

Abstract
Transcription factors of the interferon regulatory factor (IRF) family contribute to the regulation of cell proliferation and apoptosis. Here, we show that CD4+ T helper (Th) cells lacking IRF4 (IRF4−/−) are highly sensitive to apoptosis. After infection of IRF4−/− mice with the protozoan parasite Leishmania major, the lesion-draining lymph nodes developed the prototypic lymphadenopathy of wild-type mice after 4 wk, but demonstrated almost total loss of cellularity and enhanced apoptosis after 7 wk. In vitro, activation of IRF4−/− CD4+ Th cells led to greatly increased apoptosis compared with wild-type cells. Coculture of IRF4−/− and IRF4+/+ CD4+ cells did not increase survival of IRF4−/− CD4+ cells, indicating that the enhanced rate of IRF4−/− Th cell apoptosis was neither transferable nor due to lack of a cytokine. Enhanced CD4+ cell apoptosis was also observed after anti-CD95 mAb treatment, despite normal CD95 expression. Removal of endogenous cytokines, notably interleukin (IL)-4, led to increased and equally high levels of IRF4−/− and IRF4+/+ cell apoptosis, whereas the protective activity of exogenous IL-4 was reduced in IRF4−/− CD4+ cells despite normal expression of the IL-4 receptor. Therefore, IRF4 is central in protecting CD4+ cells against proapoptotic stimuli.
Keywords: Th cell, apoptosis, IRF4, CD95, Leishmania major, T helper cell, IL-4
Introduction
The IFN regulatory factor (IRF) family of transcription factors includes the IRF1-IRF7 proteins, IFN-stimulated gene factor 3 γ, and IFN consensus sequence-binding protein (ICSBP; reference 1). IRF proteins bind to regulatory elements found in the promoters of IFN-inducible genes. IRF1, IRF2, and ICSBP are mandatory for normal Th1 cell differentiation due to their ability to induce IL-12 (2–5). IRF1 and IRF2 have also been implicated in the regulation of cell proliferation (6). Recently, striking immune defects were found in mice lacking IRF4, including impaired antibody production, almost absent antiviral cytotoxic T cell responses, and deficient Th2 cell differentiation of CD4+ T cells (7–9). IRF4 may also influence the life span of lymphocytes because IRF4−/− mice develop progressive lymphadenopathy (7) and because overexpression of IRF4 leads to enhanced apoptosis in human Jurkat cells (10).
Herein, we have studied the role of IRF4 in T cell survival in more detail in vivo and in vitro using IRF4−/− mice. For the in vivo studies, mice were infected with the protozoan parasite Leishmania major. In mouse strains resistant to this infection (e.g., C57BL/6), L. major–specific Th1 cells expand and control the disease (11). We demonstrate that in L. major–infected IRF4−/− mice of the C57BL/6 genotype, initial hyperplasia is followed by strongly enhanced apoptosis in the lesion-draining LNs. This finding correlates with enhanced apoptosis of IRF4−/− Th cells after TCR or CD95 triggering, a surprising result in light of the spontaneous lymphadenopathy in IRF4−/− animals and the conflicting results reported in Jurkat cells.
Materials and Methods
Mice.
IRF4−/− mice (7) were used at the seventh backcross generation to C57BL/6. Wild-type C57BL/6, BALB/c, and C57BL/6-CD45.1 congenic mice were purchased from Charles River Laboratories. RAG-1–deficient mice were bred at Taconic. All mice used were between 6 and 12 wk of age. All experiments were conducted in accordance with German and Canadian animal protection laws.
Cell Purification and Adoptive Transfer into RAG-1−/− Mice.
C57BL/6 B cells and IRF4+/− as well as IRF4−/− CD4+ Th cells were purified by magnetic cell sorting using the MACS system (Miltenyi Biotec) as described previously (3). The purity of these cells was always 93–98%. B cells (9 × 106) and CD4+ IRF4+/− or IRF4−/− cells (6 × 106) were transferred i.p. into RAG-1−/− mice (3–4 mice per experimental group). After cell transfer, these mice were infected with L. major promastigotes as described in the next paragraph and analyzed 6 wk later. All experiments were performed at least twice with similar results.
Leishmania Infection and Apoptosis In Vivo.
Mice (3–4 per group) were infected in the right hind footpad with 2 × 107 stationary-phase promastigotes of L. major strain MHOM/IL/81/FEBNI as described previously (3). Lesion development as well as parasite burden were determined as described previously (3). Apoptosis within LNs of infected mice was investigated 5 wk after L. major infection by the immediate processing of single cell suspensions for a TdT-mediated dUTP nick-end labeling (TUNEL) reaction and flow cytometric analysis (In Situ Cell Death kit; Boehringer). LNs were fixed and processed for hematoxylin eosin (HE) staining according to standard procedures.
Th Cell Stimulation In Vitro.
Total CD4+ Th cells from IRF4+/+ or −/− mice were purified using the first steps of the multisort kit (Miltenyi Biotec) as described previously (5). 106/ml of purified Th cells were stimulated in vitro for 72 h with 5 μg/ml of immobilized anti-CD3 mAb, 2.5 μg/ml of soluble anti-CD28 mAb (BD Biosciences), or 100 U/ml of recombinant human IL-2 as described previously (5). After a resting period of 48–72 h in the absence of anti-CD3 mAb, but in the presence of IL-2, viable cells were purified on a Ficoll density gradient and restimulated in the presence of IL-2 either with anti-CD3 (immobilized at 5 μg/ml) or with 1 μg/ml of soluble anti-Fas mAb Jo2 (Becton Dickinson) together with 2 μg/ml of protein G (Sigma-Aldrich). Apoptosis was analyzed by annexin V and propidium iodide (PI) staining as described previously (12) at the indicated times after secondary stimulation. Viable as well as dead cells were included in this analysis according to forward and side scatter characteristics. The role of CD95L during apoptosis was analyzed either by staining with PE-conjugated anti-CD95L mAb (Becton Dickinson) or by inclusion of a neutralizing anti-CD95L mAb (clone K10; 10 μg/ml; Becton Dickinson) during restimulation. The CD95 molecule was stained using biotinylated mAb Jo2 detected by FITC-conjugated streptavidin (Becton Dickinson). Cells were analyzed on a FACScan™ using Lysys II software (Becton Dickinson). In experiments where IRF4−/− and CD45.1 congenic CD4+ Th cells were mixed, they were distinguished by staining with anti-CD45.2 mAb (Becton Dickinson) and PE-conjugated anti–mouse IgG Ab (Dianova).
Results and Discussion
Enhanced Apoptosis within Lesion-draining LN Cells of L. major–infected IRF4−/− Mice.
To determine the role of IRF4 for the immune response during an infection, IRF4−/− and IRF4+/− mice were infected with L. major parasites. 4 and 7 wk later, the number of viable cells in the lesion-draining popliteal LNs was determined after staining with trypan blue. In uninfected mice of either genotype, popliteal LNs contained <106 cells. At 4 wk after infection, the cell number per LN had increased equally in IRF4−/− and IRF4+/− mice to ∼30 × 106. Significantly, by 7 wk after infection, popliteal LN of IRF4+/− mice still contained ∼15 × 106 cells, whereas in popliteal IRF4−/− LNs, the cell number had virtually dropped to zero (<105 cells/LN). In contrast, cell numbers in IRF4−/− LNs not draining the infectious lesion were either comparable to the respective IRF4+/− LNs or higher as a reflection of the progressive lymphadenopathy of IRF4−/− mice (7). While our work was in progress, a similar finding was reported by another group, although without further analysis of this phenomenon (13).
The disappearance of popliteal IRF4−/− LNs was due to apoptosis, as analyzed 5 wk after L. major infection by immediate processing of LN cells for TUNEL staining. At this time, the destruction of the draining LNs was not yet complete. Cells in draining IRF4−/− LNs (Fig. 1 A) exhibited a rate of apoptosis almost six times higher than that in IRF4+/− LNs (Fig. 1 B). No enhanced rate of apoptosis was noted in the irrelevant axillary IRF4−/− LNs (Fig. 1 D). Histological examination confirmed these findings: apoptotic cells were detectable in the draining LNs of IRF4−/− (Fig. 1 E), but not of IRF4+/− mice (Fig. 1 F). LN cell apoptosis correlated with an increase in lesion size (Fig. 1 G) and with a strongly increased parasite burden as follows: 7 wk after infection, footpads of IRF4−/− mice contained log 7.3 ± 0.6 parasites versus log 2.6 ± 0.3 in IRF4+/− mice. Spleens of IRF4−/− mice harbored log 6.6 ± 0.3 parasites versus log 3.2 ± 0.4 in IRF4+/− mice.
Figure 1.
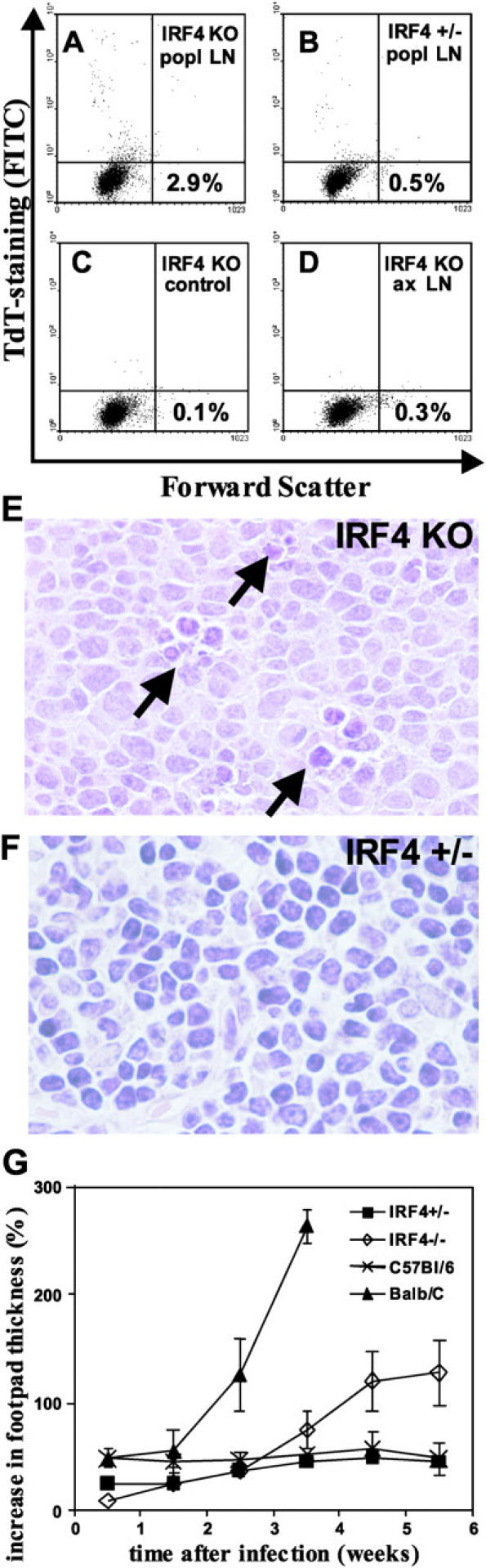
Enhanced cell apoptosis in draining LNs of L. major–infected IRF4−/− mice. (A–D) 5 wk after L. major infection, single cell suspensions of popliteal LN cells were processed for TUNEL staining and analyzed by flow cytometry. (E and F) HE-staining of popliteal LNs. (A and C–E) IRF4−/− mice. (B and F) IRF4+/− mice. (A–C, E, and F) Draining popliteal LNs. (D) Irrelevant axillary LNs. (C) Control staining for IRF4−/− mice without the TdT enzyme. (y axis) staining for apoptotic cells (FITC). Numbers indicate the percentage of apoptotic cells located in the top left quadrant. Arrows in E indicate clusters of apoptotic cells. Three independent experiments were performed with similar results. (G) Increase in lesion size: IRF4+/− and IRF4−/− as well as control susceptible BALB/c and resistant C57BL/6 mice were infected with L. major, and the increase in lesion size was monitored, as described previously (reference 3).
Because CD4+ Th cells are a major component of the hyperplastic LN after L. major infection (11), it was possible that IRF4−/− Th cells are prone to activation-induced apoptosis. To analyze this issue, we adoptively transferred purified IRF4+/+ B cells plus CD4+ T cells from either IRF4+/− or IRF4−/− mice into IRF4+/+ RAG-1–deficient mice that otherwise lack T and B cells. The recipients were infected with L. major and their popliteal LNs were analyzed 6 wk later. Although the draining LNs of mice receiving IRF4+/− CD4+ T cells contained 8.0 ± 1.7 × 105 cells, this number was <5 × 104 in each of three mice receiving IRF4−/− CD4+ T cells. Thus, even though not formally proving enhanced apoptosis, the number of IRF4−/− CD4+ T cells in vivo was strongly reduced compared with that of IRF4+/− CD4+ T cells, even in an environment in which all other cells were competent with respect to IRF4.
Enhanced Apoptosis of TCR-triggered IRF4−/− CD4+ T Cells In Vitro.
Next, we asked whether IRF4−/− Th cells exhibit alterations during activation-induced cell death (AICD; reference 14) in vitro. Total CD4+ IRF4−/− and IRF4+/+ Th cells were purified and stimulated for 72 h via their TCRs using anti-CD3 mAb in the presence of anti-CD28 mAb and IL-2. After a resting period of 72 h in the absence of anti-CD3, but with the presence of IL-2, viable cells were restimulated via anti-CD3 mAb. Apoptosis was determined 10 h later by flow cytometry using forward and side scatter characteristics as indicators of cell morphology (Fig. 2 A), as well as by annexin V and PI staining. In all experiments, both methods yielded comparable results; representative results are depicted in Fig. 2 A (bottom) and Fig. 2 B. Therefore, throughout this paper, results for only one of the two methods will be presented.
Figure 2.
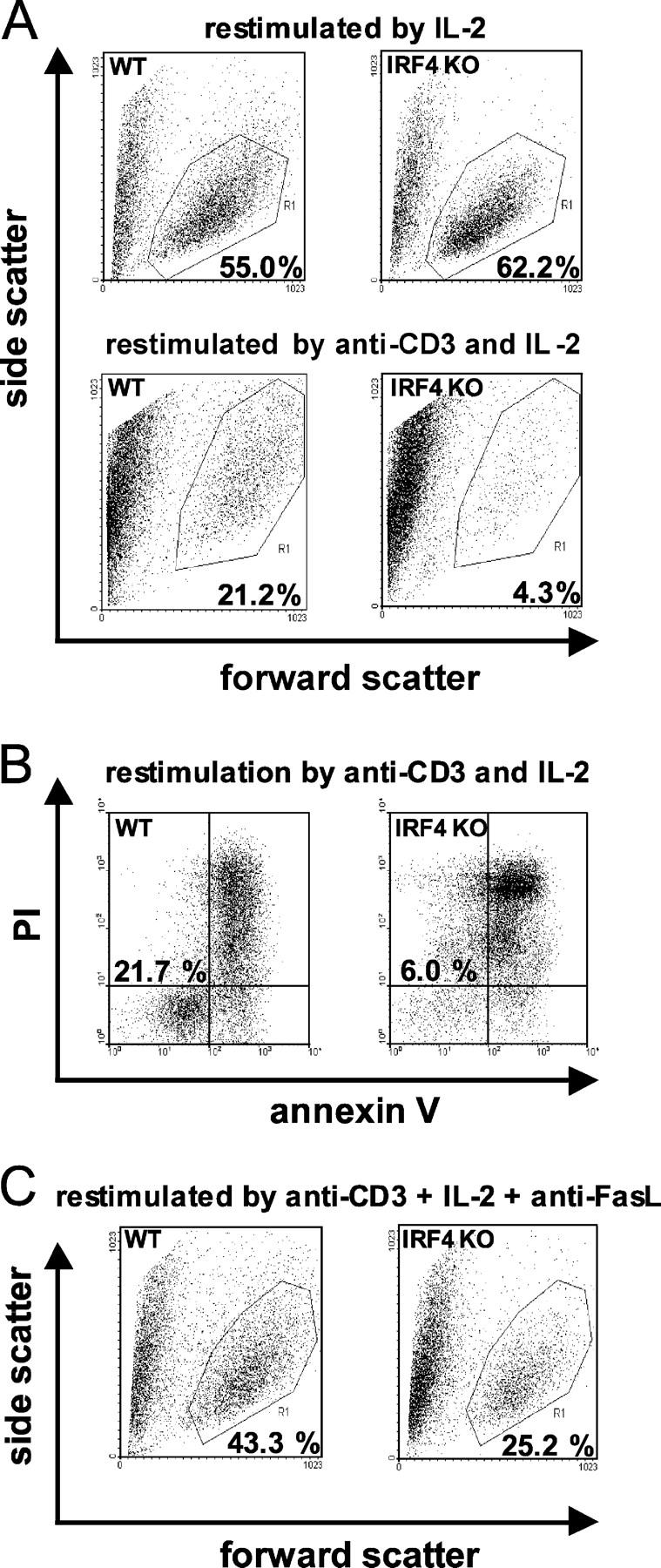
Enhanced TCR-induced apoptosis of IRF4−/− Th cells in vitro. IRF4+/+ (wt) and IRF4−/− Th cells were stimulated for 72 h with anti-CD3/CD28 mAb, rested for 72 h, and restimulated with IL-2 with or without anti-CD3 mAb, as indicated. In C, restimulation was performed in the presence of anti-CD95L mAb. After 10 h, flow cytometry was used to show cell morphology (A and C) or apoptosis after staining with annexin V and PI (B). Numbers refer to the percentage of viable cells depicted in the polygon (A and C) or bottom left quadrant (B). The results are representative of three different experiments.
Considerably more IRF4+/+ Th cells than IRF4−/− Th cells survived restimulation by anti-CD3. In contrast, the percentage of viable cells was equal in IRF4+/+ and IRF4−/− Th cell populations restimulated only with IL-2 (Fig. 2 A, top). To normalize for IL-2 concentration, saturating amounts of human IL-2 were added in all experiments. In control experiments, increased apoptosis was specific for IRF4 deficiency and not found in IRF2−/− Th cells (unpublished data).
Mediation of Enhanced IRF4−/− Th Cell Apoptosis by a Cell-intrinsic Process.
A major part of AICD is mediated through interaction of CD95 with its ligand CD95L (14). To analyze this interaction in IRF4−/− and IRF4+/+ Th cells, cells were primed, followed by a resting phase, as aforementioned. However, restimulation was performed in the presence of a neutralizing antibody directed against CD95L. Anti-CD95L mAb significantly blocked apoptosis in both cell populations, but its efficacy was much higher in IRF4−/− Th cells; the ratio of viable IRF4+/+ versus IRF4−/− Th cells shifted from 5:1 without anti-CD95L mAb to 2:1 in its presence (Fig. 2 C). At this time point, expression of CD95 and CD95L was similar in cells of both genotypes (unpublished data). These results suggest that the enhanced apoptosis of IRF4−/− Th cells is due to signaling events downstream of CD95.
Next, death was induced by restimulation with an agonistic anti-CD95 mAb and cross-linked by protein G (Fig. 3 A) in the absence of anti-CD3. As before, IRF4−/− Th cells were much more susceptible to apoptosis. This result was specific to the presence of anti-CD95 mAb, but not of protein G alone (Fig. 3 A) or of control IgG plus protein G (not depicted). The same result was obtained when the sub-G0 cell fraction was determined as a parameter of apoptosis (Fig. 3 B).
Figure 3.
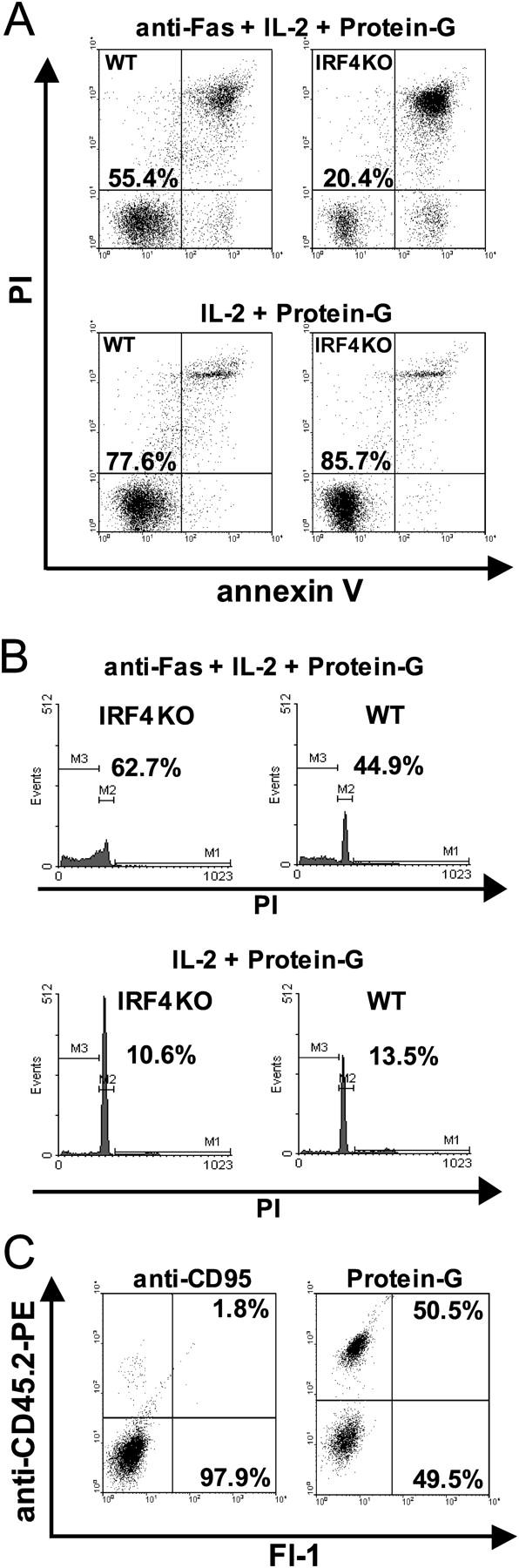
A cell-intrinsic mechanism mediates the augmented apoptosis of IRF4−/− Th cells. (A and B) IRF4−/− and IRF4+/+ Th cells were primed as in Fig. 2 and rested for 48 h. Restimulation was performed for 4 h in the presence or absence of anti-CD95 mAb. (A) Cells were analyzed as in Fig. 2 B. (B) Cell cycle analysis using propidium iodide (PI) was performed as described previously (reference 10). The percentage of dead cells in the sub-G0 fraction (located in region M3) is indicated. (C) CD45.2+IRF4−/− and CD45.1+IRF4+/+ CD4+ Th cells were purified, mixed at a ratio of 4:1, stimulated, rested, and triggered by anti-CD95 mAb as in A. After 4 h, IRF4−/− Th cells were detected using anti-CD45.2 mAb (y axis). Viable cells according to forward and side scatter characteristics were gated. Numbers indicate the percentage of viable IRF4−/− (top) or CD45.1 congenic IRF4+/+ (bottom) Th cells. For better visibility, a two-dimensional plot for fluorescence intensity in the FITC and PE channels is shown despite a one-color staining. Two experiments were performed with similar outcome.
To test if the augmentation of IRF4−/− Th cell apoptosis was due to fratricide, or to lack of a cytokine, we took advantage of Th cells of the C57BL/6 background congenic for the CD45.1 gene. Such cells can be identified by a mAb that stains the CD45.2, but not the CD45.1 allotype. CD45.2+IRF4−/− and CD45.1+IRF4+/+ CD4+ Th cells were purified, mixed, and stimulated by anti-CD3/CD28 as aforementioned. In the first experiments of this kind, it was noted that IRF4+/+ Th cells, despite comparable viability, have a 3–4-fold growth advantage over IRF4−/− Th cells. Thus, in a typical experiment, one well of IRF4−/− Th cells contained 0.80 × 106 ± 0.0106 cells after stimulation and resting, whereas a well of IRF4+/+ Th cells contained 2.40 × 106 ± 0.05 × 106. Therefore, to account for this difference in proliferation, CD45.2+IRF4−/− and CD45.1+IRF4+/+ cells were mixed at a ratio of 4:1 at the beginning of the experiment. After resting, the mixed cells were triggered via CD95 for 4 h, stained with anti-CD45.2 mAb, and gated viable cells were analyzed by flow cytometry (Fig. 3 C). Mixed control cells without anti-CD95 mAb treatment consisted of CD45.2+IRF4−/− and CD45.1+IRF4+/+ cells at a ratio of 1:1. In contrast, anti-CD95 mAb treatment led to an almost total disappearance of viable IRF4−/− Th cells. These data demonstrate that the enhanced death of IRF4−/− Th cells cannot be conferred on IRF4+/+ cells. Also, IRF4−/− Th cells are not simply defective producers of a protective cytokine. Rather, their increased apoptosis is most likely caused by a deregulated cell-intrinsic program.
Soluble Factors Including IL-4 Influence Apoptosis of WT and IRF4−/− Th Cells.
Among other factors, IL-4 protects cells from Fas-mediated apoptosis, as shown previously in B cells and thyrocytes (15, 16). Significantly, IRF4−/− Th cells display a defect in their response to IL-4, namely a dramatically reduced differentiation into Th2 cells (8, 9, 13). Thus, different levels of apoptosis in IRF4−/− and IRF4+/+ Th cells could be due to a different response to endogenous cytokines such as IL-4. To test this hypothesis, IRF4−/− and IRF4+/+ Th cells were stimulated as aforementioned. However, before transferring cells and supernatants into cultures without anti-CD3 mAb, half of the cells were washed, followed by culture with IL-2. After another 48 h, cell death was triggered by anti-CD95 mAb. Removal of supernatants strongly increased the sensitivity toward CD95-mediated death in IRF4−/− and IRF4+/+ Th cells (Fig. 4 A). Yet in most experiments, a significant difference in the respective levels of apoptosis remained. Therefore, a soluble factor protected IRF4−/− and IRF4+/+ Th cells from apoptosis, but with and without cytokines, IRF4−/− cell apoptosis was still significantly increased (unpublished data). A possible explanation for this outcome was that cytokines had already partially protected IRF4+/+ Th cells before the supernatants were removed.
Figure 4.
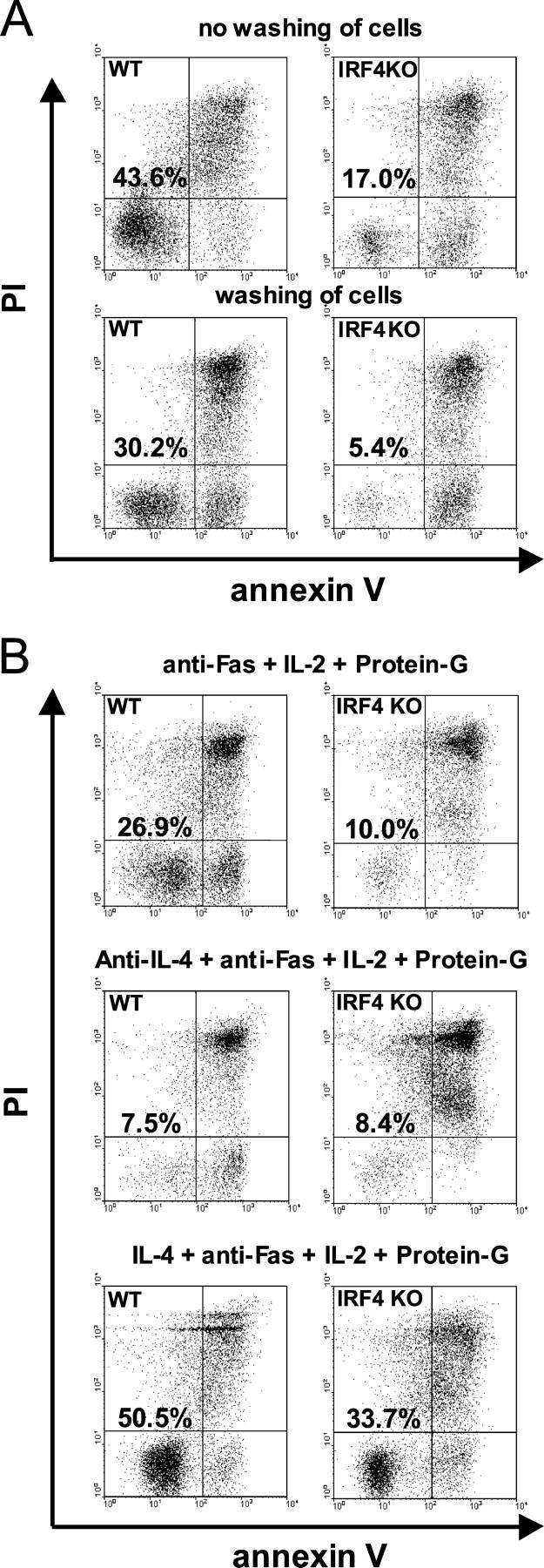
Role of IL-4 in the augmented apoptosis of IRF4−/− Th cells. (A and B) IRF4−/− and IRF4+/+ cells were primed for 72 h, rested for 48 h, and restimulated as in Fig.3 A. (A) At the start of the resting period, cells were either left unwashed or underwent extensive washing. (B) Cells were cultured with 10 ng/ml IL-4 or anti–IL-4 mAb, as indicated, for the duration of the experiment. (A and B) Apoptosis was triggered and evaluated after 4 h as in Fig. 3 A. Numbers indicate the percentage of viable cells present in the bottom left quadrant. The numbers were normalized for the percentage of viable cells cultured without anti-CD95 mAb in the respective condition. Three experiments were performed with a similar outcome.
Because IL-4 was the most likely cytokine candidate for protection, it was now either neutralized or added in saturating amounts during primary stimulation and resting, until anti-CD95 mAb was added (Fig. 4 B). Neutralization of IL-4 increased the rate of apoptosis to almost identical levels in both IRF4−/− and IRF4+/+ Th cells. In contrast, the addition of IL-4 protected IRF4+/+ Th cells from apoptosis. However, significant apoptosis still occurred in IRF4−/− Th cells, despite normal expression of the IL-4 receptor (unpublished data) and normal Stat6 phosphorylation in response to IL-4 (9).
Therefore, the increased susceptibility of IRF4−/− Th cells toward apoptosis is mediated at least in part by an altered response to IL-4, which also leads to their defective IL-4–dependent Th2 differentiation (8, 9, 13). Of course, IL-4 may not be the only protective cytokine at work in this system, although we have excluded an activity of exogenous IL-15, a known protective cytokine (17). In vivo, a reduced response to IL-4 may be exacerbated by defective Th2 cell differentiation, and this combination may explain the phenotype seen in the LNs of L. major–infected IRF4−/− mice.
Different intracellular molecules have been implicated in IL-4–mediated protection from apoptosis. Examples are c-FLIP and BCL-XL in thyrocytes and BCL-XL in B cells (15, 16). In Th cells, a candidate molecule is Gfi-1 because it is induced by IL-4 and inhibits apoptosis through unknown mechanisms (18). c-FLIP is probably not involved in the increased apoptosis of IRF4−/− Th cells because we have indications that enhanced death can be triggered not only via CD95, but also by other stimuli, such as γ-irradiation (unpublished data). This finding likely places the responsible molecules downstream of CD95 and its antiapoptotic regulator c-FLIP. Accordingly, we found similar c-FLIP expression in IRF4−/− and IRF4+/+ Th cells (unpublished data). Apoptosis of IRF4−/− Th cells was inhibited by a nonspecific caspase inhibitor, and procaspase-3 was cleaved more completely in IRF4−/− as compared with IRF4+/+ Th cells (unpublished data). Therefore, our current analysis focuses on molecules upstream of caspase-3 and present in pathways triggered by different apoptotic stimuli. However, levels of BCL-XL and BCL-2 do not explain the difference because their expression was similar in IRF4−/− and IRF4+/+ Th cells (unpublished data).
Finally, we would like to discuss a paper recently published in this journal that is in conflict with our observations (10). Fanzo et al. showed convincingly that, in human Jurkat cells, overexpressed IRF4 actually leads to enhanced apoptosis and not to protection, as would be expected from our results. The observation that IRF4 can suppress transcription when operating alone or in cooperation with ICSBP, while activating transcription in complex with different partners such as PU.1, may account for this discrepancy. Possibly, it is not IRF4 itself, but rather the availability of its diverse binding partners that determines whether IRF4 is pro- or antiapoptotic. As an example, in HTLV-I–infected leukemia cells, IRF4 is overexpressed and seems to be antiapoptotic (19), a finding that is also divergent to the report on Jurkat cells. Such considerations may also explain why IRF4−/− mice, despite their apoptotic LN cells during leishmaniasis, eventually suffer from lymphadenopathy (7), possibly as a first step of malignant transformation.
In addition, Fanzo et al. also presented studies with T cells from IRF4−/− mice. In their analysis, these T cells appeared to be protected from proapoptotic stimuli. Although we used the same mouse strain in our work, we obtained opposite results as follows: IRF4−/− Th cells showed increased apoptosis in >15 different experiments. In addition, increased resistance to apoptosis was not noted in IRF4−/− Th cells when cultured under the conditions described by Fanzo et al., as determined by annexin V staining, forward and side scatter characteristics, or by sub-G0 determination after PI staining (Fig. 3 B). In our analysis, the protocol used by this paper to prime T cells (1 μg/ml anti-CD3, no anti-CD28) left a significant fraction of IRF4−/− Th cells small and probably unstimulated (unpublished data). Likely, these cells are not as susceptible to AICD as cells primed according to our protocol (5 μg/ml anti-CD3 plus anti-CD28). Therefore, despite the different results, our data support a protective role of IRF4 during AICD of T helper cells.
Acknowledgments
We thank Dr. Moll for HE-stainings.
This work was supported by the Deutsche Forschungsgemeinschaft (grant Lo 396).
M. Lohoff and H.-W. Mittrücker contributed equally to this work.
References
- 1.Mamane, Y., C. Heylbroeck, P. Genin, M. Algarte, M.J. Servant, C. LePage, C. DeLuca, H. Kwon, R. Lin, and J. Hiscott. 1999. Interferon regulatory factors: the next generation. Gene. 237:1–14. [DOI] [PubMed] [Google Scholar]
- 2.Taki, S., T. Sato, K. Ogasawara, T. Fukuda, M. Sato, S. Hida, G. Suzuki, M. Mitsuyama, E.H. Shin, S. Kojima, et al. 1997. Multistage regulation of Th1-type immune responses by the transcription factor IRF-1. Immunity. 6:673–679. [DOI] [PubMed] [Google Scholar]
- 3.Lohoff, M., D. Ferrick, H.W. Mittrucker, G.S. Duncan, S. Bischof, M. Rollinghoff, and T.W. Mak. 1997. Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. Immunity. 6:681–689. [DOI] [PubMed] [Google Scholar]
- 4.Scharton, K.T., C. Contursi, A. Masumi, A. Sher, and K. Ozato. 1997. Interferon consensus sequence binding protein–deficient mice display impaired resistance to intracellular infection due to a primary defect in interleukin 12 p40 induction. J. Exp. Med. 186:1523–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohoff, M., G.S. Duncan, D. Ferrick, H.W. Mittrucker, S. Bischof, S. Prechtl, M. Rollinghoff, E. Schmitt, A. Pahl, and T.W. Mak. 2000. Deficiency in the transcription factor interferon regulatory factor (IRF)-2 leads to severely compromised development of natural killer and T helper type 1 cells. J. Exp. Med. 192:325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harada, H., M. Kitagawa, N. Tanaka, H. Yamamoto, K. Harada, M. Ishihara, and T. Taniguchi. 1993. Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science. 259:971–974. [DOI] [PubMed] [Google Scholar]
- 7.Mittrucker, H.W., T. Matsuyama, A. Grossman, T.M. Kundig, J. Potter, A. Shahinian, A. Wakeham, B. Patterson, P.S. Ohashi, and T.W. Mak. 1997. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. 275:540–543. [DOI] [PubMed] [Google Scholar]
- 8.Rengarajan, J., K.A. Mowen, K.D. McBride, E.D. Smith, H. Singh, and L.H. Glimcher. 2002. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J. Exp. Med. 195:1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lohoff, M., H.W. Mittrucker, S. Prechtl, S. Bischof, F. Sommer, S. Kock, D.A. Ferrick, G.S. Duncan, A. Gessner, and T.W. Mak. 2002. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc. Natl. Acad. Sci. USA. 99:11808–11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fanzo, J.C., C.M. Hu, S.Y. Jang, and A.B. Pernis. 2003. Regulation of lymphocyte apoptosis by interferon regulatory factor 4 (IRF-4). J. Exp. Med. 197:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reiner, S.L., and R.M. Locksley. 1995. The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13:151–177. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida, H., Y.Y. Kong, R. Yoshida, A.J. Elia, A. Hakem, R. Hakem, J.M. Penninger, and T.W. Mak. 1998. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 94:739–750. [DOI] [PubMed] [Google Scholar]
- 13.Tominaga, N., K. Ohkusu-Tsukada, H. Udono, R. Abe, T. Matsuyama, and K. Yui. 2003. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int. Immunol. 15:1–10. [DOI] [PubMed] [Google Scholar]
- 14.Dhein, J., H. Walczak, C. Baumler, K.M. Debatin, and P.H. Krammer. 1995. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature. 373:438–441. [DOI] [PubMed] [Google Scholar]
- 15.Stassi, G., D. Di Liberto, M. Todaro, A. Zeuner, L. Ricci-Vitiani, A. Stoppacciaro, L. Ruco, F. Farina, G. Zummo, and R. De Maria. 2000. Control of target cell survival in thyroid autoimmunity by T helper cytokines via regulation of apoptotic proteins. Nat. Immunol. 1:483–488. [DOI] [PubMed] [Google Scholar]
- 16.Wurster, A.L., V.L. Rodgers, M.F. White, T.L. Rothstein, and M.J. Grusby. 2002. Interleukin-4-mediated protection of primary B cells from apoptosis through Stat6-dependent up-regulation of Bcl-xL. J. Biol. Chem. 277:27169–27175. [DOI] [PubMed] [Google Scholar]
- 17.Bulfone-Paus, S., D. Ungureanu, T. Pohl, G. Lindner, R. Paus, R. Ruckert, H. Krause, and U. Kunzendorf. 1997. Interleukin-15 protects from lethal apoptosis in vivo. Nat. Med. 3:1124–1128. [DOI] [PubMed] [Google Scholar]
- 18.Zhu, J., L. Guo, B. Min, C.J. Watson, J. Hu-Li, H.A. Young, P.N. Tsichlis, and W.E. Paul. 2002. Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity. 16:733–744. [DOI] [PubMed] [Google Scholar]
- 19.Mariner, J.M., Y. Mamane, J. Hiscott, T.A. Waldmann, and N. Azimi. 2002. IFN regulatory factor 4 participates in the human T cell lymphotropic virus type I-mediated activation of the IL-15 receptor alpha promoter. J. Immunol. 168:5667–5674. [DOI] [PubMed] [Google Scholar]