

Abstract
Glucocorticoid-induced tumor necrosis factor receptor family-related gene (GITR) is a member of the tumor necrosis factor receptor (TNFR) family that is expressed at low levels on unstimulated T cells, B cells, and macrophages. Upon activation, CD4+ and CD8+ T cells up-regulate GITR expression, whereas immunoregulatory T cells constitutively express high levels of GITR. Here, we show that GITR may regulate alloreactive responses during graft-versus-host disease (GVHD) after allogeneic bone marrow transplantation (BMT). Using a BMT model with major histocompatibility complex class I and class II disparity, we demonstrate that GITR stimulation in vitro and in vivo enhances alloreactive CD8+CD25− T cell proliferation, whereas it decreases alloreactive CD4+CD25− proliferation. Allo-stimulated CD4+CD25− cells show increased apoptosis upon GITR stimulation that is dependent on the Fas–FasL pathway. Recipients of an allograft containing CD8+CD25− donor T cells had increased GVHD morbidity and mortality in the presence of GITR-activating antibody (Ab). Conversely, recipients of an allograft with CD4+CD25− T cells showed a significant decrease in GVHD when treated with a GITR-activating Ab. Our findings indicate that GITR has opposite effects on the regulation of alloreactive CD4+ and CD8+ T cells.
Keywords: transplantation immunology, in vivo animal models, immune regulation, lymphocyte activation, T lymphocyte subsets
Introduction
Glucocorticoid-induced tumor necrosis factor receptor family-related gene (GITR), also known as TNFRSF18, is a type I transmembrane protein with high homology to other members of the TNFR family, including 4-1BB, CD27, and OX40 (1, 2). As with other members of the TNFR family, signaling through GITR may induce cell survival or cell death. Stimulation of human and mouse T cells through GITR induces NFκB activation via the TRAF2–NIK signaling pathway (3, 4). The intracellular domain of GITR binds Siva, a cytoplasmic molecule that contains a death domain, and may signal for induction of cell death (5). Four splice variants that differ in GITR intracellular domain have been identified. These cytoplasmic distinctions may generate different signaling events, and one of them may encode a decoy receptor (6). Mouse macrophages express constitutive levels of GITR–GITR ligand (GITRL) and stimulation with soluble GITR (sGITR) leads to an increased production of nitric oxide (7), COX-2 (8), and MMP-9 (9). Macrophage stimulation with sGITR signals through the Rel–NFκB pathway, but it remains to be determined if sGITR is an agonist or antagonist of GITR signaling (10). In vitro studies using murine GITRL protein and GITRL transfectants demonstrate that GITR signaling can enhance or inhibit the proliferation of Ag-stimulated T helper 1, T helper 2, and naive CD4+ T cells from TCR transgenic mice depending on the concentration of the cognate peptide, thus suggesting that GITR can function as a costimulatory receptor for TCR activation (11). Mice deficient for GITR have normal lymphoid and T cell development (12). However, experiments with GITR−/− T cells showed hyperproliferation to TCR stimulation, increased IL-2 production, increased IL-2 receptor α chain (CD25) expression, and increased susceptibility to activation-induced cell death (AICD) (12). The human GITRL has been detected in several tissues including ovary, testis, kidney, pancreas, PBLs, lymph nodes, and human umbilical vein endothelial cells (3, 4). Mouse GITRL expression has been demonstrated in macrophages, B cells, and both immature and mature dendritic cells (11, 13, 14). Stimulation with mitogens or LPS results in a temporary increase in the expression levels of GITRL, which is regulated by the transcription factor NF-1(11).
Mouse models for autoimmune disease suggest that GITR activation may break self-tolerance and induce autoimmunity presumably by inhibition of immunoregulatory T cell suppression (15, 16). In vivo administration of the agonist anti-GITR Ab, DTA-1, does not deplete GITR-expressing cells (15), but the mechanism by which immunoregulatory T cells are inhibited by GITR activation or the effect of GITR stimulation on other cell types such as B cells has not been defined. Apart from in vitro studies suggesting a costimulatory role for GITR in CD4+ T cell activation (11, 15, 17, 18), the function of GITR on CD4+CD25− and especially CD8+CD25− remains largely unknown. Because GITR expression is up-regulated upon stimulation of T cells, we were interested in studying GITR expression and activation of alloreactive CD4+ CD25− and CD8+CD25− T cells and its effects on the development of GVHD.
Materials and Methods
Reagents and Antibodies.
The DTA-1 hybridoma was generated as described previously (15), and rat IgG control Ab was obtained from Anogen. Antimurine CD16/CD32 FcR block (2.4G2), TNFR1 (55R-593), TNF (MP6-XT3), and all of the following fluorochrome labeled and purified antibodies against murine Ag were obtained from BD Biosciences: CD4 (RM4-5), CD8 (53-6.7), CD62L (MEL-14), CD122 (TM-B1), CD44 (IM7), CD45R/B220 (RA3-6B2), Gr-1 (RB6-8C5), CD25 (PC61), CD69 (H1.2F3), H-2Kb (AF6-88.5), Ly 9.1 (30C7), Fas (JO2), FasL (MFL3), isotype controls; rat IgG2a-κ (R35-95), rat IgG2a-λ (B39-4), rat IgG2b (A95-1), hamster IgG group 1 liter (Ha4/8), streptavidin-FITC, -PE, and -PCP. Biotinylated antimurine GITR (BAF524) was obtained from R&D Systems. Carboxyfluorescein succinimidyl ester (CFSE) was obtained from Molecular Probes.
In Vitro Assays.
Tissue culture medium consisted of RPMI 1640 or DMEM supplemented with 10% heat inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 50 μM of 2-mercaptoethanol (2-ME). For Ab stimulation, 1 μg/ml anti-CD3 and anti-CD28 was used. For proliferation assays, 10 μg/ml DTA-1 or rat IgG control Ab was used. T cells were purified, and 105 cells/well were incubated for 6 d with irradiated (2,000 cGy) splenocytes as stimulators (2 × 105 cells/well) in 96-well plates. Cultures were pulsed during the final 18 h with 1 μCi/well thymidine and harvested with Topcount Harvester (19). Cell proliferation was determined as counts per minute.
BMT and T Cell Purification.
Female C57BL/6 (H-2b), BALB/c (H-2k), B10.BR (H-2k), CBA/J(H-2k), lpr (B6.MRL-Fas lpr), and C57BL/6 (Ly5.1+) were obtained from The Jackson Laboratory. GITR−/− mice (C57BL/6 × 129/SvJ) were generated at Memorial Sloan-Kettering (12). All mice were used between 8–10 wk of age. BM cells were removed aseptically from femurs and tibias and depleted of T cells by incubation with anti–Thy-1.2 for 30 min at 4°C followed by incubation with Low-TOX-M rabbit complement (Cedarlane Laboratories) for 40 min at 37°C. Cells (5 × 106 BM cells without splenic T cells) were resuspended in DMEM (Life Technologies) and transplanted by tail vein infusion (0.25 ml total volume) into lethally irradiated recipients on day 0. On day 0, before transplantation, recipients received 900 cGy (BALB/c) or 1300 cGy (CBA/J) total body irradiation (137Cs source) as a split dose with a 3-h interval between doses (to reduce gastrointestinal toxicity). T cells were obtained from spleens, purified over a nylon wool column, or positively selected with anti-CD5 magnetic beads. CD4+CD25− and CD8+CD25− T cells were purified with magnetic beads (∼90% purity; Miltenyi Biotec) or sorted with Moflow (∼98–99% purity; DakoCytomation). Experiments were performed with sorted fractions and confirmed with bead-purified T cells. In brief, CD25− T cells were obtained by negative selection of splenocytes treated with anti-CD25 PE-conjugated Ab and anti-PE microbeads. CD4+ and CD8+ fractions were separated by positive selection for anti-CD4 or anti-CD8 antibodies conjugated to microbeads. Mice were housed in sterilized micro-isolator cages and received normal chow and autoclaved hyperchlorinated drinking water (pH 3.0). All experiments were performed in accordance with our institutional guidelines.
Assessment of GVHD.
The severity of GVHD was assessed with a clinical GVHD scoring system as described previously (20). In brief, mice were individually scored every week for five clinical parameters on a scale from zero to two: weight loss, posture, activity, fur, and skin. A clinical GVHD index was generated by summation of the five criteria scores (0–10). Survival was monitored daily. Animals with scores >5 were considered moribund and were killed. GVHD organ pathology for bowel (terminal ileum and ascending colon) and liver was assessed in a blinded fashion on formalin-preserved, paraffin-embedded, hematoxylin and eosin–stained histopathology sections with a semi-quantitative scoring system. In brief, bowel and liver were scored for 18–22 parameters associated with GVHD as described previously (21, 22).
CFSE Labeling.
Cells were labeled with CFSE as described previously (23). In brief, T cells were incubated with CFSE at a final concentration of 2.5 μM in PBS at 37°C for 20 min. Cells were washed three times with PBS before i.v. injection.
Flow Cytometric Analysis.
T cells were washed in FACS® buffer (PBS with 2% FBS and 0.1% sodium azide) and incubated for 15 min at 4°C with anti-CD16/CD32 FcR block. Subsequently, cells were incubated for 30 min at 4°C with antibodies and washed twice with FACS® buffer. Stained cells were resuspended in FACS® buffer and analyzed on a FACSCalibur™ flow cytometer (Becton Dickinson) with CELLQuest™ or Flowjo software (Treestar). For annexin V analysis, after cell surface staining, the stained cells were resuspended in 100 μl annexin V binding buffer and 5 μl annexin V Ab. After a 20-min incubation at room temperature in the dark, an additional 300 μl annexin V binding buffer was added, and the cells were analyzed.
ELISA.
MLR supernatant IL-2 and IFNγ levels were performed according to the manufacturer's instructions with Quantikine M kits from R&D Systems.
DTA-1 Administration.
For GVHD studies, DTA-1 and rat isotype control was administered by i.p. at days −1, 6, and 13 (1 mg/day). For adoptive transfer experiments with CFSE-labeled T cells, the antibodies were administered at day −1 (1 mg i.p.).
Online Supplemental Material.
The opposite effects of GITR stimulation on CD4+CD25− and CD8+CD25− T cells remain even at different concentrations of DTA-1 Ab and are independent from the TNF–TNFR pathway. Supernatants from CD4+CD25− T cells activated in the presence of GITR stimulation have decreased levels of IL-2 and IFNγ. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040116/DC1.
Results
Alloreactive CD4+ and CD8+ T Cells Up-regulate GITR.
To assess whether activation of CD4+ and CD8+ T cells results in up-regulation of GITR expression, we analyzed GITR cell surface expression on activated (in vitro stimulation with anti-CD3 and anti-CD28 antibodies) and alloreactive (in vivo alloactivation after transfer into an allogeneic recipient) CD4+ and CD8+ T cells. In vitro stimulation with plate-bound anti-CD3/anti-CD28 antibodies of CD4+CD25− and CD8+CD25− T cells enhanced the expression of cell surface GITR, whereas freshly isolated immunoregulatory T cells had constitutive high levels of GITR (unpublished data). We used an MHC class I/II disparate model (C57BL/6→BALB/c) to analyze the expression of GITR on alloreactive T cells in vivo in two ways. First, we infused CFSE-labeled donor CD3+ T cells into sublethally irradiated allogeneic and syngeneic recipients and recovered the cells from the spleen 3 d after infusion (Fig. 1 A). CFSE-labeled donor T cells from syngeneic recipients expressed low levels of cell surface GITR, in contrast with fast-dividing donor alloreactive T cells recovered from allogeneic recipients that had an activated phenotype (CD25+ and CD44+) and increased levels of GITR on their surface. Second, we infused T cell–depleted (TCD) allo-BM (C57BL/6 TCD-BM) and allogeneic T cells (C57BL/6) into lethally irradiated recipients (BALB/c), and after 9 d, determined the GITR expression on donor T cells from the spleens of these recipients (Fig. 1 B). We found that these (alloreactive) donor T cells indeed had an activated phenotype (CD25+, CD44+, and CD62L−; unpublished data), and as expected, both CD4+ and CD8+ donor T cells had increased GITR expression. Therefore, we conclude that alloreactive CD4+ and CD8+ T cells up-regulate their GITR expression.
Figure 1.
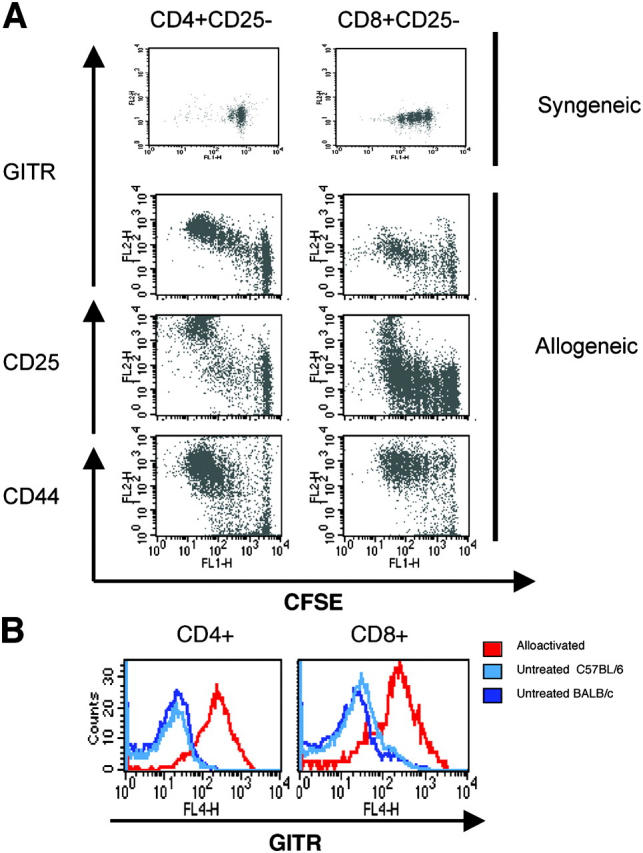
Alloactivation induces up-regulation of cell surface GITR on T cells. (A) Sublethally irradiated (750 cGy) syngeneic hosts (C57BL/6 Ly5.1) and allogeneic hosts (BALB/c) were infused with CFSE-labeled donor T cells (C57BL/6). GITR, CD25, and CD44 expression of these donor T cells was determined 3 d after infusion. (B) 9 d after BMT, GITR and CD25 expression was determined on splenic T cells from BALB/c recipients of C57BL/6 TCD-BM (5 × 106) and T cells (0.5 × 106), which were developing GVHD (red histogram). Blue histograms represent untransplanted controls (light blue, C57BL/6; dark blue, BALB/c).
Paradoxical Effect of GITR on CD4+CD25− and CD8+ CD25− T Cells.
We studied the effects of GITR stimulation on alloreactive T cells using an anti-GITR agonist mAb (DTA-1) for in vitro and in vivo experiments. To exclude the previously described effects of GITR on immunoregulatory T cells (15), we used purified C57BL/6 CD4+CD25− and CD8+CD25− T cells as effectors in MLR experiments with MHC class I/II disparate irradiated stimulators (BALB/c; Fig. 2 A). Addition of DTA-1 to the MLR resulted in an ∼2-fold decrease in proliferation of CD4+CD25− T cells, whereas CD8+CD25− T cell proliferation increased by ∼3.5-fold when compared with addition of control Ab (Fig. 2 A, top). To eliminate potentially confounding variables associated with GITR expression on immunoregulatory T cells, B cells, and macrophages present in the splenocyte population used as stimulators, splenocytes isolated from GITR+/− and GITR−/− mice were tested as stimulators. Because these mice are on a mixed H2b background (C57BL/6 × 129/SvJ), we used purified CD4+CD25− and CD8+CD25− T cells from BALB/c mice as effectors (Fig. 2 A, bottom). Again, CD4+ CD25− T cells showed decreased proliferation, whereas CD8+CD25− T cell proliferation was enhanced by the addition of the DTA-1 Ab. Because this effect was observed using GITR−/− stimulators, we conclude that GITR stimulation via DTA-1 is independent of GITR expression on the stimulator population (including immunoregulatory T cells) and has a direct effect on CD4+CD25− and CD8+CD25− effector T cells. Also, this effect was not strain specific because results generated from T cells derived from C57BL/6 and BALB/c were consistent.
Figure 2.
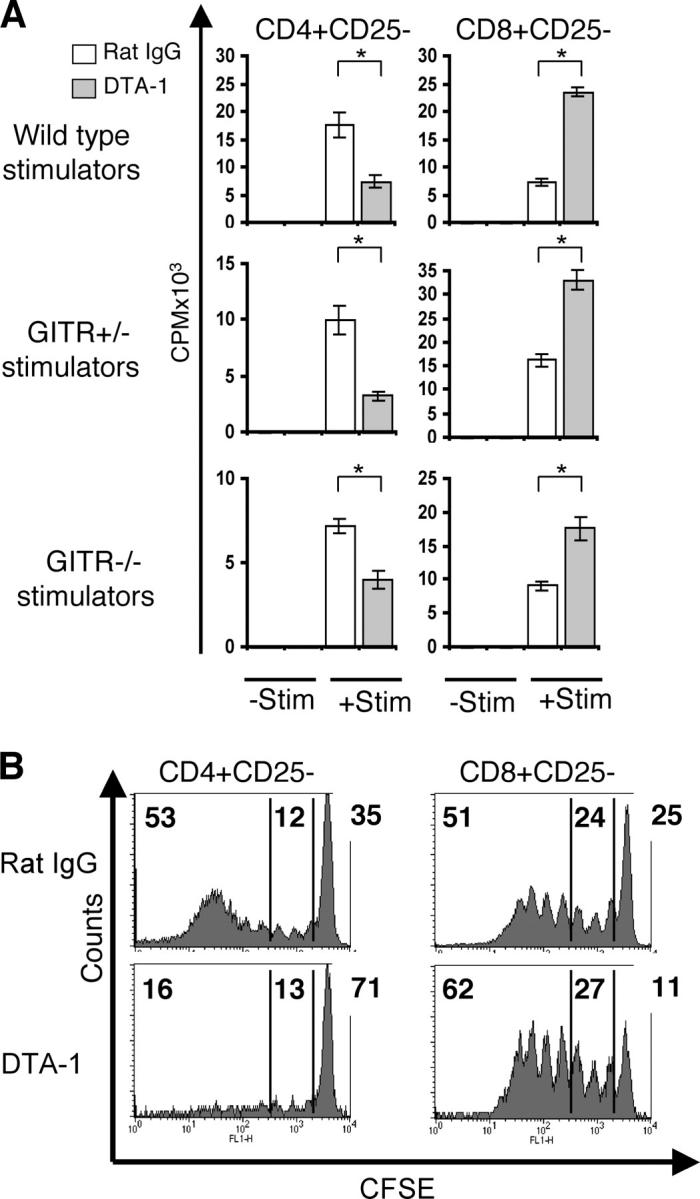
GITR stimulation induces paradoxical responses in CD4+ CD25− and CD8+CD25− T cells in vitro and in vivo. (A) An anti-GITR agonist Ab (DTA-1; 10 μg/ml) was added to MLRs of purified CD4+CD25− and CD8+CD25− splenic T cells as effector cells (105) with irradiated splenocytes as stimulators (2 × 105). (Wild-type stimulators) C57BL/6 effectors and BALB/c stimulators. (GITR+/− and GITR−/− stimulators) BALB/c effectors and GITR+/− or GITR−/− stimulators (C57BL/6 × 129/SvJ). −Stim, without stimulators. +Stim, with stimulators. *, P < 0.001. (B) Proliferative profile of recovered CFSE-labeled CD4+CD25− and CD8+CD25− T cells from sublethally irradiated hosts treated with 10 μg/ml DTA-1 or rat IgG control. These data are representative of three independent experiments. (top left) Percentage of alloreactive fast-proliferating donor T cells. (center) Percentage of slow proliferating donor T cells. (top right) Nondividing donor T cells.
Our experiments clearly show inhibition of CD4+ CD25− proliferation upon GITR stimulation, whereas other groups show enhancement of CD4+CD25− T cell proliferation. Shimizu et al. (15) demonstrated that CD4+CD25− T cells from CD28−/− were able to proliferate when stimulated via GITR. Tone et al. (11) showed that stimulation of GITR using a recombinant mouse GITRL could increase the proliferation of a Th2 clone through a wide range of cognate peptide concentration and a Th1 clone only at low peptide concentrations. Therefore, we analyzed whether the difference in proliferation of the alloreactive T cells could depend on the amount of anti-GITR agonist Ab present in the MLR. Because the Ag concentration is fixed (allorecognition), we titrated the anti-GITR Ab concentration over a 4-log range (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040116/DC1). Addition of different concentrations spanning from 0.01 to 10 μg/ml of the GITR-stimulating Ab (DTA-1) to an MLR resulted again in decreased proliferation of CD4+CD25− T cells and increased CD8+ CD25− T cell proliferation.
Our data would suggest that CD4+CD25− T cells from GITR−/− mice would show increased proliferation, whereas CD8+CD25− T cells from GITR−/− mice would have impaired proliferative capacity. Ronchetti et al. (17) have shown that, indeed, upon anti-CD3 stimulation, CD4+ but not the CD8+ GITR−/− subpopulation had a higher proliferation rate than the CD4+ GITR+/+ subpopulation. Because T cell responses to Ab stimulation may be irrelevant to our model of allo-BMT, we tested the proliferative capacity of GITR−/− and GITR+/− T cells derived from mice of a mixed background (C57BL/6 × 129/SvJ). Our preliminary results indicate that there is no difference in CD4+CD25− and CD8+CD25− T cell proliferation upon allostimulation using third party stimulators (BALB/c; unpublished data). However, these experiments were performed with cells from GITR−/− and GITR+/− on a mixed background and we cannot rule out that genetic differences (other than the presence or absence of GITR) could have affected the alloresponse. Therefore, definitive experiments will have to be deferred until the GITR−/− mice have been completely backcrossed (>N 10).
To test whether the in vitro effects of anti-GITR agonist Ab (DTA-1) on alloreactive T cells were consistent in vivo, sublethally irradiated BALB/c mice were treated with DTA-1 or rat IgG control before adoptive transfer of C57BL/6 CFSE-labeled CD4+CD25− or CD8+CD25− T cells (Fig. 2 B). 3 d after T cell infusion, donor T cells were recovered and analyzed by flow cytometry. The in vivo proliferation profile of CFSE-labeled T cells allows the discrimination of slow proliferating cells, described in some models as homeostatic expansion, versus fast-dividing alloreactive T cells (24). GITR stimulation had no impact on the proportion of slow dividing CD4+CD25− (12% in controls vs. 13% in DTA-1–treated recipients) and CD8+CD25− (24 vs. 27%) T cells. However, GITR stimulation decreased the percentage of fast-dividing alloreactive CD4+CD25− T cells (from 53 to 16%) and increased the percentage of fast-dividing alloreactive CD8+CD25− T cells (from 51 to 62%). There were more nondividing CD4+CD25− T cells in the DTA-1–treated group (71% in DTA-1–treated recipients vs. 35% in the controls), and fewer nondividing CD8+CD25− T cells in the DTA-1–treated group (11 vs. 25%) compared with the control Ab groups. These results indicate that GITR stimulation in vivo can inhibit alloreactive CD4+CD25− expansion while it enhances alloreactive CD8+CD25− expansion.
Fas–FasL Mediate GITR Inhibition of CD4+CD25− T Cell Expansion.
To determine if the Fas–FasL pathway was involved in GITR inhibition of CD4+CD25− expansion, we studied the effect of GITR stimulation on cell surface expression of Fas and FasL (Fig. 3 A). CD4+CD25− and CD8+CD25− T cells allostimulated in the presence of the agonistic anti-GITR Ab (DTA-1) did not increase Fas expression nor was there a difference in FasL expression (unpublished data), although FasL cell surface expression is notoriously difficult to demonstrate by flow cytometric analysis. To address this question in a different assay, we studied the effect of DTA-1 on T cells from Fas-deficient lpr mice (Fig. 3 B). Addition of DTA-1 to an MLR had no effect on proliferation by lpr CD4+CD25− T cells, in contrast with the inhibitory effect on wild-type CD4+CD25− T cells. Alloreactive proliferation of both wild-type and lpr CD8+CD25− T cells was increased when DTA-1 was added to the MLR. To demonstrate that GITR inhibition of CD4+CD25− proliferation was due to FasL signaling, we studied the effect of anti-FasL blocking Ab during allostimulation (Fig. 3 C). As aforementioned, CD4+CD25− T cell proliferation was impaired in the presence of DTA-1 while the addition of the FasL blocking Ab rescued CD4+CD25− proliferation, although not completely. Because CD4+CD25− proliferation is not completely restored after FasL blocking, other members of the TNFR family could be implicated. Proliferation of purified CD4+ CD25− T cells from TNF−/− and TRAIL−/− mice allostimulated in the presence of DTA-1 treatment remained impaired (Fig. 3 D). We further tested the role of TNFR1 by using an anti-TNFR1 blocking Ab. Proliferation of purified CD4+CD25− T cells with TNFR1 and TNF blocking Ab in the presence of GITR stimulation still remained impaired (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20040116/DC1). These results show that GITR-mediated inhibition of CD4+CD25− T cell expansion involves the Fas–FasL pathway and not the TNFR or TRAIL pathways, although the involvement of other members of the TNF family cannot be excluded.
Figure 3.

Fas–FasL pathway is involved in inhibition of CD4+CD25− proliferation. (A) Fas expression was determined on purified C57BL/6 CD4+CD25− and CD8+CD25− T cells 72 h after anti-GITR agonist Ab (DTA-1) was added to MLRs with irradiated BALB/c stimulators. (shaded histograms) Isotype control. (blue histograms) Rat IgG–treated MLR. (red histograms) DTA-1–treated MLR. (B) DTA-1 was added to MLRs with purified CD4+CD25− and CD8+CD25− from lpr (B6.MRL-Fas lpr) or wild-type (C57BL/6) mice as effectors and irradiated BALB/c splenocytes as stimulators. These data are representative of three experiments. *, P < 0.001. (C) Purified C57BL/6 CD4+CD25− T cells were stimulated with irradiated BALB/c splenocytes in the presence of rat IgG control Ab, DTA-1 Ab, or DTA-1 plus MFL3, a FasL-blocking Ab. These data are representative of two independent experiments. (D) CD4+CD25− T cells were purified from wild-type C57BL/6, C57BL/6-TNF−/−, and C57BL/6 TRAIL−/− mice and stimulated with irradiated BALB/c splenocytes in the presence of rat IgG control Ab or DTA-1 Ab. (unshaded bars) Rat IgG treatment. (shaded bars) DTA-1 treatment. Results are presented as percent proliferation and are representative of two independent experiments. *, P < 0.007.
We measured the amount of IL-2 production of CD4+CD25− T cells in the presence or absence of DTA-1 during allostimulation (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20040116/DC1). IL-2 was decreased in MLR supernatants of DTA-1–treated cocultures compared with control Ab–treated cocultures at days 2 and 5 of coculture (day 2, ∼1.5 fold decrease and day 5, ∼2.1-fold decrease). These decreased levels of IL-2 detected in our system may be a reflection of less viable CD4+CD25− T cells.
These experiments indicate that GITR activation can initiate Fas-mediated AICD of alloreactive CD4+CD25− T cells, whereas GITR stimulation of CD8+CD25− T cell proliferation is independent of Fas–FasL signaling.
In Vivo GITR Stimulation Increases Apoptosis of CD4+ CD25− Alloreactive T Cells.
We wanted to address in our GVHD model if GITR stimulation increased CD4+CD25− apoptosis in vivo. We infused 3 × 106 purified CD4+CD25− T cells into lethally irradiated hosts treated with DTA-1 or rat IgG control Ab and determined apoptosis of donor T cells after 3, 5, and 7 d of allo-BMT (Fig. 4 A). Donor CD4+CD25− T cells harvested from spleens of DTA-1–treated recipients showed increased annexin V staining when compared with donor T cells derived from control recipients treated with rat IgG (Fig. 4, B and C). We also studied the expression of activation markers at day 7 after BMT on CD4+CD25− T cells from the DTA-1–treated group compared with the rat IgG–treated group and detected no significant difference in the level of CD25 and CD44 expression (unpublished data). These results indicate that DTA-1 treatment induces significantly more apoptosis of donor alloreactive CD4+CD25− T cells early in the course of GVHD.
Figure 4.
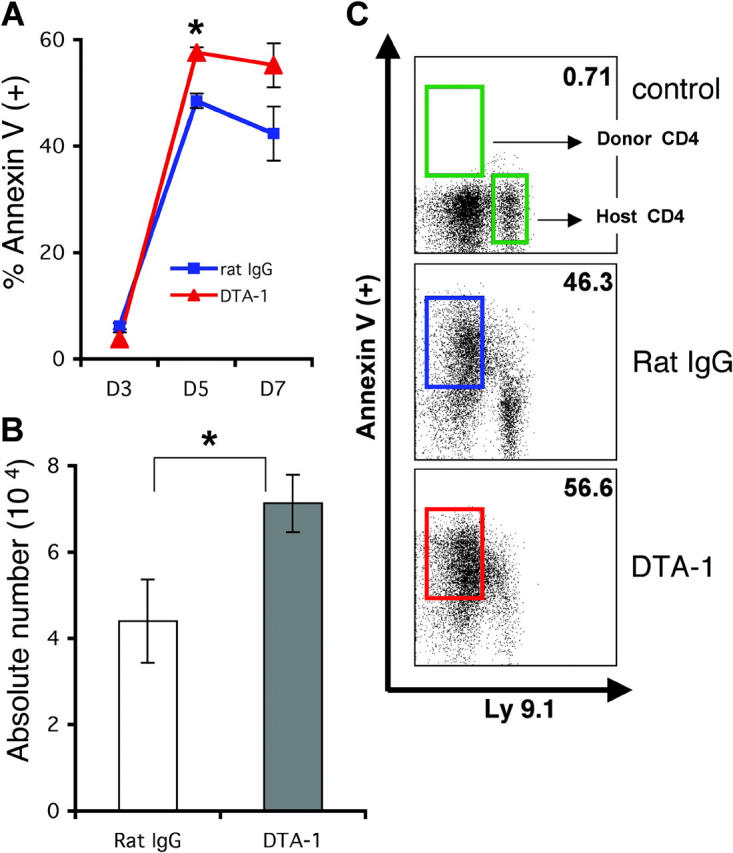
GITR stimulation induces early apoptosis of CD4+CD25− alloreactive T cells after BMT. Lethally irradiated (900 cGy) BALB/c recipients of C57BL/6 TCD-BM and C57BL/6 CD4+CD25− splenic T cells were treated with DTA-1 or control Ab (1 mg i.p. on days −1 and 6). Spleens were harvested on days 3, 5, and 7, and stained for annexin V (+) cells (n = 3 per group, per time point). (A) Time course of the percentage of donor-derived annexin V (+) cells is shown. *, P = 0.0004. (B) Absolute numbers of annexin V (+) donor CD4+ T cells at day 5. *, P = 0.05. (C) Representative FACS® analyses of annexin V (+) donor and host CD4+ T cells at day 7.
GITR Stimulation Modulates GVHD.
In the C57BL/6→BALB/c strain combination, alloreactive CD4+ T cells are more potent as GVHD effectors (25), whereas graft-versus-tumor activity is mostly dependent on alloreactive CD8+ T cells (26). We used this model to determine the effects of anti-GITR agonistic Ab (DTA-1) on alloreactive CD4+CD25− and CD8+CD25− T cells during the development of GVHD. Pilot experiments determined that DTA-1 administration beginning at day −1 was optimal. The dose and schedule were consistent with previous papers in which anti-GITR antibodies have been used in vivo (weekly administration of 1 mg i.p.; references 16, 27).
We hypothesized that DTA-1 administration to allo-BMT recipients could ameliorate GVHD mediated by CD4+CD25− T cells due to enhanced AICD of alloreactive T cells and aggravate GVHD mediated by CD8+CD25− T cells due to their enhanced proliferation by GITR stimulation. Indeed, we observed that DTA-1–treated recipients of CD4+CD25− C57BL/6 donor T cells, although not free of disease, had a significant delay and decrease in GVHD morbidity and mortality compared with recipients treated with a control Ab (Fig. 5, left).
Figure 5.
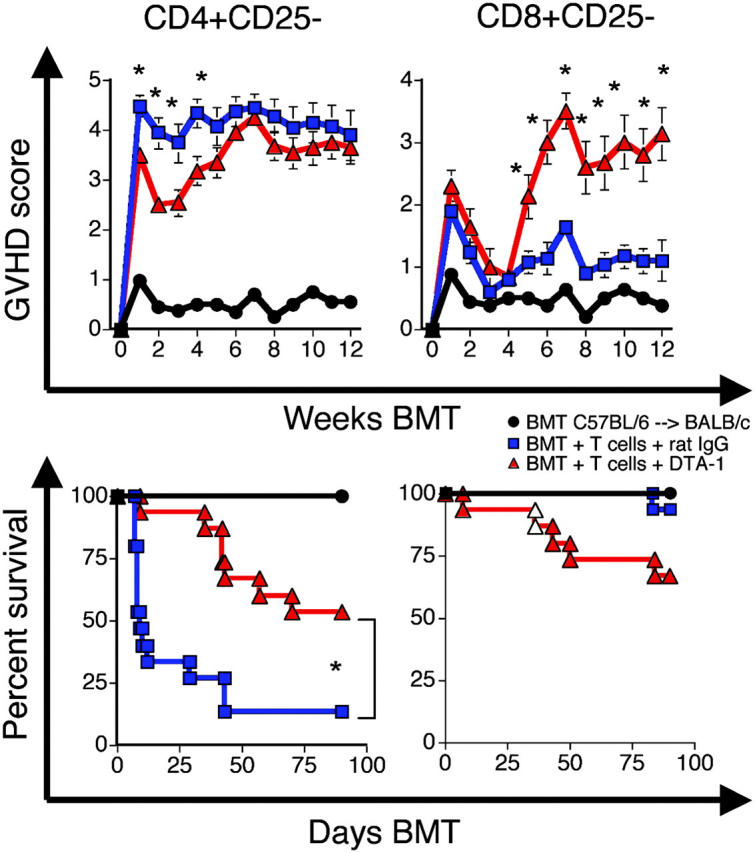
GITR stimulation decreases GVHD mediated by CD4+CD25− cells and increases GVHD mediated by CD8+CD25− T cells. Lethally irradiated (900 cGy) BALB/c recipients of C57BL/6 TCD-BM and C57BL/6 CD4+CD25− or CD8+CD25− splenic T cells were treated with DTA-1 Ab (1 mg i.p. on days −1, 6, and 13) or control Ab. (top) Mean ± SEM clinical GVHD scores. (bottom) Kaplan-Meier survival curves. Data shown are combined from three experiments, n = 15 per group. *, P ≤ 0.01.
Although alloreactive CD4+CD25− cells are more potent on a per cell basis in the C57BL/6→BALB/c model, this strain combination has a class I and class II disparity, and CD8+CD25− can induce GVHD. In other GVHD models with full MHC disparity, CD8+-mediated GVHD can always be demonstrated (28). DTA-1–treated recipients of donor BM and donor CD8+CD25− T cells had significantly increased GVHD morbidity and mortality (Fig. 5, right). Additional experiments in a MHC-matched strain combination in which GVHD is primarily dependent on alloreactive CD8+ T cells (B10.BR→ CBA/J) demonstrated that DTA-1 administration to allo-BMT recipients could aggravate GVHD (unpublished data). However, these mice were infused with unfractionated donor splenocytes; thus, increased mortality could also be due to the inhibition of suppressor function via GITR stimulation (15, 16) independent of its effect on CD8+ donor T cells.
To further assess GVHD, target organ histopathology was studied (Fig. 6). Mice transplanted and infused with donor CD4+CD25− donor T cells were killed at day 21 (Fig. 6 A). We lowered the dose of donor CD4+CD25− T cells to 0.3 × 106 and delayed the time of organ harvest to day 21 due to high mortality in the rat IgG control group (Fig. 5). We observe significantly less GVHD target organ damage in liver and intestines of DTA-1–treated recipients compared with control recipients of CD4+CD25− T cells. There was greater thymic cellularity in DTA-1–treated recipients, which is consistent with less thymi damage. Also, higher numbers of granulocytes were detected, consistent with less GVHD-associated myelosuppression. Mice transplanted and infused with CD8+CD25− donor T cells were killed, and organs were harvested at day 55, when differences in clinical scores were more pronounced (Fig. 6 B). Consistent with our previous results, GITR stimulation of donor CD8+CD25− donor T cells resulted in increased target organ damage and myelosuppression.
Figure 6.
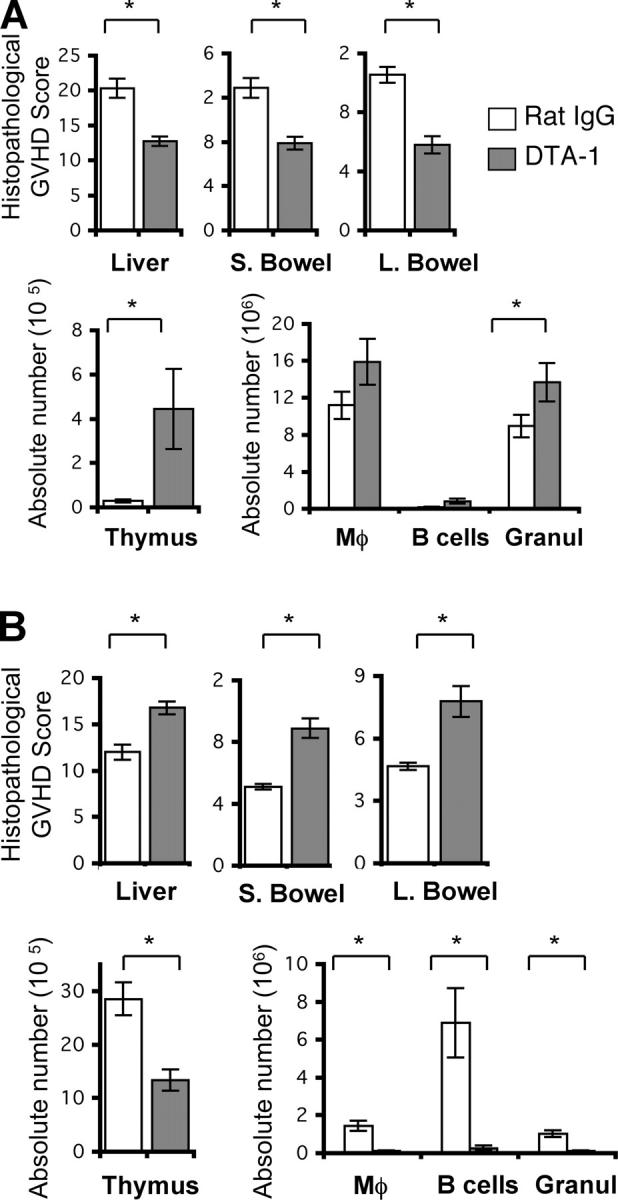
GITR stimulation decreases GVHD-associated organ damage mediated by CD4+CD25− T cells and increases GVHD-associated organ damage mediated by CD8+CD25− T cells. Lethally irradiated (900 cGy) BALB/c recipients of C57BL/6 TCD-BM and C57BL/6 CD4+CD25− cells or CD8+CD25− splenic T cells were treated with DTA-1 Ab or control Ab (1 mg i.p. on days −1, 6, and 13). Recipients of CD4+CD25− T cells were killed, and organs were harvested on day 21 (A). Recipients of CD8+CD25− T cells were killed, and organs were harvested on day 55 (B). (top) A semi-quantitative histopathological analysis for GVHD in liver, small bowel, and large bowel. (bottom left) The absolute number of thymocytes. (bottom right) The absolute numbers of splenic Mac-1+ (Mφ), B cells, and granulocytes (Granul) were analyzed by flow cytometry. Data shown are representative of one experiment, n = 9–10 per group. *, P ≤ 0.05.
Discussion
Previous experiments have indicated that in vitro stimulation of GITR on immunoregulatory T cells inhibits their suppressive effect and that in vivo GITR stimulation induced development of autoimmunity presumably due to immunoregulatory T cell inhibition (15, 16). The effects of GITR stimulation on CD4+ and CD8+ have not been fully addressed, especially in vivo. Here, we demonstrate that GITR stimulation in vitro and in vivo has an important role in the costimulation of alloreactive CD4+CD25− and CD8+ CD25− T cells independent of its effect on immunoregulatory T cells. Stimulation with a GITR-activating Ab inhibited CD4+CD25− proliferation and decreased GVHD, whereas it enhanced alloreactive CD8+CD25− T cell proliferation and increased GVHD. Our data indicate that GITR-mediated inhibition of CD4+CD25− T cell expansion involves the Fas–FasL pathway, suggesting that GITR activation can initiate Fas-mediated AICD of alloreactive CD4+CD25− T cells, whereas GITR stimulation of CD8+CD25− T cell proliferation is independent of Fas–FasL signaling. Our results are consistent with the notion that GITR stimulation could lower the threshold for T cell activation inducing increased AICD in CD4+CD25−, but not in CD8+CD25− T cells, where it induces proliferation. Thus, GITR stimulation of alloreactive CD4+CD25− T cells in vivo can provide a novel strategy to prevent or treat GVHD.
Other laboratories have described GITR signaling as costimulatory for CD4 and CD8 (15, 17). Experiments using polyclonal stimulation with low concentrations of plate-bound or soluble anti-CD3 Ab (0.1–0.5 μg/ml; references 15, 17, 18) demonstrate that anti-GITR increases proliferation, but in some experiments, this costimulation is lost at higher concentrations of anti-CD3 Ab (1–3 μg/ml; references 15, 18). Our experiments addressing the role of GITR stimulation using Ab stimulation showed no difference in the proliferation of CD4+CD25− cells stimulated with 10 μg/ml of soluble anti-CD3 and soluble anti-GITR agonistic Ab (DTA-1). CD8+CD25− T cell proliferation under the same conditions was slightly increased (unpublished data). However, experiments addressing the effects of GITR stimulation upon Ag-specific recognition, a more representative model of allo-specific responses, show results consistent with ours. Because GVHD is a Th1-mediated complication of BMT (29, 30), experiments by Tone et al. (11) with a Th1 clone derived from TCR transgenic mice using a rGITRL for GITR stimulation are more relevant to our model. When the Th1 clone is stimulated with a low concentration of the cognate peptide (1 nM), rGITRL proliferation is enhanced. In contrast, when the Th1 clone is stimulated in the presence of rGITRL and higher peptide concentrations (10–100 nM), proliferation is inhibited. The same results were observed using naive T cells derived from the same transgenic mice where the presence of GITR stimulation at high peptide concentrations inhibited proliferation (11).
Furthermore, Shimizu et al. (15) have shown that xenostimulated CD4+CD25− T cells have decreased proliferation in the presence of anti-GITR agonistic Ab (DTA-1). Murine CD4+CD25− T cells were stimulated with rat APCs. The authors demonstrate that DTA-1 has no cross-reactivity with rat APCs, indicating that any effect observed by the addition of the agonist Ab will be a result of its direct effect on the murine cells. When CD4+CD25− T cells were xenostimulated in the presence of a control Ab, they proliferate extensively, whereas addition of DTA-1 to the same culture resulted in a marked decrease in proliferation. These experiments that analyze Ag-dependent T cell activation are very similar to our results, suggesting that during allo- and xenostimulation the addition of GITR stimulation may induce a potent costimulation, which can inhibit proliferation at higher Ag concentration. Our experiments indicate that this inhibitory effect on proliferation could be due to AICD.
We believe that the overall effect of in vivo GITR stimulation needs to be reconsidered because GITR stimulation can have a differential and/or paradoxical effect on regulatory T cells, CD4+ effector T cells, and CD8+ effector T cells. Our data in clinically relevant models for GVHD suggest that in vivo GITR stimulation holds therapeutic promise for the separation of CD8-mediated graft-versus-tumor activity from CD4-mediated GVHD activity.
Acknowledgments
S.J. Muriglan would like to dedicate this paper to Donald Holmquist.
A.N. Houghton and T. Ramirez-Montagut were supported by the National Institutes of Health (grants CA59350, CA58621, and CA33049) and Swim Across America. This work was supported by RO1 grants HL69929 and HL72412 from the National Institutes of Health (to M.R.M. van den Brink). M.R.M. van den Brink is the recipient of a Damon Runyon Scholar Award of the Cancer Research Fund.
S.J. Muriglan and T. Ramirez-Montagut contributed equally to this work.
This work was presented at the American Society of Hematology Annual Meeting in 2003, the American Society for Blood and Marrow Transplantation in 2004, the American Association for Cancer Research Annual Meeting in 2004, and the American Association for Immunologists in 2004 in abstract form.
Abbreviations used in this paper: AICD, activation-induced cell death; BMT, BM transplantation; CFSE, carboxyfluorescein succinimidyl ester; GITR, glucocorticoid-induced tumor necrosis factor receptor family-related gene; GITRL, GITR ligand; sGITR, soluble GITR; TCD, T cell–deleted.
References
- 1.Nocentini, G., L. Giunchi, S. Ronchetti, L.T. Krausz, A. Bartoli, R. Moraca, G. Migliorati, and C. Riccardi. 1997. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA. 94:6216–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nocentini, G., A. Bartoli, S. Ronchetti, L. Giunchi, A. Cupelli, D. Delfino, G. Migliorati, and C. Riccardi. 2000. Gene structure and chromosomal assignment of mouse GITR, a member of the tumor necrosis factor/nerve growth factor receptor family. DNA Cell Biol. 19:205–217. [DOI] [PubMed] [Google Scholar]
- 3.Gurney, A.L., S.A. Marsters, R.M. Huang, R.M. Pitti, D.T. Mark, D.T. Baldwin, A.M. Gray, A.D. Dowd, A.D. Brush, A.D. Heldens, et al. 1999. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Curr. Biol. 9:215–218. [DOI] [PubMed] [Google Scholar]
- 4.Kwon, B., K.Y. Yu, J. Ni, G.L. Yu, I.K. Jang, Y.J. Kim, L. Xing, D. Liu, S.X. Wang, and B.S. Kwon. 1999. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J. Biol. Chem. 274:6056–6061. [DOI] [PubMed] [Google Scholar]
- 5.Spinicelli, S., G. Nocentini, S. Ronchetti, L.T. Krausz, R. Bianchini, and C. Riccardi. 2002. GITR interacts with the pro-apoptotic protein Siva and induces apoptosis. Cell Death Differ. 9:1382–1384. [DOI] [PubMed] [Google Scholar]
- 6.Nocentini, G., S. Ronchetti, A. Bartoli, S. Spinicelli, D. Delfino, L. Brunetti, G. Migliorati, and C. Riccardi. 2000. Identification of three novel mRNA splice variants of GITR. Cell Death Differ. 7:408–410. [DOI] [PubMed] [Google Scholar]
- 7.Shin, H.H., M.H. Lee, S.G. Kim, Y.H. Lee, B.S. Kwon, and H.S. Choi. 2002. Recombinant glucocorticoid induced tumor necrosis factor receptor (rGITR) induces NOS in murine macrophage. FEBS Lett. 514:275–280. [DOI] [PubMed] [Google Scholar]
- 8.Shin, H.H., B.S. Kwon, and H.S. Choi. 2002. Recombinant glucocorticoid induced tumour necrosis factor receptor (rGITR) induced COX-2 activity in murine macrophage Raw 264.7 cells. Cytokine. 19:187–192. [DOI] [PubMed] [Google Scholar]
- 9.Lee, H.S., H.H. Shin, B.S. Kwon, and H.S. Choi. 2003. Soluble glucocorticoid-induced tumor necrosis factor receptor (sGITR) increased MMP-9 activity in murine macrophage. J. Cell. Biochem. 88:1048–1056. [DOI] [PubMed] [Google Scholar]
- 10.Shin, H.H., H.W. Lee, and H.S. Choi. 2003. Induction of nitric oxide synthase (NOS) by soluble glucocorticoid induced tumor necrosis factor receptor (sGITR) is modulated by IFN-gamma in murine macrophage. Exp. Mol. Med. 35:175–180. [DOI] [PubMed] [Google Scholar]
- 11.Tone, M., Y. Tone, E. Adams, S.F. Yates, M.R. Frewin, S.P. Cobbold, and H. Waldmann. 2003. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA. 100:15059–15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronchetti, S., G. Nocentini, C. Riccardi, and P.P. Pandolfi. 2002. Role of GITR in activation response of T lymphocytes. Blood. 100:350–352. [DOI] [PubMed] [Google Scholar]
- 13.Yu, K.Y., H.S. Kim, S.Y. Song, S.S. Min, J.J. Jeong, and B.S. Youn. 2003. Identification of a ligand for glucocorticoid-induced tumor necrosis factor receptor constitutively expressed in dendritic cells. Biochem. Biophys. Res. Commun. 310:433–438. [DOI] [PubMed] [Google Scholar]
- 14.Kim, J.D., B.K. Choi, J.S. Bae, U.H. Lee, I.S. Han, H.W. Lee, B.S. Youn, D.S. Vinay, and B.S. Kwon. 2003. Cloning and characterization of GITR ligand. Genes Immun. 4:564–569. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3:135–142. [DOI] [PubMed] [Google Scholar]
- 16.Uraushihara, K., T. Kanai, K. Ko, T. Totsuka, S. Makita, R. Iiyama, T. Nakamura, and M. Watanabe. 2003. Regulation of murine inflammatory bowel disease by CD25+ and CD25- CD4+ glucocorticoid-induced TNF receptor family-related gene+ regulatory T cells. J. Immunol. 171:708–716. [DOI] [PubMed] [Google Scholar]
- 17.Ronchetti, S., O. Zollo, S. Bruscoli, M. Agostini, R. Bianchini, G. Nocentini, E. Ayroldi, and C. Riccardi. 2004. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 34:613–622. [DOI] [PubMed] [Google Scholar]
- 18.Kohm, A.P., J.S. Williams, and S.D. Miller. 2004. Cutting edge: ligation of the glucocorticoid-induced TNF receptor enhances autoreactive CD4(+) T cell activation and experimental autoimmune encephalomyelitis. J. Immunol. 172:4686–4690. [DOI] [PubMed] [Google Scholar]
- 19.Alpdogan, O., S.J. Muriglan, B.J. Kappel, E. Doubrovina, C. Schmaltz, R. Schiro, J.M. Eng, A.S. Greenberg, L.M. Willis, J.A. Rotolo, et al. 2003. Insulin-like growth factor-I enhances lymphoid and myeloid reconstitution after allogeneic bone marrow transplantation. Transplantation. 75:1977–1983. [DOI] [PubMed] [Google Scholar]
- 20.Cooke, K.R., L. Kobzik, T.R. Martin, J. Brewer, J. Delmonte, Jr., J.M. Crawford, and J.L. Ferrara. 1996. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 88:3230–3239. [PubMed] [Google Scholar]
- 21.Crawford, J.M. 1997. Graft-versus-host disease of the liver. Graft-vs.-Host Disease. J.L.M. Ferrara, H.J. Deeg, and S.J. Burakoff, editors. Marcel Dekker, New York. 315–336.
- 22.Hill, G.R., J.M. Crawford, K.R. Cooke, Y.S. Brinson, L. Pan, and J.L. Ferrara. 1997. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 90:3204–3213. [PubMed] [Google Scholar]
- 23.Lyons, A.B., and C.R. Parish. 1994. Determination of lymphocyte division by flow cytometry. J. Immunol. Methods. 171:131–137. [DOI] [PubMed] [Google Scholar]
- 24.Alpdogan, O., S.J. Muriglan, J.M. Eng, L.M. Willis, A.S. Greenberg, B.J. Kappel, and M.R. van den Brink. 2003. IL-7 enhances peripheral T cell reconstitution after allogeneic hematopoietic stem cell transplantation. J. Clin. Invest. 112:1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann, P., J. Ermann, M. Edinger, C.G. Fathman, and S. Strober. 2002. Donor-type CD4+CD25+ regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J. Exp. Med. 196:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edinger, M., P. Hoffmann, J. Ermann, K. Drago, C.G. Fathman, S. Strober, and R.S. Negrin. 2003. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 9:1144–1150. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu, J., and E. Moriizumi. 2003. CD4+CD25− T cells in aged mice are hyporesponsive and exhibit suppressive activity. J. Immunol. 170:1675–1682. [DOI] [PubMed] [Google Scholar]
- 28.Schmaltz, C., O. Alpdogan, K.J. Horndasch, S.J. Muriglan, B.J. Kappel, T. Teshima, J.L. Ferrara, S.J. Burakoff, and M.R. van den Brink. 2001. Differential use of Fas ligand and perforin cytotoxic pathways by donor T cells in graft-versus-host disease and graft-versus-leukemia effect. Blood. 97:2886–2895. [DOI] [PubMed] [Google Scholar]
- 29.Krenger, W., and J.L. Ferrara. 1996. Graft-versus-host disease and the Th1/Th2 paradigm. Immunol. Res. 15:50–73. [DOI] [PubMed] [Google Scholar]
- 30.Krenger, W., G.R. Hill, and J.L. Ferrara. 1997. Cytokine cascades in acute graft-versus-host disease. Transplantation. 64:553–558. [DOI] [PubMed] [Google Scholar]