

Abstract
The leukocyte integrin lymphocyte function-associated antigen 1 (LFA-1) and its endothelial ligand intercellular adhesion molecule (ICAM)-1 play an important role in transmigration as demonstrated by in vivo and in vitro models of inflammation. Despite the prominent role, little is known concerning the distribution and dynamic behavior of these adhesion molecules during leukocyte transmigration. Therefore, we examined the spatial and temporal distribution of LFA-1 on neutrophils actively transmigrating tumor necrosis factor-α–activated human umbilical vein endothelial monolayers under shear flow. Upon neutrophil arrest, LFA-1 was evenly distributed. However, once neutrophils initiated transmigration, LFA-1 rapidly redistributed to form a ringlike cluster at the neutrophil–endothelial junctional interface through which transmigration occurred. As transmigration was completed, LFA-1 redistributed to the neutrophil uropod. Endothelial ICAM-1 and JAM-A both colocalized with the ringlike LFA-1 cluster. Further analysis of PMA-stimulated neutrophils, which increase mobility of LFA-1, showed a rapid redistribution of LFA-1 and ICAM-1, but not endothelial JAM-A. Thus, endothelial JAM-A does not appear to contribute to adhesion or transmigration in this system. This is the first demonstration that neutrophil LFA-1 rapidly redistributes to form a ringlike structure that coclusters with endothelial ICAM-1 as the neutrophil transmigrates.
Keywords: inflammation, diapedesis, integrins, adhesion molecules, imaging
Introduction
A critical event during an inflammatory response is the recruitment of blood leukocytes to the site of injury, immune response, or infection. This process involves a sequential, multistep adhesion cascade between leukocyte and endothelial cell adhesion molecules that mediates leukocyte rolling, firm adhesion, and transmigration of the endothelium (1). Most in vitro experimental systems have found that the majority of transmigration events occur at endothelial junctions (2, 3). Transmigrating leukocytes encounter multiple junctional molecules and molecular complexes. CD99 and PECAM-1 are implicated in the transmigration of some but not all leukocyte types (4). Several groups have also examined how endothelial cell adherens junctions and tight junctions regulate passage of blood leukocytes (for review see reference 5). More recently, our laboratory showed that adherent neutrophils and monocytes triggered focal gaps in VE-cadherin-GFP (VE-cadGFP) through which the leukocytes migrated (3, 6). Transmigration also occurred at sites of small preexisting gaps in VE-cadGFP, which confirmed a previous paper (7, 8). These findings provide a mechanism for trafficking through endothelial adherens junctions and raise the question of how other molecules on both leukocytes and endothelial cells behave during transmigration.
Although previous in vivo and in vitro studies have revealed a critical role for the LFA-1–intercellular adhesion molecule (ICAM)-1 ligand pair in leukocyte transmigration (9–11), little is known concerning the physical location of this adhesion pair during leukocyte transmigration. Here, we investigated the distribution of neutrophil LFA-1 and endothelial ICAM-1 in leukocyte transmigration by directly visualizing these molecules in live cells under flow. These techniques preserve the temporal sequence of events and the spatial organization that can otherwise become altered in tissues subjected to fixation, permeabilization, and staining with antibodies. This strategy also avoids postfixation proteolysis of susceptible endothelial junctional proteins. This paper is the first demonstration of changes in leukocyte LFA-1 and endothelial cell ICAM-1 distribution during neutrophil transmigration using live cell imaging. Our data show that neutrophil LFA-1 undergoes rapid and dynamic redistribution during the process of transmigration. LFA-1 is rapidly redistributed to the site of contact at endothelial cell junctions, where it forms a distinct ringlike cluster. Endothelial ICAM-1 also becomes enriched at the LFA-1 rings, and this LFA-1–ICAM-1 cluster remains around the neutrophil as it transmigrates. Our data suggest that the changes in LFA-1 distribution reflect the molecular changes in LFA-1 affinity and avidity known to occur in integrin activation, and adhesion to ligands such as ICAM-1 (12–14), and represent their physiological correlates during transmigration.
Materials and Methods
Materials.
DPBS (Dulbecco's PBS) with Ca2+ and Mg2+ (DPBS+) or without (DPBS−) and M199 were purchased from BioWhittaker Bioproducts. Recombinant hTNF-α was purchased from Genzyme. The working solution that gave the maximal response (25 ng/ml) contained <10 pg/ml of endotoxin (3). Human serum albumin (25% albuminate) was obtained from Baxter Healthcare Corp. All other chemicals were of the highest grade available from Baker Chemical.
Human Cell Culture and Leukocyte Isolation.
Pooled human umbilical vein endothelial cells (HUVECs) were isolated and cultured as described previously (15) with the following modifications: 2 mM glutamax (l-alanyl-l-glutamine; Invitrogen) was substituted for glutamine and culture media (M199) was supplemented with 100 μM ascorbic acid and 100 nM hydrocortisone (Sigma-Aldrich). The latter two components were supplemented to physiological plasma concentrations and improved endothelial cell barrier function (unpublished data). HUVEC monolayers (subculture 2) were allowed to reach confluence and used in experiments 48 h later. Human neutrophils were isolated from anticoagulated whole blood obtained from healthy volunteer donors as described previously (16). The Brigham and Women's Hospital Human Use Committee Board have approved all protocols involving human subjects.
mAbs.
The following mAbs have been reported previously and were used as purified IgG:nonblocking anti–JAM-A (1H2A9; IgG1; reference 17); nonblocking anti–ICAM-1 mAb CL23.4 was provided by S. Ferrone (Roswell Park Cancer Institute, Buffalo, NY; reference 18); nonblocking anti-CD11a/LFA-1 mAb TS2/4 (IgG1) was a gift from L. Klickstein (Brigham and Women's Hospital, Boston, MA; reference 19); anti–MHC-I (W6/32; IgG2a; reference 20); blocking anti–VE-cadherin, clone 75 (Transduction Labs), and nonblocking control VE-cadherin, TEA 1/31 were purchased commercially (Beckman Coulter). 1H2A9, W6/32, CL23.4, and TS2/4 mAb were conjugated to fluorescent Alexa Fluor-488 or Alexa Fluor-568 using protein labeling kits as described by the manufacturer (Molecular Probes). To stain live endothelial cells, confluent HUVEC monolayers were incubated with Alexa-labeled mAb (0.5–2 μg per 25-mm diameter glass coverslip) at 37°C for 5 min, rinsed immediately, and inserted into the flow chamber. This mAb concentration resulted in uniform labeling and had no effect on leukocyte adhesion or transmigration, and did not alter endothelial cell permeability (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040965/DC1). For labeling leukocytes, labeled mAb (1–2 μg/ml) was added to leukocytes (5 × 106 cells/ml), incubated for 10 min at 8°C, and washed once before use.
Endothelial Cell Permeability Assay.
HUVECs were plated onto gelatin-coated Costar transwell filters (2.5 × 104 cells/filter) and grown to confluence (4 d), and the permeability was assessed using FITC-dextran (70 kD; anionic; Molecular Probes) as described previously (15).
Leukocyte Transmigration under Flow.
The parallel plate flow chamber used for leukocyte transmigration assays has been described previously (21). Confluent HUVEC monolayers were treated with 25 ng/ml TNF-α for 4–6 h and inserted into the flow chamber for adhesion assays. Freshly isolated neutrophils (unstained or stained with Alexa dye-labeled mAb) were diluted to 5 × 105 cells/ml and drawn across HUVEC monolayer at 0.25 ml/min (calculated shear stress of 0.5 dynes/cm2), which avoids leukocyte string formation and high density leukocyte adhesion that otherwise hamper detailed analysis (20). The chamber temperature was maintained at 37°C, and transmigration events were monitored using the digital recording system described in the next paragraph.
Live Cell Fluorescent Imaging of Leukocyte–Endothelial Transmigration under Flow.
Live cell imaging was performed using MetaMorph 4.6 software (Universal Imaging Corp.) to control a digital imaging system coupled to an inverted microscope (model TE2000; Nikon). Separate excitation and emission filter wheels (Sutter Instrument Co.) combined with a [Q]polychroic beamsplitter (Chroma Technology Corp.) allow for rapid acquisition of different fluorescence channels. Chroma filters with appropriate excitation and emission maxima were used to eliminate overlap between dyes (excitation 580/20, emission 630/40 for Alexa Fluor-568; excitation 492/18, emission 530/40 for Alexa Fluor-488). Transmitted and fluorescent light illumination was controlled by either a Uniblitz electronic shutter (Vincent Associates) or a shutter built in the excitation filter wheel. A high-sensitivity cooled CCD camera (ORCA-ER; Hamamatsu Co.) acquired images via the MetaMorph software. Live cell imaging under flow was performed with PlanApo 20× or 40× differential interference contrast (DIC) objectives. For time lapse two-color experiments, sequential red, green, and DIC images were collected every 5–15 s from the same field. In single color experiments, only red/green channel and DIC images were collected. Focus was manually maintained at the level of the endothelial junctions. Images were analyzed and processed using MetaMorph software, v4.6, and ImageJ v1.31 (http://rsb.info.nih.gov/ij). Images were subjected to 20 blind deconvolutions (v.9.2.1; AutoQuant Imaging, Inc.). Images presented are representative of multiple experiments on different days with leukocytes isolated from different donors.
Ratio Imaging.
Live cell imaging of neutrophil transmigration through TNF-α–activated HUVECs was performed as described before. For each pair of fluorochromes, backgrounds were digitally subtracted, and signal intensity was normalized on adherent cells before transmigration so that red and green channels had approximately the same maximum and minimum intensity levels. The normalized images were digitally divided using the Metamorph stack arithmetic divide function and the resultant ratio image was multiplied by an arbitrary scalar to ensure nonfractional values. All red, green, or ratio images have been manipulated as a group to allow direct comparison. The linescan data represent average normalized intensity along a five-pixel-wide line between the asterisks shown in the green channel (see Fig. 2). Images are representative of multiple leukocytes.
Figure 2.

LFA-1 ratio imaging. (left) A neutrophil labeled with nonblocking mAb against LFA-1 (red channel) and MHC-I (green channel) at initial adhesion (t = 0) and during transmigration (t = 1:00). (white arrow) The portion of the neutrophil that has penetrated the endothelium. (third row) The digital ratio of red/green channels; (white areas) more LFA-1; (darker areas) more MHC-I. Note the LFA-1 ring visible in the second column ratio image. (right) Comparison of leukocyte JAM-A and MHC-I did not show any specific structures associated with transmigration. (bottom row) A linescan of red and green channels between the points represented by asterisks. Note the peaks of LFA-1 visible where the linescan bisects the ringlike structure (black arrowheads). Bar, 10 μm.
Laser Scanning Confocal Microscopy of Neutrophil Transmigration under Static Conditions.
HUVECs (SC-2) were plated on fibronectin-coated, glass-bottom 35-mm cell culture plates (MatTek) and allowed to grow to confluence. Confluent HUVEC monolayers were stimulated with TNF-α (4–6 h, 25 ng/ml), coincubated with neutrophils (0.5 × 106 cells/ml) under static conditions for 4 min at 37°C, washed to remove nonadherent neutrophils, and fixed for 5 min at room temperature with 4% neutral buffered formalin to avoid movement artifact during confocal scanning. Surface molecules on HUVEC monolayers and neutrophils were immunofluorescently labeled as described before, and neutrophils in the process of transmigration were observed using a laser scanning confocal head (Radiance 2100; Bio-Rad Laboratories) attached to an inverted microscope (model TE2000; Nikon) through a 60× oil immersion objective (PlanApo, 1.4 NA; Nikon). The confocal iris was set to provide a 0.5-μm slice thickness and a transmigration event was scanned along the Z-axis from basolateral to apical aspects in 0.5-μm steps. Serial sections were deconvolved to remove out of focus light (v.9.2.1; AutoQuant Imaging, Inc.). A three-dimensional reconstruction of the scanned volume was obtained and digitally sliced in the XZ plane through the widest point of the region of transmigration to indicate a profile view of the distribution of LFA-I or MHC-I on a transmigrating neutrophil (see Fig. 3). Images were analyzed in Lasersharp 2000 v4.3 (Bio-Rad Laboratories) and ImageJ v1.31 and figures were generated in Photoshop v.7.0.
Figure 3.

Confocal imaging of neutrophils during transmigration. Neutrophils were labeled with nonblocking mAb for LFA-1 (left) or MHC class I (right) and incubated with 4–6-h TNF-α–stimulated HUVECs under static conditions. Cells were formalin (10%) fixed and imaged by confocal microscopy. (top) Profile slices (XZ plane) of transmigrating neutrophils, demonstrating the concentration of LFA-1 at the neutrophil–endothelial cell interface (left) in contrast with the relatively uniform distribution of MHC-I (right). The solid line is positioned along the endothelial cell interface, and the broken line is positioned across the portion of the neutrophil that has yet to transmigrate. The intensity profiles of these lines (bottom) show peaks of high fluorescence intensity corresponding to LFA-1 concentration at the site of transmigration along the endothelial surface. LFA-1 on the surface of the nontransmigrated portion of the neutrophil (broken line) shows low intensity peaks. In contrast, MHC class I distribution (right) shows similar peaks corresponding to the neutrophil surface at both the endothelial cell interface (solid line) and the nontransmigrated portion (broken line). Bar, 5 μm.
Quantification of JAM-A, ICAM-1, and MHC-I under PMA-activated Neutrophils.
Neutrophils were activated with 100 ng/ml PMA (5 min at 37°C), washed once in flow buffer, and resuspended in flow buffer without PMA. Activated neutrophils were allowed to interact with TNF-α–treated HUVECs in a flow chamber and imaged as described before. For endothelial JAM-A, ICAM-1, or MHC-I imaging, red and green channels were normalized as described before and overlaid with DIC. A mask corresponding to the DIC outline of each adherent neutrophil in a field of view was generated. This was used to quantify the red and green fluorescence associated with adherent neutrophils. Neutrophil-associated fluorescence was divided by the total fluorescence in a field of view to obtain a percent value. Data are representative of three independent experiments.
Online Supplemental Material.
Fig. S1 documents that the JAM-A mAb does not alter neutrophil adhesion or transmigration, nor affect endothelial monolayer permeability. Figs. S2 and S3 are additional examples of neutrophil LFA-1 redistribution and ICAM-1 redistribution during neutrophil transmigration, respectively. The accompanying videos show the behavior of ICAM-1 and LFA-1 during transmigration. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040965/DC1.
Results
Live Cell Time Lapse Visualization of LFA-1 Revealed Redistribution and Accumulation around the Site of Transmigration.
LFA-1 dynamics in live neutrophils were monitored by immunolabeling with a fluorescent-tagged nonblocking anti-LFA-1 mAb, TS 2/4. Neutrophils were perfused across TNF-α–activated HUVEC monolayers under defined shear flow using the live-cell, time lapse microscopy system described in Materials and Methods. In this system, typically 50% of adherent neutrophils transmigrated over a time period of several minutes. Analysis from paired DIC and fluorescence images from multiple experiments using neutrophils and HUVECs from multiple donors was performed. Fig. 1 demonstrates the typical sequence of events during neutrophil transmigration. Neutrophils briefly roll and come to a stable arrest at, or very close to, an endothelial junction, in agreement with previous papers (22). The time lapse acquisition system used here was not suitable to resolve the fast motion of rolling and, hence, we have focused on the events that follow at and after stable arrest (Fig. 1, t = 0:00). During this period of early adhesion, LFA-1 was relatively evenly distributed on the neutrophil surface; although some unevenness in LFA-1 distribution was observed, no consistent accumulations or structures were apparent at this early time point. On leukocytes that were subsequently going to transmigrate, a small cluster of LFA-1 appeared, apparently at the contact surface of the neutrophil with the endothelium (Fig. 1, outline column, t = 0:20, arrow, and see Fig. 3; elements of this image may be better appreciated when viewed on screen in the online version, where subtle differences in fluorescence intensity are more visible). This cluster grew in intensity as LFA-1 was depleted from the rest of the leukocyte surface to contribute to it (Fig. 1, t = 0:35 and 0:45, arrows). The center portion of the LFA-1 cluster appeared to be relatively deficient in LFA-1, leading to a ringlike appearance. These rings were not always complete, but at least partial rings were observed in >95% of transmigration events (see Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20040965/DC1, for several examples from a single coverslip).
Figure 1.
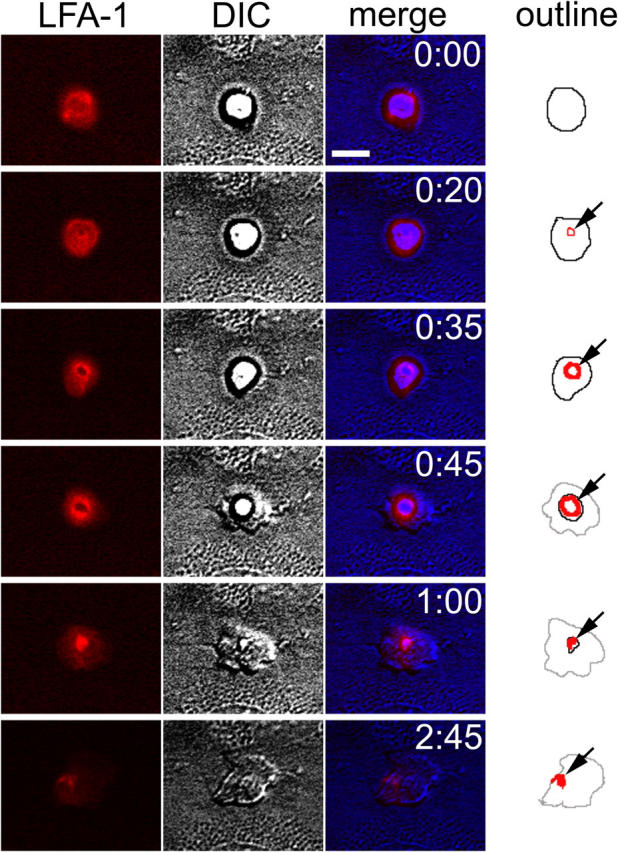
LFA-1 redistribution during neutrophil transmigration. Time lapse imaging of neutrophils transmigrating through 4–6-h TNF-α–activated HUVECs. (first column, red) LFA-1 distribution shown by fluorochrome-conjugated nonblocking mAb; (second column, gray) matched DIC images; (third column) overlay of fluorescence and DIC (DIC channel has been digitally mapped to the blue channel, to ensure clear separation from red); and (fourth column) the outline of significant features in fluorescence and DIC channels. (black line) The portion of the neutrophil above the endothelium; (gray lines) transmigrated portion of neutrophil under the endothelium; and (red lines) LFA-1 relocalization. Elements of this image may be better appreciated when viewed on screen in the online version, where subtle differences in fluorescence intensity are more visible. Time is shown in min:s. Bar, 10 μm.
As neutrophil transmigration progressed, a pseudopod was extended through the LFA-1 ring like structure and through the junction of tightly apposed endothelial cells (t = 0:35 and 0:45). The neutrophil cell body squeezed through the LFA-1 ring, which grew in diameter to accommodate it. The ring remained at the level of the endothelial surface, even while the bulk of the neutrophil had transmigrated through it. Notably, little LFA-1 staining was detected on the pseudopod thrust beneath the endothelium as compared with MHC-I (Figs. 1 and 2). Eventually, the bulk of the neutrophil passed through the endothelial junction, and the LFA-1 ring condensed to a small bright patch at its tail, or uropod (Fig. 1, t = 1:00, arrow). This uropodal accumulation of LFA-1 persisted even after transmigration was complete, and the neutrophil was migrating under the endothelium, away from the site of transmigration (Fig. 1, t = 2:45, arrow).
As a control, next we examined the behavior of leukocyte MHC-I and JAM-A, neither of which are thought to be involved in transmigration (20, 23). Redistribution to a ringlike structure during transmigration was specific for LFA-1; neither neutrophil MHC-I nor JAM-A showed a similar behavior (Fig. 2). Early after adhesion, LFA-1 colocalized with MHC-I over the surface of the neutrophil (Fig. 2, t = 0, left). 1 min later, the neutrophil had partly transmigrated beneath the endothelium (Fig. 2, t = 1:00, arrow). As before, LFA-1 was now largely concentrated in the ringlike structure, and was largely depleted from the rest of the cell body above or beneath the endothelium. In contrast, MHC-I was more evenly distributed over the penetrating pseudopod and also over the apical portion that had yet to pass through. Although a slight ring of MHC-I was visible, this was variable and relatively much less visible than the redistribution of LFA-1 as quantified by image analysis (Fig. 2, bottom two rows, linescans). This weak accumulation of MHC-I may be due to membrane ruffling or microdomain formation at the site of transmigration. Another possibility is that the apparent accumulation of MHC-I was partly due to tangential viewing of the leukocyte edge from the microscope objective below. To confirm that the LFA-1 ring represented a specific clustering rather than an edge effect, ratio imaging of LFA-1 and MHC-I was performed. Here, the brightness value of the LFA-1 signal was divided by that of MHC-I for every pixel. At any location where LFA-1 was brighter, the corresponding ratio image pixel showed a bright signal. Where MHC-I was brighter, the ratio image showed a darker signal. At t = 0, ratio imaging confirmed uniform distribution of LFA-1. But during transmigration, LFA-1 was concentrated in a ring shape relative to MHC-I. This observation is graphically shown in the line trace (Fig. 2, bottom). The pixel intensity of a line between the asterisks was plotted against the position. At t = 0, the LFA-1 and MHC-I traces largely overlap. However, during transmigration, LFA-1 forms two peaks corresponding to where the line intersects the LFA-1 ring. Quantitatively, MHC-I rings are less distinct. Leukocyte JAM-A and MHC-I were also compared during early adhesion (Fig. 2, right). Both molecules appeared to be evenly distributed before and during transmigration by direct imaging as well as by quantitative analysis (t = 0 and 1:00). Thus, the LFA-1 ring structure described previously is specific.
To more directly visualize LFA-1 redistribution in comparison with control MHC-I, serial Z sections of fixed neutrophils in the process of transmigration were acquired using confocal microscopy. A vertical slice of two representative transmigrating neutrophils are shown in Fig. 3. In the bottom panel, two line traces are shown for each molecule. One line shows intensity through the cell body (Fig. 3, dashed line, noncontact region), and the second line runs through the contact surface (Fig. 3, solid line). LFA-1 was enriched severalfold at the contact surface with the endothelial monolayer, whereas MHC-I remained evenly distributed over the neutrophil surface.
Endothelial ICAM-1 Redistributes under the LFA-1 Ring.
Endothelial cell ICAM-1 is essential for neutrophil arrest and transmigration (for review see reference 1). From recent studies, ICAM-1 has been shown to form “docking structures” (endothelial microvilli enriched in ICAM) that appear to surround and engulf adherent T lymphoblasts (24, 25). Here, we tested whether neutrophils form similar structures in a model of robust transmigration under flow. TNF-α–induced HUVECs were immunolabeled with directly conjugated nonblocking anti–ICAM-1 mAb CL23.4 (18). LFA-1–immunolabeled neutrophils were perfused across the endothelium, and two-color live cell, time lapse immunofluorescence was performed. A typical sequence of events is depicted in Fig. 4. ICAM-1 is significantly up-regulated on HUVECs after 4-h treatment with TNF-α and is evenly distributed across much of the apical surface, but appears to be somewhat excluded from the junctional area (Fig. 4, t = 0:00; note broken white line in merge panel where ICAM-1 redistribution will occur later), in keeping with previous papers (26). Neutrophils come to stable arrest at or close to endothelial junctions (t = 0:50). As seen before, LFA-1 remains relatively evenly distributed across the leukocyte surface, and ICAM-1 distribution remains unchanged. The earliest sign of LFA-1 redistribution is usually accompanied by a gap in the ICAM-1 staining (Fig. 4, arrow, t = 1:15). During transmigration, as the LFA-1 ring grows in diameter and intensity, ICAM-1 also tends to show some relocalization to surround the site of penetration (Fig. 4, t = 1:55). Compared with leukocyte LFA-1, the redistribution of ICAM-1 is less robust. Nevertheless, there was frequently appreciable codistribution between the LFA-1 ring and the underlying ICAM-1 (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20040965/DC1). The ICAM-1 staining closes around the neutrophil when transmigration is complete. Interestingly, when the neutrophil is migrating beneath the endothelium, there is no colocalization between uropodal LFA-1 and endothelial ICAM-1, suggesting that LFA-1 is not binding to ICAM-1 ligand at this stage and that other integrins are involved. Confocal microscopy corroborated the clustering of ICAM-1 during transmigration (Fig. 4 B) and, interestingly, showed no evidence of ICAM-1–rich microvilli engulfing the leukocyte as reported recently (25).
Figure 4.
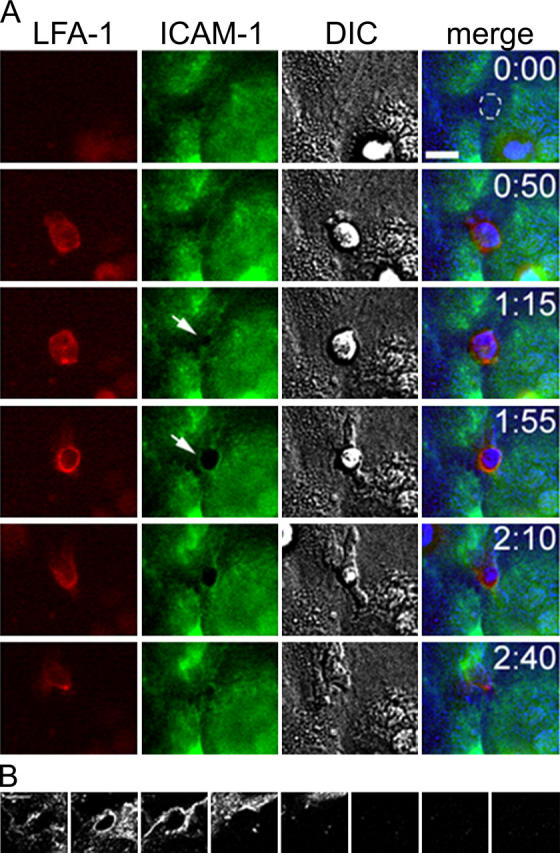
ICAM-1 redistribution during neutrophil transmigration. (A, red channel) Neutrophil LFA-1; (green channel) endothelial ICAM-1; and (gray channel) DIC. In the merged image, DIC images have been assigned to the blue channel to avoid overlap with red and green channels. At t = 0:00, ICAM-1 is largely absent from the junctional region, and the position of the future ICAM-1 clustering (from t = 1:55) has been marked with a broken white line. At t = 1:15, white arrow shows gap in ICAM-1 corresponding to newly formed LFA-1 ring. At t = 1:55, ICAM-1 redistribution is marked by white arrow. Bar, 10 μm. (B) Representative confocal scan of endothelial ICAM-1 during PMN transmigration shows the localization of ICAM-1 in a thin section. The endothelial monolayer was scanned in 0.5-μm steps starting at the basolateral plane and progressing up through the apical surface and beyond (left to right). Confocal images demonstrate the clustering of ICAM-1 during transmigration in a region ∼1 μm in thickness (starting from left, panels 2 and 3). Bar, 5 μm.
Endothelial JAM-A Also Redistributes with ICAM-1 under the LFA-1 Ring.
Because LFA-1 is reported to recognize endothelial JAM-A, and antibodies against JAM-A block a small but significant proportion of T cell transmigration (23), we monitored endothelial ICAM-1 and JAM-A simultaneously with directly conjugated nonblocking mAb using two-color time lapse, live cell immunofluorescence (Fig. 5). As reported previously, JAM-A was enriched at endothelial junctions in our assay system (15). During neutrophil transmigration, this junctional JAM-A was redistributed around the site of penetration, forming a complete or partial ring that colocalized with the ICAM-1 ring (Fig. 5, t = 2:30, arrows). Thus, both JAM-A and ICAM-1 redistribute during neutrophil transmigration. However, these data do not indicate whether neutrophil LFA-1 actually engages endothelial JAM-A during the process of transmigration.
Figure 5.
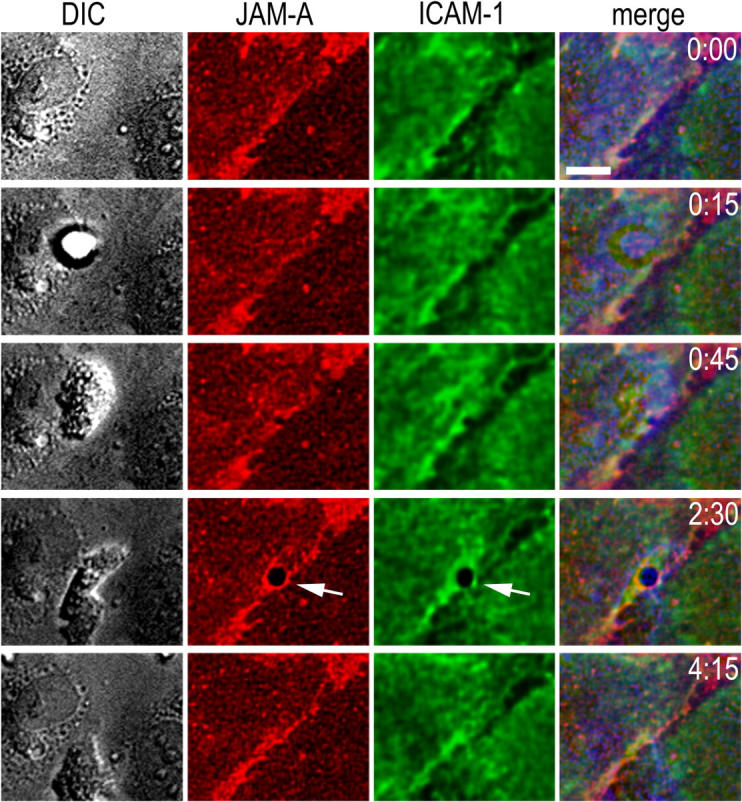
Endothelial JAM-A redistribution during neutrophil transmigration. (gray channel) DIC images; (red channel) endothelial JAM-A; and (green channel) ICAM-1. As in Fig. 4, in the merged images DIC has been assigned to the blue channel to avoid overlap. JAM-A and ICAM-1 redistribution is shown by white arrows at t = 2:30. Bar, 10 μm.
PMA-activated Leukocytes Cause Redistribution of ICAM-1 But Not JAM-A.
We demonstrate in the previous paragraphs that neutrophil LFA-1 redistributes around the site of neutrophil penetration, along with both ICAM-1 and JAM-A at the endothelial junction. A large increase in mobility of LFA-1 has been documented after PMA activation of lymphoblasts (12) and after IL-8 stimulation of neutrophils (27). Although ICAM-1 is a well-characterized ligand for LFA-1 and plays a critical role in neutrophil adhesion and transmigration during inflammation (9, 10, 28), much less information is available for JAM-A. Recently, endothelial JAM-A was also reported to be a ligand for LFA-1 on T cells (23). To test whether activated LFA-1 itself redistributes or causes redistribution of its ligands in the current system, PMA activation was used (see Materials and Methods for activation protocol).
Untreated neutrophils came to a stable arrest at or near the junctions of TNF-α–treated endothelium and transmigrated the endothelium (∼50% of adherent cells transmigrated). In sharp contrast, PMA-treated neutrophils adhered randomly across the endothelium, but failed to transmigrate. Like untreated neutrophils, PMA-treated neutrophils showed relatively even distribution of LFA-1 upon early adherence (unpublished data). However, if the microscope objective was focused on the adherent neutrophils at the neutrophil–endothelial cell interface, one could observe that over a time frame of a few minutes, LFA-1 clustered at the contact surface with the endothelium (Fig. 6, left column). This clustering took the form of a broad patch with indistinct outlines; a few puncta of brighter fluorescence were visible, but ringlike structures were never observed in our studies.
Figure 6.

PMN-activated neutrophils cause LFA-1 and ICAM-1 redistribution. Neutrophils were pretreated with 100 ng/ml PMA as described in Materials and Methods and allowed to interact with 4–6-h TNF-α–activated HUVECs under flow. (left) Two-color imaging of LFA-1 and ICAM-1; (middle) endothelial JAM-A and ICAM-1; and (right) endothelial JAM-A and MHC class I. To compare JAM-A and ICAM-1 fluorescence, neutrophil-associated red and green fluorescence from 365 adherent leukocytes was quantified and compared with the total fluorescence of the field of view to obtain a normalized intensity percent value. For JAM-A and MHC-I, 378 adherent neutrophils were quantified. Statistical significance was calculated using a paired Student's t test. Bar, 10 μm.
Interestingly, ICAM-1 also showed clustering under the flattened, PMA-activated neutrophils. To measure the specificity of ligand clustering, the studies were repeated using the two-color live cell immunofluorescence system. Endothelial cells were labeled with directly conjugated anti–ICAM-1 or anti–JAM-A and anti–MHC-I, using nonoverlapping fluorochromes. Neutrophil adherence caused a slight but distinct concentration of ICAM-1, but not JAM-A or MHC-I (Fig. 6). The degree of fluorescence concentration relative to the fluorescence of the entire field of view was quantified for the JAM-A/ICAM-1 or JAM-A/MHC-I combinations. Neither JAM-A nor MHC-I showed a significant increase under PMA-treated neutrophils, as compared with the overall levels, but ICAM-1 showed a statistically significant enrichment (P < 0.0001). The magnitude of this ICAM-1 enrichment was ∼20% greater that MHC-I or JAM-A. Compared with LFA-1 clustering on the leukocyte, the relatively weak ICAM-1 enrichment even after this strong stimulus may correlate with the variable appearance of ICAM-1 relocalization during transmigration in this system.
Discussion
This paper describes the clustering of neutrophil LFA-1 into a ringlike structure at the contact surface with endothelial cells, through which diapedesis occurs. On the endothelial side, ICAM-1 also clustered around the site of transmigration, although this clustering was less robust. To resolve the temporal and spatial dynamics of this process, we used live cell epifluorescence imaging with nonblocking antibodies directed toward neutrophil LFA-1 and endothelial ICAM-1. The sequence of events is depicted in Fig. 7. Neutrophils roll on the endothelium and come to firm adherence at or very near endothelial junctions in this model (steps 1 and 2), in agreement with prior studies (22). At this phase, LFA-1 is relatively evenly distributed on the neutrophil, ICAM-1 is expressed on the endothelial apical surface, and endothelial JAM-A is diffuse and junctional. LFA-1 appears to concentrate at the contact surface with endothelium, forming a small ringlike structure (step 3). This ring grows in diameter and intensity, whereas LFA-1 from the rest of the neutrophil is recruited toward it, and a pseudopod is inserted between the endothelial junctions as transmigration begins (step 4). Interestingly, little if any LFA-1 was detected on the pseudopod thrust between the two tightly apposed endothelial cells. On the endothelial side, a roughly matching ICAM-1 cluster is formed around the site of penetration (not depicted for clarity). The neutrophil cell body squeezes across the cell border at the ring, and LFA-1 eventually condenses into the uropod, before transmigration is quite complete (Figs. 1 and 4). Lastly, the neutrophil retracts its uropod under the endothelium, and migrates away from the junction, with LFA-1 remaining in the uropod (step 6). Concomitantly, the ICAM-1 clustering on the endothelial cell dissipates.
Figure 7.

Schematic outline of neutrophil transmigration through TNF-α–stimulated HUVECs. (top) Side view and (bottom) bottom view as might be seen through inverted microscope. Neutrophil is shown as a pink ball with LFA-1 as the red outline. Endothelial cells are shown as flattened pale green shapes with ICAM-1 shown as a dark green outline.
Redistribution of LFA-1 into a ringlike structure appears to be necessary for leukocyte transmigration as it was always observed in transmigrating neutrophils, but not in cells that remained stably adhered to the apical endothelial surface. In addition, PMA-stimulated neutrophils, adherent to the endothelial surface, exhibited LFA-1 clustering only and did not transmigrate or form ringlike LFA-1 structures. It is likely that the LFA-1 ring represents a population of active integrin molecules because of the redistribution and enrichment of its endothelial ligand ICAM-1, in close apposition. Changes in affinity and avidity of LFA-1 have been documented in different systems, typically studying adhesion to purified ligands or endothelial cells (12, 14; for review see references 13, 27). In T cells, PKC-β (28), the small GTPase Rap1, and its effector RapL (29–31) can regulate cellular morphology and the surface distribution of LFA-1. No such studies have been performed in neutrophils to date. By extension, reorganization of LFA-1 into a ring has been described during the formation of the T cell–APC immune synapse (13), although the time scale and functional significance of immune synapse formation and the neutrophil transmigration are distinct. Nevertheless, similar mechanisms may potentially account for LFA-1 redistribution in both cases, and similar downstream signaling events may occur as a consequence. Additional studies are necessary to dissect whether the aforementioned molecules and/or other pathways such as the phosphatidyl-inositol 3-kinase (14) control neutrophil LFA-1 during transmigration.
The rapid redistribution of surface LFA-1 described herein most likely represents changes of LFA-1 affinity and avidity in neutrophils undergoing robust and active transmigration through TNF-α–activated endothelial cells. Functionally, the LFA-1 ring interacting with ICAM-1 may provide a stable foundation from which neutrophil pseudopods erupt to probe the endothelial cell–cell junctions. By extension, LFA-1 redistribution into focal ring structures (but not to pseudopods projected beneath the endothelium) may also serve to direct leukocyte actomyosin transmigration machinery toward the endothelium, resulting in the propulsion of the neutrophil through the monolayer. Previous reports have discussed mechanisms to differentiate cell movement on a flat surface versus a three-dimensional matrix and proposed an integrin-independent means of locomotion in restricted spaces termed “chimneying” (32). Our observations extend this discussion in the context of neutrophil transmigration by arguing for the establishment of a highly adhesive “ring” of contact at the site of transmigration. The redistribution of LFA-1 and ICAM-1 could function as an anchor or fulcrum used in the movement of a transmigrating neutrophil across the endothelium, notably in a direction perpendicular to any prevailing shear forces exerted by blood flow. An additional function of such adhesion molecule ringlike structures may involve the retention of monolayer seal between the transmigrating leukocyte and endothelium to prevent loss of barrier function. Indeed, prior reports using in vivo (33) and in vitro (7, 34) models have suggested that little change occurs in vessel and endothelial cell barrier function during transmigration of neutrophils. It will be of interest to study whether other leukocyte types use LFA-1–ICAM-1 or other adhesion molecule combinations (e.g., VLA-4–VCAM-1) in ringlike structures to transmigrate across endothelium.
Both potential ligands for LFA-1, endothelial ICAM-1 and JAM-A, colocalized under the LFA-1 ringlike structure. However, ICAM-1 appeared to be actively recruited toward the junctions because it was not enriched at junctions before transmigration. In contrast, JAM-A is constitutively present at junctions, and appeared to remain localized around the penetrating neutrophil pseudopod, contrary to the behavior of another junctional molecule, VE-cadherin (3, 6). Both ICAM-1 and JAM-A may potentially interact with LFA-1; however, our attempts to manipulate JAM-A function by antibody blockade or adenoviral overexpression did not significantly change the rate or extent of neutrophil transmigration under flow (unpublished data). To gain further insight into this process, we used PMA-activated neutrophils. PMA treatment is known to dramatically increase LFA-1 affinity and avidity (12). Significantly, activation of neutrophils by PMA did recruit endothelial cell ICAM-1, but not JAM-A, to the site of adhesion. This result suggests that it is a change in the LFA-1 activation state that triggers subsequent changes in ICAM-1. Furthermore, the PMA activation data suggest either that JAM-A is not a LFA-1 ligand or that there is some other difference in activation or signaling that occurs during transmigration but not during PMA-triggered adhesion. Thus, the role for JAM-A in neutrophil transmigration remains unclear at this time.
Previous studies have characterized the behavior of endothelial and leukocyte adhesion molecules during adhesion. ICAM-1 and VCAM-1 have been reported to cluster with cytosolic linker proteins in docking structures that enveloped leukocytes and leukocyte cell lines after prolonged adhesion (24). In another paper, ICAM-1 clustered in actin-rich microvilli-like membrane projections that colocalized with LFA-1 during leukocyte adhesion (25). These authors speculated that docking structures mediate leukocyte engulfment by the endothelial cell, although a direct association with leukocyte transmigration was not established. In our assay system, we focused on neutrophils actively transmigrating at junctions of endothelial cells rather than adherent leukocytes, transfected cell lines, and exogenously added chemoattractants. Strikingly, we did not observe leukocyte engulfment by ICAM-1–containing docking structures or microvilli-like projections containing ICAM-1 emanating from endothelium during transmigration at junctions of 4-h TNF-α–activated HUVECs. Rather, we observed the coordinated remodeling of both leukocyte and endothelial adhesion molecules within a narrow plane at endothelial cell–cell junctions (Fig. 4 B). Other likely explanations for these differences reside in the use of the chemoattractant, platelet-activating factor in the latter study and the use of T lymphoblasts in the former study, which have a low rate of transmigration relative to neutrophils and a prolonged mobility on the apical surface that precedes transmigration. Whether or not the docking structure and the LFA-1–ICAM-1 transmigration complex are functionally related, the extensive cellular and molecular remodeling that occurs in both would imply an intimate cross talk between both cell types during transmigration.
In summary, the current findings show that neutrophils transiently form a ringlike cluster of LFA-1 on the leukocyte contact surface with the endothelium as they rapidly transmigrate. Simultaneously, an ICAM-1 cluster forms in close apposition on the endothelial surface. It is possible that other endothelial and leukocyte surface molecules are also recruited to these clusters. We hypothesize that this complex serves to orchestrate the endothelial and leukocyte adhesion molecules necessary for the intricate process of transmigration to occur.
Acknowledgments
The authors thank T. Betz for assistance in transmigration assays; the Center for Newborn at the Brigham and Women's Hospital and the Birthing unit at South Shore Hospital for providing umbilical cords; and K. Case, V. Davis, and D. Lamont for well-characterized cultures of HUVECs. The authors gratefully acknowledge technical assistance of C. Felts of Research Precision Instruments in the setup of the MetaMorph imaging system; our colleagues Drs. R. Lee and H. Huang at Brigham and Women's Hospital for assistance with the deconvolution software; our colleagues Drs. A.H. Lichtman (Brigham and Women's Hospital) and R. Alon (Weizmann Institute of Science) for many thoughtful and helpful discussions during the course of these studies.
This work was supported by National Institutes of Health grant nos. HL-53995 (to F.W. Luscinskas); DK62894 (to Y. Liu); and HL54229, DK61379, HL58467, DK64399, and HL72124 (to C.A. Parkos).
The authors have no conflicting financial interests.
S.K. Shaw and S. Ma contributed equally to this work.
Abbreviations used in this paper: DIC, differential interference contrast; HUVEC, human umbilical vein endothelial cell; ICAM, intercellular adhesion molecule.
References
- 1.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 2.Luscinskas, F.W., S. Ma, A. Nusrat, C.A. Parkos, and S.K. Shaw. 2002. The role of endothelial cell lateral junctions during leukocyte trafficking. Immunol. Rev. 186:57–67. [DOI] [PubMed] [Google Scholar]
- 3.Shaw, S.K., P.S. Bamba, B.N. Perkins, and F.W. Luscinskas. 2001. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J. Immunol. 167:2323–2330. [DOI] [PubMed] [Google Scholar]
- 4.Muller, W.A. 2003. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 24:327–334. [DOI] [PubMed] [Google Scholar]
- 5.Luscinskas, F.W., S. Ma, A. Nusrat, C.A. Parkos, and S.K. Shaw. 2002. Leukocyte transendothelial migration: a junctional affair. Semin. Immunol. 14:105–113. [DOI] [PubMed] [Google Scholar]
- 6.Allport, J.R., W.A. Muller, and F.W. Luscinskas. 2001. Monocytes induce reversible focal changes in vascular endothelial cadherin complex during transendothelial migration under flow. J. Cell Biol. 148:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns, A.R., D.C. Walker, E.S. Brown, L.T. Thurmon, R.A. Bowden, C.R. Keese, S.I. Simon, M.L. Entman, and C.W. Smith. 1997. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J. Immunol. 159:2893–2903. [PubMed] [Google Scholar]
- 8.Burns, A.R., R.A. Bowden, S.D. MacDonell, D.C. Walker, T.O. Odebunmi, E.M. Donnachie, S.I. Simon, M.L. Entman, and C.W. Smith. 2000. Analysis of tight junctions during neutrophil transendothelial migration. J. Cell Sci. 113:45–57. [DOI] [PubMed] [Google Scholar]
- 9.Smith, C.W., S.D. Marlin, R. Rothlein, C. Toman, and D.C. Anderson. 1989. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Invest. 83:2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding, Z.M., J.E. Babensee, S.I. Simon, H. Lu, J.L. Perrard, D.C. Bullard, X.Y. Dai, S.K. Bromley, M.L. Dustin, M.L. Entman, et al. 1999. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163:5029–5038. [PubMed] [Google Scholar]
- 11.Henderson, R.B., L.H.K. Lim, P.A. Tessier, F.N.E. Gavins, M. Mathies, M. Perretti, and N. Hogg. 2001. The use of lymphocyte function-associated antigen (LFA)-1–deficient mice to determine the role of LFA-1, Mac-1, and α4 integrin in the inflammatory response of neutrophils. J. Exp. Med. 194:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kucik, D.F., M.L. Dustin, J.M. Miller, and E.J. Brown. 1996. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J. Clin. Invest. 97:2139–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dustin, M.L., T.G. Bivona, and M.R. Philips. 2004. Membranes as messengers in T cell adhesion signaling. Nat. Immunol. 5:363–372. [DOI] [PubMed] [Google Scholar]
- 14.Lum, A.F.H., C.E. Green, G.R. Lee, D.E. Staunton, and S.I. Simon. 2002. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J. Biol. Chem. 277:20660–20670. [DOI] [PubMed] [Google Scholar]
- 15.Shaw, S.K., B.N. Perkins, Y.C. Lim, Y. Liu, A. Nusrat, F.J. Schnell, C.A. Parkos, and F.W. Luscinskas. 2001. Reduced expression of junctional adhesion molecule and platelet/endothelial cell adhesion molecule-1 (CD31) at human vascular endothelial junctions by cytokines tumor necrosis factor-alpha plus interferon-gamma Does not reduce leukocyte transmigration under flow. Am. J. Pathol. 159:2281–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allport, J.R., H. Ding, A. Ager, D.A. Steeber, T.F. Tedder, and F.W. Luscinskas. 1997. L-selectin shedding does not regulate human neutrophil attachment, rolling, or transmigration across human vascular endothelium in-vitro. J. Immunol. 158:4365–4372. [PubMed] [Google Scholar]
- 17.Barton, E.S., J.C. Forrest, J.L. Connolly, J.D. Chappell, Y. Liu, F.J. Schnell, A. Nusrat, C.A. Parkos, and T.S. Dermody. 2001. Junction adhesion molecule is a receptor for reovirus. Cell. 104:441–451. [DOI] [PubMed] [Google Scholar]
- 18.Matsui, M., M. Temponi, and S. Ferrone. 1989. Characterization of a monoclonal antibody-defined human melanoma-associated antigen susceptible to induction by immune interferon. J. Immunol. 139:2088–2095. [PubMed] [Google Scholar]
- 19.Klickstein, L.B., and T.A. Springer. 2004. Adhesion structure subpanel 1, E rosetting/GPI anchor: CD2, CD48, CD55, CD58, CD59, CD99, and CDw108. Leukocyte Typing V. White Cell Differentiation Antigens. S.F. Schlossman and L. Boumsell, editors. Oxford University Press, New York, NY. 1468–1472.
- 20.Lim, Y.C., K. Snapp, G.S. Kansas, R. Camphausen, H. Ding, and F.W. Luscinskas. 1998. Important contributions of P-selectin glycoprotein ligand-1-mediated secondary capture to human monocyte adhesion to P-selectin, E-selectin, and TNF-alpha-activated endothelium under flow in vitro. J. Immunol. 161:2501–2508. [PubMed] [Google Scholar]
- 21.Luscinskas, F.W., G.S. Kansas, H. Ding, P. Pizcueta, B.E. Schleiffenbaum, T.F. Tedder, and M.A. Gimbrone Jr. 1994. Monocyte rolling, arrest and spreading on IL-4-activated vascular endothelium under flow is mediated via sequential action of L-selectin, β1-integrins, and β2-integrins. J. Cell Biol. 125:1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gopalan, P.K., A.R. Burns, S.I. Simon, S. Sparks, L.V. Mcintire, and C.W. Smith. 2000. Preferential sites for stationary adhesion of neutrophils to cytokine-stimulated HUVEC under flow conditions. J. Leukoc. Biol. 68:47–57. [PubMed] [Google Scholar]
- 23.Ostermann, G., K.S. Weber, A. Zernecke, A. Schroder, and C. Weber. 2002. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat. Immunol. 3:151–158. [DOI] [PubMed] [Google Scholar]
- 24.Barreiro, O., M. Yanez-Mo, J.M. Serrador, M.C. Montoya, M. Vicente-Manzanares, R. Tejedor, H. Furthmayr, and F. Sanchez-Madrid. 2002. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol. 157:1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carman, C.V., C.D. Jun, A. Salas, and T.A. Springer. 2003. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J. Immunol. 171:6135–6144. [DOI] [PubMed] [Google Scholar]
- 26.Bradley, J.R., and J.S. Pober. 1996. Prolonged cytokine exposure causes a dynamic redistribution of endothelial cell adhesion molecules to intercellular junctions. Lab. Invest. 75:463–472. [PubMed] [Google Scholar]
- 27.Carman, C.V., and T.A. Springer. 2003. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr. Opin. Cell Biol. 15:547–556. [DOI] [PubMed] [Google Scholar]
- 28.Volkov, Y., A. Long, S. McGrath, E.D. Ni, and D. Kelleher. 2001. Crucial importance of PKC-beta(I) in LFA-1-mediated locomotion of activated T cells. Nat. Immunol. 2:508–514. [DOI] [PubMed] [Google Scholar]
- 29.Katagiri, K., M. Shimonaka, and T. Kinashi. 2004. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-gamma1. J. Biol. Chem. 279:11875–11881. [DOI] [PubMed] [Google Scholar]
- 30.Shimonaka, M., K. Katagiri, T. Nakayama, N. Fujita, T. Tsuruo, O. Yoshie, and T. Kinashi. 2003. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J. Cell Biol. 161:417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katagiri, K., A. Maeda, M. Shimonaka, and T. Kinashi. 2003. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 4:741–748. [DOI] [PubMed] [Google Scholar]
- 32.Malawista, S.E., and C.A. De Boisfleury. 1997. Random locomotion and chemotaxis of human blood polymorphonuclear leukocytes (PMN) in the presence of EDTA: PMN in close quarters require neither leukocyte integrins nor external divalent cations. Proc. Natl. Acad. Sci. USA. 94:11577–11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng, M., H. Zhang, C. Lowell, and P. He. 2002. Tumor necrosis factor-alpha-induced leukocyte adhesion and microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 283:H2420–H2430. [DOI] [PubMed] [Google Scholar]
- 34.Huang, A.J., M.B. Furie, S.C. Nicholson, J. Fischbarg, L.S. Liebovitch, and S.C. Silverstein. 1988. Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J. Cell. Physiol. 135:355–366. [DOI] [PubMed] [Google Scholar]