukemia and myeloproliferative disorder in transgenic mice expressing p210bcr/abl: a novel transgenic model for human Ph1-positive leukemias. Blood. 91:2067–2075. [PubMed] [Google Scholar]
- 16.Honda, H., T. Ushijima, K. Wakazono, H. Oda, Y. Tanaka, S. Aizawa, T. Ishikawa, Y. Yazaki, and H. Hirai. 2000. Acquired loss of p53 induces blastic transformation in p210bcr/abl-expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood. 95:1144–1150. [PubMed] [Google Scholar]
- 17.Di Cristofano, A., M. De Acetis, A. Koff, C. Cordon-Cardo, and P.P. Pandolfi. 2001. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet. 27:222–224. [DOI] [PubMed] [Google Scholar]
- 18.Di Cristofano, A., P. Kotsi, Y.F. Peng, C. Cordon-Cardo, K.B. Elkon, and P.P. Pandolfi. 1999. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 285:2122–2125. [DOI] [PubMed] [Google Scholar]
- 19.de Rooij, J., and J.L. Bos. 1997. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene. 14:623–625. [DOI] [PubMed] [Google Scholar]