

Abstract
Amino acid residues 111–129 represent an immunodominant epitope of myelin basic protein (MBP) in humans with human leukocyte antigen (HLA)-DRB1*0401 allele(s). The MBP 111–129–specific T cell clone MS2-3C8 was repeatedly isolated from a patient with multiple sclerosis (MS), suggesting an involvement of MS2-3C8 T cells in the pathogenesis. To address the pathogenic potential of the MS2-3C8 T cell clone, we generated transgenic (Tg) mice expressing its T cell receptor and restriction element, HLA-DRB1*0401, to examine the pathogenic characteristics of MS2-3C8 Tg T cells by adoptive transfer into HLA-DRB1*0401 Tg mice. In addition to the ascending paralysis typical of experimental autoimmune encephalomyelitis, mice displayed dysphagia due to restriction in jaw and tongue movements and abnormal gait. In accordance with the clinical phenotype, infiltrates of MS2-3C8 Tg T cells and inflammatory lesions were predominantly located in the brainstem and the cranial nerve roots in addition to the spinal cord and spinal nerve roots. Together, these data suggest a pathogenic role of MBP-specific T cells in inflammatory demyelination within the brainstem and cranial nerve roots during the progression of MS. This notion may help to explain the clinical and pathological heterogeneity of MS.
Keywords: HLA-DRB1*0401, myelin basic protein, transgenic mouse, experimental autoimmune encephalomyelitis, multiple sclerosis
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disorder of the central nervous system (CNS). The severity of clinical symptoms, the extent of demyelination, and the location of lesions can vary considerably, suggesting a high degree of heterogeneity in MS pathogenesis (1). Although the etiology of MS is unknown, the study of experimental autoimmune encephalomyelitis (EAE), an animal model of MS, and the immunological assessments of MS patients support the concept that MS is an autoimmune disease mediated by autoreactive CD4+ T cells with specificity for myelin antigens (2, 3).
MS has been shown to be a polygenic disease with a strong genetic link to the human leukocyte antigen (HLA) complex (4–7). HLA-DR2 is the allele most often associated with MS in Caucasians (8), especially in the relapsing/remitting type of MS (9, 10). HLA-DR4 genes have been found primarily associated with the chronic progressive type of MS and have been found at higher prevalence in MS patients from various ethnic groups, particularly from the Mediterranean area (11–13). Hence, it appears that certain HLA-DR molecules, besides conferring disease susceptibility, may also affect phenotypic characteristics in MS including disease course. This is consistent with findings in EAE, where lesion distribution and disease course, i.e., chronic relapsing versus acute, vary with MHC class II background and the antigen used for disease induction (2). Thus, both the target myelin antigen for autoreactive T cells and the MHC/HLA class II context are important determinants for disease expression. Immunodominant epitopes of suspect autoantigens are largely determined by the HLA-DR allotypes expressed by the patient: whereas myelin basic protein (MBP) 83–99 is an immunodominant epitope in DRB1*1501- and DRB5*0101-positive individuals (14, 15), MBP 111–129 is immunodominant in the context of HLA-DRB1*0401, the most common HLA-DR4 allele (16). Interestingly, whereas MBP 83–99 is encephalitogenic in several EAE-susceptible mouse and rat strains (17), MBP 111–129 is a less common encephalitogen (18–20).
To assess the encephalitogenic potential of MBP 111–129–specific T cells, we selected T cell clone (TCC) MS2-3C8, which had been isolated from the peripheral blood of a patient with relapsing/remitting MS (16). MBP-specific T cell lines were established from peripheral blood obtained from the patient at two time points 25 mo apart (Fig. 1). The vast majority of MBP-responsive cultures at each time point (92 and 80%, respectively) recognized single epitopes within MBP 111–129 in the context of HLA-DRB1*0401 and were found to be clonal T cell populations by TCR sequence analysis. Cloned TCR sequences identical to MS2-3C8 were overrepresented and detected at both time points. The repeated isolation of these clonal T cells indicated a persistent expansion and suggested relevance to the disease process. Previously, humanized TCR transgenic (Tg) mouse models were found suitable to assess the pathogenic potential of human TCR (21); thus, we chose the MS2-3C8 TCR for the generation of humanized TCR Tg mice to examine whether human MBP 111–129–specific and DRB1*0401-restricted T cells have encephalitogenic potential. To explore the pathogenic potential of MS2-3C8 T cells in a murine model, one must consider that murine MBP 111–129 differs from the human sequence by a conservative Arg to Lys substitution in position 122, which might affect T cell recognition. Indeed, whereas MS2-3C8 TCC responded to both human and murine peptides, we found that some MBP 111–129–specific human TCCs did not cross-react with murine MBP 111–129 and therein serve as experimental controls.
Figure 1.

Relationship between the clinical course of patient with MS and the isolation of MS2-3C8 TCR. (A) The clinical course of the MS patient from whom MS2-3C8 T cell clone was isolated. The onset of clinical symptoms that led to the diagnosis of MS preceded by 16 mo the initial visit at the Neuroimmunology Branch, National Institutes of Health (indicated as Start on panel A, horizontal axis). The subsequent follow-up time points are expressed in months from the start. Clinical exacerbations that required i.v. steroid treatment are indicated by black bars. The text boxes with black arrows indicate two clinical episodes experienced by the patient involving cranial nerves, including tongue motor or sensory function, salivatory function, and taste. Limiting dilution assays (LDA) were performed from PBMC obtained at two time points (I and II) as indicated by the white arrows. (B) Persistent presence of MS2-3C8 T cell clone in the MS patient shown in A. T cell clone MS2-3C8 was isolated from LDA I. TCR sequences identical to MS2-3C8 were obtained from T cell clones isolated from LDA II, showing the persistence of the MS2-3C8 TCC for at least 26 mo of follow-up.
Materials and Methods
Animals.
HLA-DRB1*0401 Tg mice lacking the endogenous MHC class II, IA, and IE were purchased from Taconic Farms or bred in our facility. Rag 1−/− Tg mice were purchased from Jackson Laboratory. Mice were housed in specific pathogen-free facilities at the National Institutes of Health.
Generation of Human TCR–HLA-DRB1*0401–IA−/− Tg Mice.
Rearranged MS2-3C8 TCR Vα (AV4S1AJ32) and Vβ (BV2S1BJ1S6) chains were amplified from genomic DNA using primer pairs located in introns upstream and downstream of the V-(D)-J regions. The rearranged Vα and Vβ segments were subcloned into a pair of cassette vectors, pTα and pTβ, respectively (22). The chimeric gene constructs were microinjected into C57BL/6 fertilized eggs at SAIC. Using the same approach, chimeric TCR Tg mice were also generated from the healthy donor-derived human MBP 111–129–specific HLA-DRB1*0401-restricted HD4-1C2 T cell clone (16) that is unresponsive to murine MBP 111–129.
FACS® Analysis.
Anti–HLA-DR (FITC) and anti–human TCR Vβ 2 (PE/FITC/biotinylated) were purchased from Coulter-Immunotech. Other fluorescently labeled antibodies were purchased from BD Biosciences. FACS® analysis was performed on a FACSCalibur using CellQuest software (Becton Dickinson).
Tissue Culture.
Purified human and murine MBP 111–129 (Hu-MBP 111–129, LSRFSWGAEGQRPGFGYGG; Mu-MBP 111–129, LSRFSWGAEGQKPGFGYGG) synthetic peptides were purchased from the Stanford Pan Facility. Murine IL-12 and IL-2 were purchased from Peprotech. CD4+ or CD8+ splenic T cells (>95% pure by FACS® analysis) obtained by positive selection using anti-CD4– or anti-CD8–coated microbeads (Miltenyi Biotec) were cultured with human and murine MBP 111–129. 4 × 104 Tg T cells and 4 × 105 irradiated (3,000 rad) spleen cells from HLA-DRB1*0401–IA−/− Tg mice were incubated at 37°C for 72 h and 1 μCi [3H]thymidine (Dupont) was added to each well for the last 12 h of culture. Quantitation of secreted cytokines was measured by Duoset Murine ELISA kits (R&D Systems). The proliferation of human T cell clones were analyzed as described previously (16).
Induction of EAE.
CD4+ MS2-3C8 and HD4-1C2 Tg Th1 cells were established by cultivation of Tg spleen cells with Hu-MBP 111–129 (10 μg/ml) in the presence of IL-12 (3 ng/ml) as described previously (23). Once cells had rested from the antigen and IL-12–driven Th1 differentiation (typically day 10), cells were again stimulated with MBP 111–129 at 10 μg/ml without IL-12. After 3–4 d, 10–20 million Tg T cells were transferred into irradiated (350 rad) HLA-DRB1*0401–IA−/− Tg mice (10–12 wk old). Pertussis toxin (200 ng) was administered i.v. on days 0 and 2. Mice were weighed daily and assigned clinical scores of ascending EAE paralysis as follows: 0, no symptoms; 1, limp tail; 2, hindlimb weakness; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis.
Histological Examination.
Deeply anesthetized mice were perfused with 30 ml of cold PBS followed by 30 ml of PBS containing 4% paraformaldehyde. The brain and spinal column were carefully removed and post-fixed in 4% formaldehyde for up to 7 d, at which time the spinal cord was removed from the spinal column and embedded in paraffin for sectioning in selected animals. The cranium and vertebral column tissues were trimmed with the brain and spinal cord in place after the fixed cranial and vertebral tissues were routinely decalcified. The select tissue blocks containing cranial tissues were serially sectioned with sections taken at 25-μm intervals for staining. Alternatively, PBS-perfused tissues were cut and prepared for frozen sections. Immunohistochemical analysis of the frozen sections was performed by BD Biosciences.
Results
Development of Humanized Tg T Cells in MS2-3C8 TCR–HLA-DRB1*0401–IA−/− Tg Mice.
MS2-3C8 chimeric TCR α and β gene constructs containing the human TCR Vα4.1-J31/Vβ2.1-D-J1.6 and mouse TCR constant regions were microinjected into C57BL/6 fertilized eggs, and the TCR Tg offspring were crossed with HLA-DRB1*0401–IA−/− Tg mice, which were deficient in the expression of endogenous mouse MHC class II (22). Since neither a MS2-3C8 TCR-specific clonotypic antibody nor a Tg human TCR Vα 4 mAb were available, a human TCR Vβ2 mAb was used for the analysis of Tg TCR Vβ expression. The Tg TCRVβ2 chain was equally expressed on both CD4+ and CD8+ T cell subsets in the Tg mice on the C57BL/6 background (not depicted). However, the Vβ2+ Tg T cells skewed to the CD4+ T cell subset and Vβ2+ Tg T cells represented >80% of CD4+ CD3+ T cells when these Tg mice were crossed with HLA-DRB1*0401–IA−/− Tg mice (Fig. 2, A and B), suggesting that HLA-DRB1*0401 mediates positive thymic selection of HLA-DRB1*0401–restricted Tg T cells. This was confirmed in Tg mice lacking the expression of both MHC class II molecules, HLA-DRB1*0401 and mouse MHC class II. In these Tg mice, CD4+ Vβ2+ Tg T cells did not develop in the thymus, whereas CD8+ Vβ2+ Tg T cells were still positively selected (not depicted). To eliminate the influence of endogenous TCR on thymic selection, we crossed the MS2-3C8 TCR–HLA-DRB1*0401–IA−/− Tg mice with Rag-1−/− mice. We observed a large reduction in Tg T cells in the thymus and spleen of MS2-3C8 TCR–HLA-DRB1*0401–IA−/−–Rag-1−/− Tg mice. CD8+ Tg T cells could not be detected in the spleen. In contrast, a small number of CD4+ Tg T cells could still develop but only when HLA-DRB1*0401 was expressed (Fig. 2 C).
Figure 2.





Development of MS2-3C8 TCR Tg T cells in the thymus and spleen. (A) Thymic expression of the Tg TCR. Thymocytes from MS2-3C8 TCR and non–MS2-3C8 TCR Tg mice (7 wk old) were stained with anti-CD4 mAb, anti-CD8 mAb, and TCR Vβ2 mAb. Genotypes of the Tg mice are described at the top. Histograms were gated on the CD4+CD8− (CD4 SP), CD4−CD8+ (CD8 SP), CD4+CD8+ (DP), and CD4−CD8−(DN) T cells and analyzed for Vβ2 expression (filled histograms) and isotype controls (open histograms). Thymocyte counts: non-TCR Tg mouse, 2.7 × 108; TCR Tg mouse, 3.6 × 107. (B) Splenic expression of the Tg TCR. The spleen cells from the Tg mice analyzed in A were stained by anti-CD3 mAb and anti-TCRVβ2 mAb (top), anti-CD4 mAb, anti-CD3 mAb, and anti-Vβ2 mAb (middle), and anti-CD8 mAb, anti-CD3 mAb, and anti-Vβ2 mAb (bottom). Histograms were gated on the CD4+CD3+ and CD8+CD3+ and analyzed for Vβ2 expression (filled histograms) compared with isotype controls (open histograms). The percentage of cells in each quadrant/gate are indicated. Splenocyte counts: non-TCR Tg mouse, 1.5 × 108; TCR Tg mouse, 6.7 × 107. (C) Thymic selection and splenic expression of MS2-3C8 (B6) TCR Tg T cells in Tg mice on the Rag-1−/− background. Numbers indicate the percentage of cells in each quadrant. The cell numbers are as follows: MS2-3C8 TCR–HLA-DRB1*0401–IA−/−–Rag-1−/− Tg mouse, thymocytes, 3.1 × 107 and splenocytes, 2.6 × 106; MS2-3C8 TCR–HLA-DRB1*0401−/−–IA−/−–Rag-1−/− Tg mouse, thymocytes 3.3 × 107 and splenocytes 2.0 × 106. (D) Phenotypic characterization of CD4+ MS2-3C8 Tg T cells. Expression of phenotypic markers on naïve splenic T cells isolated from MS2-3C8 TCR–HLA-DRB1*0401–IA−/− Tg mice and MS2-3C8 Tg splenic cells activated in vitro with human MBP 111–129. Histograms were gated on Vβ2+ CD4+ cells or those stained after 48 h of activation. (E) Cytokines produced by CD4+ or CD8+ Tg T cells isolated from MS2-3C8 TCR–HLA-DRB1*0401–IA−/− Tg mice. 48-h supernatants were collected from human MBP 111–129–stimulated (10 μg/ml) cultures of CD4+ or CD8+ T cells, as described for the activation marker analysis, and analyzed by ELISA.
Splenic Vβ2-expressing peripheral CD4+ T cells were of naïve phenotype with low levels of CD44, high levels of L selectin and CD45RB (Fig. 2 D), and showed a predominantly Th0 helper profile with negligible amounts of IFN-γ or IL-10 and low to moderate amounts of the cytokines IL-4, TNF-α, and IL-2 (Fig. 2 E). In contrast, CD8+ T cells produced large quantities of IFN-γ with low to moderate production of IL-10 and IL-4.
Prior to determining the capacity of these CD4+ Tg T cells to induce disease, we assessed their specificity. The original CD4+ human T cell clone, MS2-3C8, proliferated in response to both human and mouse MBP 111–129 with consistently higher sensitivity to the human epitope (Fig. 3). This dual specificity and the pattern of increased relative sensitivities to the human epitope (the EC50 was typically one half log greater for human MBP 111–129) were preserved in the CD4+ Tg T cells. As a negative control, we generated HD4-1C2 Tg mice in which Tg T cells responded only to human MBP 111–129 in the context of HLA-DRB1*0401. The murine MBP 111–129 epitope failed to elicit any proliferative response at all (Fig. 3).
Figure 3.

The antigenic specificity of the original human TCRs was preserved in the humanized TCR Tg mice. Shown are the proliferative responses of the MS2-3C8 and HD4-1C2 human TCCs, mouse splenic CD4+ MS2-3C8 and HD4-1C2 TCR Tg T cell lines (TCL), and splenic CD4+ MS2-3C8 TCR Tg and HD4-1C2 TCR Tg T cells to human MBP 111–129 (•) and mouse MBP 111–129 (○).
MS2-3C8 CD4+ Tg T Cells Induce Atypical EAE Signs Besides Typical Ascending Paralysis.
We isolated CD4+ MS2-3C8 TCR Tg T cells from spleens and examined their potential to induce EAE in adoptive transfer experiments. Since EAE is a prototypic Th1-mediated autoimmune disease, we generated Th1 T cells from the spleen cells of two different Tg founder lines, B6 and G2 (Table I) , and transferred them into irradiated HLA-DRB1*0401–IA−/− Tg mice. The MS2-3C8 (B6) Tg T cell line exhibited signs of typical ascending paralysis, correlating with hindlimb weakness and tail limpness averaging a grade 2 on the EAE clinical assessment scale in irradiated HLA-DRB1*0401–IA−/− Tg mice (Fig. 4 and Table II) . In addition to the typical ascending paralysis, several EAE mice spun briskly when hung by their tails and later developed circulating movement or head-tilt (Table II). Of further interest, several mice exhibited difficulty swallowing and lingual paralysis due to restricted jaw and/or tongue movements (Table II). Swallowing difficulties were typically observed by days 14–18 and subsequently progressed in several mice to lingual paralysis. By days 16–20, mice had lost >20% of their body weight after the onset of typical ascending EAE symptoms and eventually appeared moribund (Fig. 4). According to the Animal Care and Use Committee guidelines for maximal allowed weight loss, these mice had to be killed after unsuccessful efforts to gavage mice and slow weight loss. Although typically ascending EAE symptoms had usually not progressed beyond grade 2 by the time of sacrifice, the complications of the lingual paralysis did not allow us to follow mice for evidence of neurological or CNS disease worsening. To exclude the possibility that these clinical characteristics are caused by silencing or induction of genes upon transgene integration, we examined CD4+ MS2-3C8 Tg T cells from another Tg founder line, MS2-3C8 (G2). Similar EAE symptoms were observed with CD4+ MS2-3C8 (G2) TCR Tg T cells (Fig. 4 and Table II); however, circulating movement or head-tilt were less frequently induced than with CD4+ MS2-3C8 (B6) Tg T cells. To confirm that this EAE phenotype is caused by the specificity of the MS2-3C8 Tg TCR, we transferred CD4+ MS2-3C8 Tg Th1 cells established from MS2-3C8 (B6) TCR–HLA-DRB1*0401–IA−/−–Rag-1−/− Tg mice into irradiated HLA-DRB1*0401–IA−/− Tg mice. We observed earlier disease onset with ascending paralysis followed by dysphagia attributable to lingual paralysis (Fig. 4 and Table II), supporting that the MS2-3C8 TCR specificity is involved in this phenotypic presentation. Control experiments with CD4+ HD4-1C2 Tg T cells, which do not respond to murine MBP 111–129, failed to induce any EAE signs (Fig. 4 and Table II).
Table I.
Cytokine Production of Tg T Cell Lines
| IFN-γ
|
TNF-α
|
IL-10
|
IL-4
|
|||||
|---|---|---|---|---|---|---|---|---|
| Tg T cell line | Mu | Hu | Mu | Hu | Mu | Hu | Mu | Hu |
| MS2-3C8(B6) | 26.6 | 52.6 ± 1.1 | 4.2 ± 0.1 | 7.0 ± 0.2 | 0.1 | 0.1 | 0.6 | 0.8 |
| MS2-3C8(G2) | 22.2 ± 1.8 | 50.8 ± 7.1 | 2.8 ± 0.1 | 4.8 ± 0.3 | 1.0 | 1.3 | 1.3 | 1.8 |
| HD4-1C2 | 0 | 156.4 ± 7.2 | 0 | 4.8 ± 0.7 | 0 | 0.3 | 0 | 0.8 |
Splenic MS2-3C8 and HD4-1C2 Th1 cell lines were confirmed by high IFN-γ and TNF-α production after culture with 1 μg/ml of human MBP 111–129 (Hu) or murine MBP 111–129 (Mu) for 72 h. The unit of cytokine production is indicated as ng/ml ± SD.
Figure 4.

Development of ascending paralysis and weight loss after transferring CD4+ Tg Th1 cells into irradiated HLA-DRB1*0401–IA−/− Tg mice (four to five mice for each group). Adoptive transfer of Tg Th1 cells established from MS2-3C8 (G2) Tg and MS2-3C8 (B6) Tg–Rag-1+/+ or Tg–Rag-1−/− mice recognizing murine MBP 111–129 induced disease. HD4-1C2 Tg Th1 cells, nonresponsive to murine MBP 111–129, gave rise to no clinical symptoms. Mice which lost >20% of their body weight (typically attributed to lingual paralysis or swallowing difficulties observed) were killed per the Animal Care and Use Committee guidelines.
Table II.
Development of EAE by Adoptive Transfer with Tg Th1 Cell Lines
| Ascending paralysis a | Dysphagia b | Abnormal gait c | ||||||
|---|---|---|---|---|---|---|---|---|
| Tg T cells | Incidence | Severity | Onset | Incidence | Weight loss (%) | Onset | Incidence | Onset |
| MS2-3C8 (B6) | 11/19 | 1.8 ± 0.8 | 16 ± 4 | 10/19 | 23 ± 6 | 19 ± 3 | 10/19 | 18 ± 3 |
| MS2-3C8 (B6)–Rag-1−/− | 13/13 | 2.0 ± 0.8 | 8 ± 2 | 8/13 | 27 ± 4 | 11 ± 1 | 0/13 | – |
| MS2-3C8 (G2) | 7/9 | 2.1 ± 0.4 | 14 ± 6 | 5/9 | 21 ± 5 | 20 ± 5 | 1/9 | 25 |
| HD4-1C2 | 0/9 | – | – | 0/9 | – | – | 0/9 | – |
The source of the Th1 cell line and the number of mice out of each experiment showing any sign of disease are indicated as well as the mean day of onset ± SD. 10–20 million cells were transferred i.v. after 4 d of in vitro stimulation with human MBP 111–129. Pertussis toxin was administered i.v. on days 0 and 2. Clinical symptoms were divided into three groups to distinguish aascending paralysis from the atypical symptoms of bdysphagia with accompanying weight loss (>20% body weight) and difficulties swallowing or cabnormal gait including spinning, head-tilt and loss of coordinated movement. Ascending paralysis on a scale of 0–4: 0, no symptoms; 1, limp tail; 2, hindlimb weakness; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis. Dysphagia described as the percentage of weight loss compared with peak body weight over course ± SD. Results were pooled from two to five independent experiments.
Inflammatory Lesions Are Observed in the Brainstem and Cranial Nerves and in the Spinal Cord and Spinal Nerve Roots.
Histological analysis demonstrated moderate to severe infiltration of inflammatory cells into the meninges and parenchyma of the spinal cord and brainstem (Fig. 5, A and D, and Table III). Although the predominant cell infiltrate was mononuclear cells consisting of CD4+ lymphocytes and macrophages, in some areas polymorphonuclear leukocyte (Ly-6G+) infiltration was prominent (Fig. 6). CD8+ lymphocytes (Fig. 6) and B cells (not depicted) were not apparent. The inflammatory infiltrate was commonly perivascular, but inflammatory cells were scattered throughout the affected area (Fig. 5, A and D). In some animals, demyelination was noted by pallor of myelin and Luxol Fast blue–positive debris in the tissue and within macrophages (Fig. 5, A–C). In some areas, severe inflammation was associated with tissue vacuolation and necrosis. In many areas, especially in proximity to necrotic areas, numerous axonal spheroids indicated axonal damage, and microthrombi were occasionally observed in blood vessels within inflamed areas (not depicted). Inflammation affected both cerebellum and cerebral hemispheres less frequently, however, than the spinal cord and brainstem (Table III).
Figure 5.
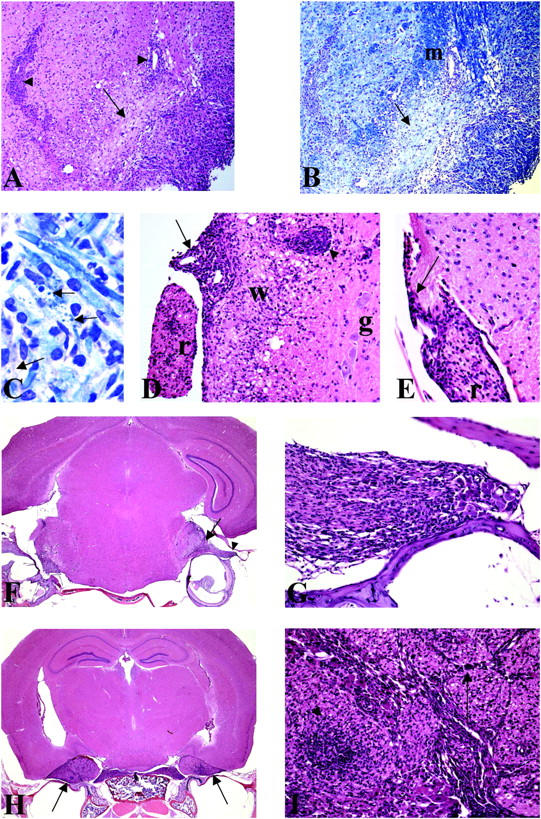
Histopathological analysis of the brain, spinal cord, and cranial nerves of mice exhibiting symptoms of EAE induced with MS2-3C8 Tg Th1 cells. (A) A section of brainstem from an irradiated HLA-DRB1*0401–IA−/− Tg mouse with EAE (grade 2) induced by MS2-3C8 (B6) Tg T cells. There is a heavy inflammatory infiltrate around blood vessels (arrowheads) and lying free in the parenchyma, and in the cochlear nucleus (bottom right corner). The arrow points to an area of pallor, seen to better effect in B. Hematoxylin and eosin, ×75. (B) Adjacent section to that depicted in A stained for myelin. The arrow shows a demyelinated area in the inferior cerebellar peduncle. The normally myelinated peduncle is seen above (m). Luxol Fast blue, ×75. (C) Magnification of the section depicted in B, showing active demyelination with Luxol Fast blue debris (arrows) lying free or within macrophage cells. Luxol Fast blue, ×817. (D) A section of spinal cord from an animal, experimentally identical to that depicted in A. White matter and grey matter are indicated as (w) and (g), respectively. A heavy infiltrate of chronic inflammatory cells in seen in the meninges (arrow) and in the perivascular space of the lateral column white matter (arrowhead). The bundle of spinal roots (r) seen on the left side of the picture also shows a marked inflammatory infiltrate. Hematoxylin and eosin, ×144. (E) Spinal cord section from an animal with an experimental paradigm the same as the animal depicted in D. In this section, the spinal cord parenchyma shows no evidence of inflammation. The peripheral nerve bundle lying in the meninges (r) shows cellular inflammatory infiltration, which is also seen in the meningeal sheath. Of interest is the junction between central and peripheral myelin, shown here by the arrowhead. The central cone of the emerging root is unaffected, whereas the peripheral nerve component is infiltrated. Hematoxylin and eosin, ×213. (F) A cross section of cranial contents from an irradiated HLA-DRB1*0401–IA−/− Tg mouse with EAE (grade 2) induced by MS2-3C8 (G2) Tg T cells. Cranial tissues from this animal were decalcified, and serial sections were stained with hematoxylin and eosin (×12). There is a marked inflammatory cell infiltrate in the cranial nerve root entry/exit zones of brainstem (arrow) and cranial nerve roots (arrowhead). (G) High magnification of the area marked by an arrowhead in F. There is a marked inflammatory cell infiltration of both the cranial nerve roots and the cranial nerve ganglia. The roots and ganglia lie between two bony spicules. Hematoxylin and eosin, ×200. (H) A cross section of cranial contents from an animal, experimentally identical to that depicted in F. Tissue section contains the brain, a branch of the trigeminal cranial nerve and the tympanic cavity of the middle ear. There is a marked inflammatory cell infiltration of trigeminal nerve (arrows). Hematoxylin and eosin, ×12. (I) High magnification of the area marked by the arrow in H showing marked leukocytic infiltration of the trigeminal nerve ganglia (arrowhead) and the peripheral nerve bundle (arrow). Hematoxylin and eosin, ×200.
Table III.
The Location and Severity of Inflammatory Lesions
| T cell lines | MS2-3C8 (B6) | MS2-3C8 (B6)/Rag1−/− | MS2-3C8 (G2) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ascending paralysis |
0 | 2 | 3 | 2 | 3 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 |
| Weight loss (%) | 0 | 22 | 17 | 32 | 15 | 32 | 10 | 25 | 32 | 23 | 22 | 22 | 14 |
| CNS | |||||||||||||
| Spinal cord | – | ++++ | +++++ | +++/ ++++ |
+++ | ++ | +++ | ++++ | +++++ | +++/ ++++ |
+++/ +++++ |
+++/ ++++ |
++/ ++++ |
| Brainstem | – | ++++ | +++/ ++++ |
++/ +++ |
++++ | +++ | ++ | ++ | +++ | +++ | +++/ ++++ |
++/ +++ |
+++ |
| Cerebellum | – | – | – | + | – | – | – | – | + | – | – | – | + |
| Cerebrum | – | – | – | + | + | – | – | ++ | + | ++ | + | + | + |
| PNS | |||||||||||||
| Spinal root | – | – | ++ | – | ++ | – | ++ | ++ | – | +++ | ++ | + | ++ |
| Sciatic and axillary nerves |
– | – | + | – | + | – | – | – | – | + | – | + | – |
| Cranial nerves | – | +++ | +++ | ++/ +++ |
++++ | ++ | ++ | ++ | +++ | ++/ +++ |
++/ +++ |
+++ | ++/ +++ |
Th1 cells from the Tg mice listed were isolated and transferred into irradiated HLA-DRB1*0401–IA−/− Tg mice. The EAE score and percentage of weight loss are listed for 13 animals which were examined histologically. For comparative purposes, a visual grading scheme was assigned to the lesions in each of the animals examined. The histological score of inflammatory lesions is as follows: +, occasional inflammatory cell cuffing in meninges; ++, mild lesions in a few sections (<50%); +++, mild or moderate lesions in many sections (>50%); ++++, severe or moderate changes in a few sections (<50%); +++++, severe or moderate changes in many sections (>50%). Necrosis and axonal degeneration were observed in most lesions of grade 4 to 5 (++++ → +++++) severity. Demyelination, as observed in Luxol Fast blue–stained slides, was seen in the spinal cord and brainstem in 6 of 12 mice examined.
Figure 6.
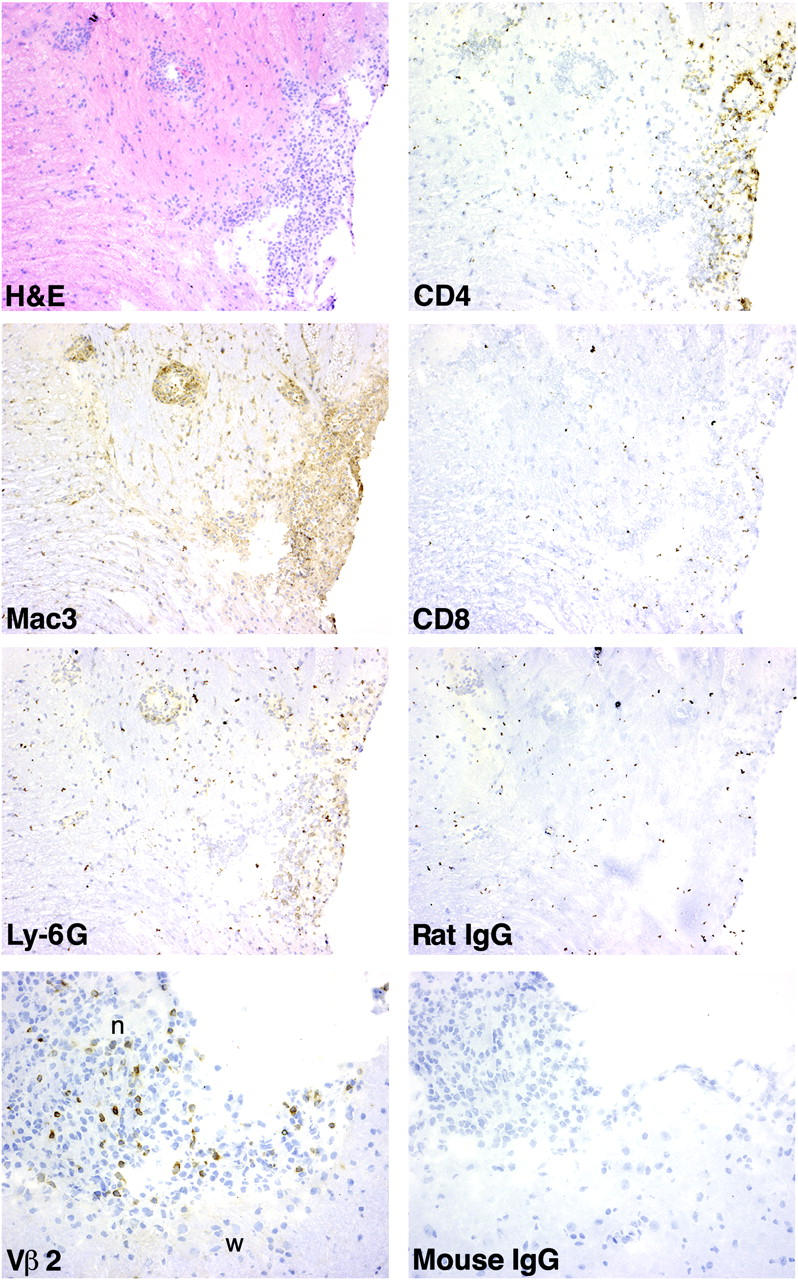
Immunohistochemical analysis of inflammatory infiltrates in brainstem and cranial nerve root. Frozen sections of brainstem from an irradiated Tg mouse with EAE (grade 2) induced by MS2-3C8 (B6) Tg T cells were stained for hematoxylin and eosin, CD4, Mac3, CD8, Ly-6G, and isotype control mAbs. Cranial nerve root (n) and brainstem (w) were stained with anti-Vβ2 and isotype control mAbs.
Of particular interest was the finding of cellular inflammation within the proximal peripheral nervous system (PNS) and CNS tissues. In some instances, PNS and CNS involvement coincided (Fig. 5 D), whereas in other instances, the inflammatory process appeared to selectively involve the PNS (Fig. 5 E) and was most intense in the nerve root between the spinal cord and ganglion. There was little evidence for inflammation along the peripheral nerve beyond the extradural space (Table III). However, occasional inflammatory infiltration of the peripheral nerves was seen in a few sections (Table III). In mice that developed dysphagia, particular attention was given to inflammatory lesions in the cranial nerves, particularly the root entry areas. In these animals, the brains were left in the cranium, and subsequent to decalcification serial sections were made to evaluate the extent of inflammation along the path of the cranial nerve roots. Inflammation was observed in the vestibulo-cochlear (Fig. 5 F) and the trigeminal nerves (Fig. 5 H). There were severe infiltrates in the ganglia and roots of these cranial nerves (Fig. 5, G and I) and entry/exit zones of brainstem (Fig. 5 F). The inflammation was greatly decreased in more distal sections of the cranial nerves (not depicted). Immunohistochemical analysis revealed infiltrates of Vβ2+ Tg T cells in the lesions of the cranial nerve roots and the brainstem (Fig. 6). These histological analyses suggest the involvement of MS2-3C8 Tg T cells in the development of neurological damage of the brainstem and cranial nerves with subsequent clinical signs such as difficulty of tongue and jaw movements in animals showing atypical EAE.
Discussion
Classical selection theories suggest that antigen expression in the thymus should lead to central deletion of T cells encountering their cognate antigen. However, there are several possible explanations for the survival of MS2-3C8 Tg TCR T cells. Golli-MBP, the isoform of MBP expressed in the thymus, does not contain MBP 111–129, and therefore deletion would not necessarily be expected (24, 25). Even if classical MBP were to enter the thymus from the periphery and be presented by thymic APCs, the low binding affinity of MBP 111–129 to DRB1*0401 (16) may allow their escape from deletion. This rationale might also explain the survival of Tg TCR T cells in other myelin protein–specific TCR Tg mice, humanized or not (21, 26–28). Except for MBP 83–99, each of these myelin peptides is either a poor MHC binder or not expressed in the thymus, e.g., MOG 35–55 and PLP 139–154 (29, 30). However, we observed a marked reduction in MS2-3C8 Tg T cells when the mice were crossed to Rag-1−/− mice due to negative selection or lack of positive selection. Analysis of thymocyte cellularity and CD5 expression on CD4+CD8+ double positive thymocytes suggested the reduction is caused by thymic negative selection (unpublished data). Moreover, Tg T cells that escaped thymic deletion may have expressed endogenous mouse TCR together with the Tg α/β TCR heterodimer; thus, dual TCR expression on the Tg T cells may have led to their survival.
In MS2-3C8 TCR–HLA-DRB1*0401 Tg mice, Tg T cells were not activated in vivo (Fig. 2 D) and mice did not develop spontaneous EAE (unpublished data). Since the MS2-3C8 human TCC is a CD4+ T cell, we investigated the pathogenic characteristics of CD4+ Tg T cells. To eliminate the influence of CD8+ and DN Tg T cells on the induction of EAE, we isolated CD4+ Tg T cells from spleen cells and transferred CD4+ Th1 Tg cells into irradiated HLA-DRB1*0401 Tg mice. For comparison with the MS2-3C8 TCR, we generated HD4-1C2 TCR Tg mice from a human T cell clone which does not cross-react to the mouse epitope. We show that MS2-3C8 Tg CD4+ Th1 cells induced EAE, whereas HD4-1C2 Tg CD4+ Th1 cells did not, indicating that EAE induction is mediated by MS2-3C8 TCR recognition of the murine MBP 111–129–HLA-DRB1*0401 complex. This was confirmed using MS2-3C8 TCR–Rag-1−/− Tg T cells which lack endogenous mouse TCRs (Fig. 4 and Table II). Since the CD8+ Tg T cells expressed CD28 and produced high amounts of IFN-γ in response to MBP 111–129, we examined the pathogenic potential of CD8+ Tg T cells by adoptive transfer into irradiated HLA-DRB1*0401–IA−/− Tg mice. We did not observe any signs of EAE in animals receiving 10–20 million CD8+ Tg T cells, nor did the cotransfer of 10 million CD8+ Tg T cells at a 1:1 ratio with CD4+ Tg T cells suppress or worsen passive EAE (unpublished data). Active EAE could not be induced despite natural processing of MBP 111–129 and the stimulation of Tg T cells in vivo (unpublished data). Peripheral mechanisms of tolerance were not apparent as the explanation for ineffective active disease induction, since Tg T cells isolated from the spleen or lymph node proliferated in response to MBP 111–129, unaffected by addition of IL-2 (not depicted). Other mechanisms involved in active EAE suppression are currently under investigation.
MS presents with substantial clinical and pathologic heterogeneity (1). Previous data from genetic studies on HLA-DR associations with MS (4–7) and EAE experiments in animals with different MHC class II backgrounds and different encephalitogens (2) suggest the clinical phenotype of MS–EAE is in part caused by the expression of different HLA-DR–MHC class II alleles and the T cell response to specific myelin peptides. Since the HLA-DR2 haplotype is strongly associated with MS, and HLA-DRB1*1501 and HLA-DRB5*0101 genes are coexpressed in humans carrying the HLA-DR2 allele, HLA-DRB1*1501-restricted MBP 83–99–specific TCR Tg mice were generated. The Tg mice developed spontaneous ascending paralysis (21). Recently, we have developed a HLA-DRB5*0101-restricted MBP 83–99–specific human TCR Tg mouse (TL3A6 Tg mice). The TL3A6 Tg Th1 cells induced severe ascending paralysis, although they do not induce lingual paralysis in spite of weight loss (unpublished data). In accordance with the clinical symptoms, the lesions were predominantly observed in the spinal cord, and less inflammatory cells infiltrated into the cranial nerves (unpublished data). Therefore, lingual paralysis associated with the inflammation of brainstem and cranial nerves was a unique feature of MS2-3C8 Tg mice.
The patient from whom the MS2-3C8 TCC was derived did not suffer from chronic persisting dysphagia; however, the patient did present two episodes, one consisting of disturbances in tongue movement impairing swallowing of fluids and speech articulation and a second episode consisting of tongue numbness and dysgeusia (Fig. 1). These episodes were comprised between the two time points at which MS2-3C8 TCRs were isolated. We are well aware that the relationship between MBP 111–129–specific T cell responses (or the presence of a specific TCR) and the manifestation of clinical signs and symptoms should be interpreted with caution; however, the phenotypic characteristics of this particular EAE model and the similarities in the patient's clinical history are intriguing. Interestingly, ∼30-40% of MS patients develop dysphagia (31–33), albeit typically late in disease.
Clearly, several factors can contribute to the clinical manifestations of EAE in any one particular model, perhaps the two greatest contributors being the genetic background and the immunizing myelin antigen or the target autoantigen. It has been reported that the antigenic epitope specificity of myelin antigens influences the phenotype of EAE and location of the lesions. In Balb/c (IAd, IEd) mice, IAd-restricted MBP 59–76–specific T cells induce classical EAE with characteristic inflammation and demyelination of the CNS (34). In contrast, IEd-restricted MBP 151–168–specific T cells induce inflammation and demyelination of preferentially PNS myelin including nerves in the hind leg and spinal roots. The authors suggested two possibilities to explain their result. The first is that the distinct inflammation sites may be attributable to qualitative differences in MBP isoform composition in the CNS and PNS. The MBP 151–168 epitope, which is encoded by exons 6 and 7, exists only in the 18.5- and 21-kD MBP isoforms, whereas the MBP 59–76 epitope exists in all of the 14-, 17-, 18.5-, and 21-kD isoforms. The minimal epitope of MBP recognized by the MS2-3C8 TCR, MBP 116–123 (16), is encoded by exons 5 and 6. Therefore, similar to the MBP 151–168 epitope recognition described above, only the 18.5- and 21-kD isoforms are recognized by MS2-3C8 Tg T cells. Although the quantitative distribution of these isoforms is unknown at the present, the unique EAE phenotype inducible by MS2-3C8 Tg T cells may be caused by the distinct distribution of MBP isoforms in the CNS and PNS. Second, the authors suggested that high versus low affinity interactions in the complex of MBP–IA or –IE molecules may generate distinct downstream signaling events after TCR engagement. Interestingly, MBP 59–76 strongly binds to IAd molecules, whereas MBP 151–168 weakly binds to IEd molecules. Such differences are likely to affect the expression of adhesion and costimulatory molecules involved in transmigration through perivascular endothelial cells of either the blood-brain or blood-nerve barrier, and thus influence factors key to T cell encephalitogenicity. This hypothesis was supported by EAE experiments in CH3 congenic mice where immunization of H2q congenic C3H mice with PLP 103–116 induces typical ascending EAE. In contrast, immunization of H2p congenic C3H mice with the same antigen induces atypical EAE, ataxia, and imbalance without any symptoms of ascending paralysis (35). The binding affinity of PLP 103–116 to IAp was lower than that to IAq, suggesting that antigen binding affinity of myelin antigens to MHC class II molecules could influence the phenotype of EAE. Moreover, signals through TCR and costimulatory molecules could lead to the expression of adhesion molecules and chemokine production that are involved in the infiltration of pathogenic T cells. EAE experiments that used CD28-deficient mice supported this hypothesis. When the CD28-deficient mice were immunized with MOG 35–55, inflammatory cells predominantly infiltrated into the leptomeninges of the brain although immunization of wild-type mice with the same antigen induced typical ascending paralysis with the infiltrates in the parenchyma of the CNS (36). Similarly, the poor binding of MBP 111–129 to HLA-DRB1*0401 molecules and the signaling events subsequent to TCR engagement may shape the unique EAE phenotype exhibited by the MS2-3C8 Tg TCR.
Properties of individual TCR, the pattern of migration of individual T cell clones, or the localization/abundance of antigens are all likely to come into play, as was suggested in the recent report of MOG Tg TCR mice. T cells specific for MOG35-55 gave rise to spontaneous optic neuritis, most often in the absence of typical EAE symptoms (37). Moreover, in each model of EAE the overwhelming presentation of symptoms considered to be typical may mask those symptoms occurring less frequently, such as those described here. In this regard, TCR transgenic mice may be valuable to glean antigen/MHC-specific pathology from the otherwise heterogeneous presentation of disease that is observed in other models. With respect to the simultaneous occurrence of cranial nerve deficits and the typical sensorimotor signs, there is precedence in MS patients who not uncommonly develop trigeminal neuralgia or more rarely involvement of other cranial nerves.
Although it is difficult to extrapolate to human disease clinical findings from experimental animal models, our results show that inflammatory responses of MS2-3C8 Tg T cells directed against MBP 111–129 can induce unique clinical and pathological features of damage to the nervous system that are distinct from previously described Tg models (21) based on TCR recognizing different MBP peptide–MHC complexes. Such correlations between specificity and restriction of myelin-specific T cells may provide important insights into the immunopathological heterogeneity of MS. Clearly, humanized Tg mice expressing a single TCR are biased and may not represent all clinical features of the MS patient from whom the Tg TCR had been derived. However, humanized TCR Tg mouse models could be an exemplary way to dissect the phenotypic heterogeneity linked to a T cell response against defined myelin peptides in the context of a disease-associated HLA-DR allele in the development of MS.
Acknowledgments
We would like to thank Drs. William Biddison, Steve Jacobson, Alfred Singer, Georgina Miller, and Michael P. Pender for helpful discussion and also thank Dr. Terri Clark and her staff for excellent animal care. We also thank Dr. Diane Mathis (Harvard Medical School, Boston, MA) for generously providing the TCR cassette vectors.
J.A. Quandt was supported by fellowships from the Canadian and National Multiple Sclerosis Societies, and Y.K. Kawamura was also supported by the National Multiple Sclerosis Society. M. Baig was supported by Howard University M.D., Ph.D. program and the Gustavus and Louise Pfeiffer Research Foundation.
J. Quandt and M. Baig contributed equally to this work.
Abbreviations used in this paper: CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; HLA, human leukocyte antigen; MBP, myelin basic protein; MS, multiple sclerosis; PNS, peripheral nervous system; TCC, T cell clone; Tg, transgenic.
References
- 1.Lassmann, H. 1998. Neuropathology in multiple sclerosis: new concepts. Mult. Scler. 4:93–98. [DOI] [PubMed] [Google Scholar]
- 2.Martin, R., H.F. McFarland, and D.E. McFarlin. 1992. Immunological aspects of demyelinating diseases. Annu. Rev. Immunol. 10:153–187. [DOI] [PubMed] [Google Scholar]
- 3.Steinman, R.M. 1996. Dendritic cells and immune-based therapies. Exp. Hematol. 24:859–862. [PubMed] [Google Scholar]
- 4.Hauser, S.L., E. Fleischnick, H.L. Weiner, D. Marcus, Z. Awdeh, E.J. Yunis, and C.A. Alper. 1989. Extended major histocompatibility complex haplotypes in patients with multiple sclerosis. Neurology. 39:275–277. [DOI] [PubMed] [Google Scholar]
- 5.Sawcer, S., H.B. Jones, R. Feakes, J. Gray, N. Smaldon, J. Chataway, N. Robertson, D. Clayton, P.N. Goodfellow, and A. Compston. 1996. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat. Genet. 13:464–468. [DOI] [PubMed] [Google Scholar]
- 6.Sadovnick, A.D., G.C. Ebers, D.A. Dyment, and N.J. Risch. 1996. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet. 347:1728–1730. [DOI] [PubMed] [Google Scholar]
- 7.Haines, J.L., M. Ter-Minassian, A. Bazyk, J.F. Gusella, D.J. Kim, H. Terwedow, M.A. Pericak-Vance, J.B. Rimmler, C.S. Haynes, A.D. Roses, et al. 1996. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group. Nat. Genet. 13:469–471. [DOI] [PubMed] [Google Scholar]
- 8.Jersild, C., B. Dupont, T. Fog, P.J. Platz, and A. Svejgaard. 1975. Histocompatibility determinants in multiple sclerosis. Transplant. Rev. 22:148–163. [DOI] [PubMed] [Google Scholar]
- 9.Olerup, O., J. Hillert, S. Fredrikson, T. Olsson, S. Kam-Hansen, E. Moller, B. Carlsson, and J. Wallin. 1989. Primarily chronic progressive and relapsing/remitting multiple sclerosis: two immunogenetically distinct disease entities. Proc. Natl. Acad. Sci. USA. 86:7113–7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uria, D.F., V. Gutierrez, B.B. Menes, J.M. Arribas, and C. Lopez-Larrea. 1993. HLA class II susceptibility and resistance genes in patients with multiple sclerosis from northern Spain, by DNA-RFLP genotyping. J. Neurol. Neurosurg. Psychiatry. 56:722–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grasso, M.G., F. Cutrupi, S. Bernardi, S. Trabace, C. Pozzilli, S. Cappellacci, and C. Fieschi. 1992. HLA determinants in familial multiple sclerosis. Neuroepidemiology. 11:85–89. [DOI] [PubMed] [Google Scholar]
- 12.Marrosu, M.G., F. Muntoni, M.R. Murru, G. Spinicci, M.P. Pischedda, F. Goddi, P. Cossu, and M. Pirastu. 1988. Sardinian multiple sclerosis is associated with HLA-DR4: a serologic and molecular analysis. Neurology. 38:1749–1753. [DOI] [PubMed] [Google Scholar]
- 13.Kurdi, A., I. Ayesh, A. Abdallat, and U. Maayta. 1977. Different B lymphocyte alloantigens associated with multiple sclerosis in Arabs and North Europeans. Lancet. 1:1123–1125. [DOI] [PubMed] [Google Scholar]
- 14.Ota, K., M. Matsui, E.L. Milford, G.A. Mackin, H.L. Weiner, and D.A. Hafler. 1990. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. 346:183–187. [DOI] [PubMed] [Google Scholar]
- 15.Chou, Y.K., P. Henderikx, M. Vainiene, R. Whitham, D. Bourdette, C.H. Chou, G. Hashim, H. Offner, and A.A. Vandenbark. 1991. Specificity of human T cell clones reactive to immunodominant epitopes of myelin basic protein. J. Neurosci. Res. 28:280–290. [DOI] [PubMed] [Google Scholar]
- 16.Muraro, P.A., M. Vergelli, M. Kalbus, D.E. Banks, J.W. Nagle, L.R. Tranquill, G.T. Nepom, W.E. Biddison, H.F. McFarland, and R. Martin. 1997. Immunodominance of a low-affinity major histocompatibility complex-binding myelin basic protein epitope (residues 111-129) in HLA-DR4 (B1*0401) subjects is associated with a restricted T cell receptor repertoire. J. Clin. Invest. 100:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin, R., and H.F. McFarland. 1995. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit. Rev. Clin. Lab. Sci. 32:121–182. [DOI] [PubMed] [Google Scholar]
- 18.Hashim, G., A.A. Vandenbark, D.P. Gold, T. Diamanduros, and H. Offner. 1991. T cell lines specific for an immunodominant epitope of human basic protein define an encephalitogenic determinant for experimental autoimmune encephalomyelitis-resistant LOU/M rats. J. Immunol. 146:515–520. [PubMed] [Google Scholar]
- 19.Westall, F.C., A.B. Robinson, J. Caccam, J. Jackson, and E.H. Ylar. 1971. Essential chemical requirements for induction of allergic encephalomyelitis. Nature. 229:22–24. [DOI] [PubMed] [Google Scholar]
- 20.Teitelbaum, D., C. Webb, R. Arnon, and M. Sela. 1977. Strain differences in susceptibility to experimental allergic encephalomyelitis and the immune response to the encephalitogenic determinant in inbred guinea pigs. Cell. Immunol. 29:265–271. [DOI] [PubMed] [Google Scholar]
- 21.Madsen, L.S., E.C. Andersson, L. Jansson, M. Krogsgaard, C.B. Andersen, J. Engberg, J.L. Strominger, A. Svejgaard, J.P. Hjorth, R. Holmdahl, et al. 1999. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat. Genet. 23:343–347. [DOI] [PubMed] [Google Scholar]
- 22.Kouskoff, V., K. Signorelli, C. Benoist, and D. Mathis. 1995. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 180:273–280. [DOI] [PubMed] [Google Scholar]
- 23.Lafaille, J.J., F.V. Keere, A.L. Hsu, J.L. Baron, W. Haas, C.S. Raine, and S. Tonegawa. 1997. Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J. Exp. Med. 186:307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitamura, K., S.L. Newman, C.W. Campagnoni, J.M. Verdi, T. Mohandas, V.W. Handley, and A.T. Campagnoni. 1990. Expression of a novel transcript of the myelin basic protein gene. J. Neurochem. 54:2032–2041. [DOI] [PubMed] [Google Scholar]
- 25.Fritz, R.B., and I. Kalvakolanu. 1995. Thymic expression of the golli-myelin basic protein gene in the SJL/J mouse. J. Neuroimmunol. 57:93–99. [DOI] [PubMed] [Google Scholar]
- 26.Lafaille, J.J., K. Nagashima, M. Katsuki, and S. Tonegawa. 1994. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell. 78:399–408. [DOI] [PubMed] [Google Scholar]
- 27.Goverman, J., A. Woods, L. Larson, L.P. Weiner, L. Hood, and D.M. Zaller. 1993. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 72:551–560. [DOI] [PubMed] [Google Scholar]
- 28.Waldner, H., M.J. Whitters, R.A. Sobel, M. Collins, and V.K. Kuchroo. 2000. Fulminant spontaneous autoimmunity of the central nervous system in mice transgenic for the myelin proteolipid protein-specific T cell receptor. Proc. Natl. Acad. Sci. USA. 97:3412–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruno, R., L. Sabater, M. Sospedra, X. Ferrer-Francesch, D. Escudero, E. Martinez-Caceres, and R. Pujol-Borrell. 2002. Multiple sclerosis candidate autoantigens except myelin oligodendrocyte glycoprotein are transcribed in human thymus. Eur. J. Immunol. 32:2737–2747. [DOI] [PubMed] [Google Scholar]
- 30.Klein, L., M. Klugmann, K.A. Nave, V.K. Tuohy, and B. Kyewski. 2000. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6:56–61. [DOI] [PubMed] [Google Scholar]
- 31.Thomas, F.J., and C.M. Wiles. 1999. Dysphagia and nutritional status in multiple sclerosis. J. Neurol. 246:677–682. [DOI] [PubMed] [Google Scholar]
- 32.Hartelius, L., and P. Svensson. 1994. Speech and swallowing symptoms associated with Parkinson's disease and multiple sclerosis: a survey. Folia Phoniatr. Logop. 46:9–17. [DOI] [PubMed] [Google Scholar]
- 33.De Pauw, A., E. Dejaeger, B. D'Hooghe, and H. Carton. 2002. Dysphagia in multiple sclerosis. Clin. Neurol. Neurosurg. 104:345–351. [DOI] [PubMed] [Google Scholar]
- 34.Yoshizawa, I., R. Bronson, A. Ben-Nun, J.R. Richert, M.E. Dorf, and S. Abromson-Leeman. 1998. Differential recognition of MBP epitopes in BALB/c mice determines the site of inflammatory disease induction. J. Neuroimmunol. 89:73–82. [DOI] [PubMed] [Google Scholar]
- 35.Kjellen, P., L. Jansson, M. Vestberg, A. Andersson, R. Mattsson, and R. Holmdahl. 2001. The H2-Ab gene influences the severity of experimental allergic encephalomyelitis induced by proteolipoprotein peptide 103-116. J. Neuroimmunol. 120:25–33. [DOI] [PubMed] [Google Scholar]
- 36.Perrin, P.J., E. Lavi, C.A. Rumbley, S.A. Zekavat, and S.M. Phillips. 1999. Experimental autoimmune meningitis: a novel neurological disease in CD28-deficient mice. Clin. Immunol. 91:41–49. [DOI] [PubMed] [Google Scholar]
- 37.Bettelli, E., M. Pagany, H.L. Weiner, C. Linington, R.A. Sobel, and V.K. Kuchroo. 2003. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]