

Abstract
We explored the role of Peyer's patch (PP) dendritic cell (DC) populations in the induction of immune responses to reovirus strain type 1 Lang (T1L). Immunofluorescence staining revealed the presence of T1L structural (σ1) and nonstructural (σNS) proteins in PPs of T1L-infected mice. Cells in the follicle-associated epithelium contained both σ1 and σNS, indicating productive viral replication. In contrast, σ1, but not σNS, was detected in the subepithelial dome (SED) in association with CD11c+/CD8α−/CD11blo DCs, suggesting antigen uptake by these DCs in the absence of infection. Consistent with this possibility, PP DCs purified from infected mice contained σ1, but not σNS, and PP DCs from uninfected mice could not be productively infected in vitro. Furthermore, σ1 protein in the SED was associated with fragmented DNA by terminal deoxy-UTP nick-end labeling staining, activated caspase-3, and the epithelial cell protein cytokeratin, suggesting that DCs capture T1L antigen from infected apoptotic epithelial cells. Finally, PP DCs from infected mice activated T1L-primed CD4+ T cells in vitro. These studies show that CD8α−/CD11blo DCs in the PP SED process T1L antigen from infected apoptotic epithelial cells for presentation to CD4+ T cells, and therefore demonstrate the cross-presentation of virally infected cells by DCs in vivo during a natural viral infection.
Keywords: antigen processing; apoptosis; epithelial cells; immunity, mucosal; reovirus infection
Introduction
Distinct populations of DCs have been identified in lymphoid and nonlymphoid tissues of humans and other mammals. Significant efforts are now being made to understand the ontogeny of these DC populations and their functional significance in the development of immunity to pathogens, tolerance to self-antigens, and induction of organ-specific autoimmunity. We study DC subsets in Peyer's patches (PPs), which represent primary sites for the induction of T and B cell immune responses at mucosal surfaces. In PPs, DCs are separated from the intestinal lumen by only a single layer of cells, the follicle-associated epithelium (FAE), which contains antigen-transporting microfold (M) cells (1). In addition, a small number of DCs are located directly within the FAE (2). Thus, PP DCs are constantly exposed to food antigens, normal bacterial flora (in the terminal ileum), and pathogenic microorganisms. Therefore, these cells likely play a crucial role in the induction of oral tolerance and immunity to infectious agents.
In previous studies, we defined three distinct subsets of CD11c+ PP DCs (1–3). CD8α−/CD11bhi DCs produce high levels of IL-10 and induce differentiation of IL-4– and IL-10–producing Th2 cells in vitro (2). In contrast, CD8α+/CD11blo and CD8α−/CD11blo DCs produce low levels of IL-10, high levels of IL-12, and induce only IFN-γ–producing Th1 cells (2). Spatial as well as functional differences define the three DC subsets. The DC-rich region of the subepithelial dome (SED) underlying the FAE contains both CD8α−/CD11bhi and CD8α−/CD11blo DCs, the T cell–rich intrafollicular region contains both CD8α+/CD11blo and CD8α−/CD11blo DCs, and the B cell follicle contains only CD8α−/CD11blo DCs (3). Although these initial studies allow the formulation of models for how PP DC populations are involved in the induction of oral tolerance and immunity to pathogens (3, 4), the validity of these models has not been tested in vivo.
To address the function of specific DC subsets in vivo, we studied mammalian reovirus infection of adult mice, an important model of mucosal virus infection (5). After peroral inoculation of mice with reovirus strain type 1 Lang (T1L), the outer capsid of the virus is processed by luminal proteases, resulting in generation of infectious subvirion particles that specifically enter PPs via M cells in the FAE (6–8). Once inside the PP, T1L productively infects columnar epithelial cells overlying both the PP (in the FAE) and lamina propria via receptors on the epithelial cell basolateral membrane (9, 10). In addition, early in infection, viral antigen is detectable in a few, poorly characterized mononuclear cells within the SED (8, 10). Later in infection, the virus spreads to systemic sites, including the spleen and mesenteric lymph nodes (11). Infection of adult mice with T1L is short-lived, and virus is usually cleared within 2 wk of inoculation by local and systemic immunity (5). The anti-reovirus immune response is characterized by production of IgA in the mucosa and development of Th1 cell and CTL responses at both local and systemic sites (12–14).
CD8α−/CD11bhi and CD8α−/CD11blo DCs in the SED are in close proximity to M cells in the FAE and are therefore poised to capture both soluble antigens and pathogens, like reovirus. In support of this possibility, Listeria monocytogenes and Salmonella typhimurium are detectable in PP DCs after peroral inoculation (15, 16). In addition, oral administration of virus-sized fluorescent polystyrene microparticles to mice results in detection of fluorescent signal in CD8α−/CD11blo DCs in the SED (17). However, little is known about DC capture and processing of enteric viral pathogens.
In this study, we found that all DC populations are resistant to productive infection by T1L after peroral inoculation, yet CD8α−/CD11blo DCs in the SED of PPs avidly take up viral antigen. In addition, our results indicate that viral antigen colocalizes with epithelial cell–derived cytokeratin, and markers of apoptosis in both tissue sections and purified CD11c+ DCs harvested from infected mice. Finally, we demonstrate that PP DCs purified from infected mice present T1L antigens to primed CD4+ T cells in vitro. These findings suggest that CD8α−/CD11blo DCs in the PP SED capture and process viral antigens from productively infected apoptotic epithelial cells for presentation to CD4+ T cells.
Materials and Methods
Cells and Virus.
Murine L929 (L) cells were grown in either suspension or monolayer cultures in Joklik's modified Eagle's minimal essential medium (Irvine Scientific) supplemented to contain 5% FBS (Biosource International), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin (Irvine Scientific). Reovirus strain T1L is a laboratory stock. Purified virion preparations were made by using second passage L cell lysate stocks of twice plaque-purified reovirus as described previously (18). Virus was released from infected cells by two cycles of freezing and thawing and sonication, recovered from lysates after two Freon (trichlorotrifluoroethane) extractions, and purified by cesium chloride gradient centrifugation. The virus band was removed, dialyzed exhaustively against dialysis buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris, pH 7.4) at 4°C, and stored in dialysis buffer at 4°C. The concentration of virus particles was calculated from protein concentration (19), and the concentration of infectious virus was determined by plaque assay (5). Reovirus was inactivated by UV irradiation as described previously (20).
Animals.
Female BALB/c mice were obtained from the National Cancer Institute. Mice were maintained in accordance with institutional guidelines for animal welfare and used at 6–12 wk of age. Groups of 5–15 mice were inoculated perorally with 108 PFUs of reovirus T1L in 100 μl of borate-buffered saline (0.13 M NaCl, 0.25 mM CaCl2, 1.5 mM MgCl2 · 6H2O, 20 mM H3BO3, 0.15 mM Na2B4O7 · 10H2O) containing 5 g/liter gelatin.
Antibodies.
DC subsets were identified by flow cytometry using anti-CD11c (N418), anti-CD11b (M1/70), anti-CD8α (Ly-2), anti-CD19 (1D3), or the appropriate isotype-matched control antibodies. Antibodies were labeled with FITC, PE, CyChrome, or allophycocyanin. Before staining, cells were incubated with anti–mouse CD16/CD32 antibody (2.4G2) to block Fc receptors (FcγRIII/II). Antibodies were purchased from BD Biosciences. The T1L σ1 protein was detected by immunofluorescence in tissue sections by using murine monoclonal antibody 5C6 (21). The T1L σNS protein was detected by using a rabbit polyclonal antiserum (22). A rabbit anti-cytokeratin polyclonal antiserum (DakoCytomation), a rabbit polyclonal antiserum specific for the activated form of caspase-3 (BD Biosciences), and an anti-B220 antibody (clone RA3-6B2; BD Biosciences) were used to stain antigens in cryosections and cytospin preparations.
DC Preparation and Purification.
DCs were prepared from PPs as described previously (3). Dissected PPs were treated with DTT and EDTA to remove the epithelium. PPs were digested with Liberase CI (Roche Applied Science) and DNase, followed by incubation with 5 mM EDTA to recover CD8α+ DCs. Cells were incubated with anti–mouse CD11c-coated magnetic beads (Miltenyi Biotec) and selected on separation columns using an AUTOMACS machine (Miltenyi Biotec). To increase the purity of the DC preparation, cells were incubated with FITC-labeled anti-CD11c and PE-labeled anti-CD19 antibodies. CD11c+/CD19− cells were isolated by FACS using a FACS Vantage™ SE sorter (BD Biosciences). DC subsets were separated by incubating cells with FITC-labeled anti-CD11c, PE-labeled anti-CD11b, CyChrome-labeled anti-CD8α, and allophycocyanin-labeled anti-CD19 antibodies. CD11c+/CD19−/CD8α+/CD11b−, CD11c+/ CD19−/CD8α−/CD11bhi, and CD11c+/CD19−/CD8α−/CD11blo were isolated by flow cytometry. Sorted DCs were typically 94–98% pure.
Reovirus Infection of L Cells and DCs In Vitro.
L cells were seeded in six-well plates (Costar) at 5 × 105 cells/well in a volume of 3 ml Eagle's minimal essential medium (Biosource International), supplemented to contain 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 mM Hepes. After incubation at 37°C for 24 h, when cells were 50–60% confluent, the medium was removed and cells were adsorbed with reovirus at a multiplicity of infection of 5 PFUs/cell in a volume of 200 μl at 37°C for 1 h. The virus inoculum was removed and 3 ml of fresh medium was added to each well. After incubation at 37°C overnight, cells were washed with PBS and fixed in acetone at −20°C before immunofluorescence staining.
FACS®-purified DC populations (106 DCs/ml) in RPMI 1640 (Biosource International) supplemented to contain 10% FBS, 2 mM l-glutamine, 100 μg/ml penicillin, 10 μg/ml streptomycin, 5% Medium NCTC-109 (Invitrogen Corporation), 15 mM Hepes, and 0.005 mM β-mercaptoethanol were adsorbed with reovirus at a multiplicity of infection of 500 PFUs/cell in 14-ml round-bottom polypropylene tubes (BD Biosciences) at 37°C for 1 h. Cells were washed, resuspended in fresh RPMI, and incubated at 37°C overnight. DCs were washed twice in PBS and centrifuged onto glass slides using a cytospin. Cells were dried at room temperature for 24 h and fixed in acetone at −20°C before immunofluorescence staining.
Immunofluorescence Staining.
PPs were dissected from the small intestines of BALB/c mice 24 h after peroral inoculation with T1L or from uninfected control mice and frozen in OCT embedding medium (Sakura Finetek). 8-μm thick frozen sections were fixed in acetone at −20°C and immunofluorescence was performed using the tyramide amplification method (TSA-Direct kit; PerkinElmer) as described previously (3). Immunofluorescence staining using the same techniques was also performed on cytospin preparations of FACS®-purified DCs and L cells that were grown on glass coverslips. Nuclei were identified by staining cells (Hoechst 33258; Sigma-Aldrich) before mounting with mounting medium (Fluoromount G; Southern Biotechnology Associates, Inc.). Fluorescent terminal deoxy-UTP nick-end labeling (TUNEL) staining of frozen sections was performed using an ApopTag-Fluorescein kit (Intergen) according to the manufacturer's instructions. TUNEL-stained sections were then stained with rabbit anti-reovirus serum using the tyramide system as described above.
Confocal Microscopy.
Confocal microscopic images of stained sections, cytospins, and L cell monolayers were collected on a confocal microscope (TCS-NT/SP; Leica) with 40× NA 1.25 or 63× NA 1.32, zoom 4, oil immersion objectives. Fluorochromes were excited using an argon laser at 488 nm for FITC and Cy3, a helium-neon laser at 633 nm for Cy5, and an argon laser (Enterprise model 651; Coherent Inc.) at 364 nm for Hoechst. Detector slits were configured to minimize diffusion of signal between channels. Alternatively, images were collected separately and later superimposed. Z-stacks (optical sections) of the images were collected with an optical thickness of 0.2 μm. Images were processed using the Leica TCS-NT/SP software (version 1.6.587), Imaris 3.1.1 (Bitplane AG), and Adobe Photoshop 6.0.
Preparation of Purified CD4+ T Cells from Reovirus-primed Mice.
Mice were inoculated in the rear footpad with an emulsion containing CFA and UV-inactivated reovirus (∼1010 particles/mouse) or CFA and PBS as a control. 7 d later, mice were killed and draining popliteal and inguinal lymph nodes were removed. Lymph node CD4+ T cells were isolated by negative selection on a CD4+ T cell isolation kit (MACS; Miltenyi Biotec). CD4+ T cell preparations were typically 96–98% pure.
Stimulation of Reovirus-primed CD4+ T Cells by PP APCs.
CD11c+/CD19− PP DCs and CD11c−/CD19+ PP B cells were isolated from mice 24 h after peroral inoculation with 108 PFUs of T1L or from uninfected control mice. APCs were γ irradiated with 2,000 rads to prevent proliferation. Purified T cells (typically 3 × 105 cells) from reovirus-primed or control mice were cocultured in triplicate with irradiated DCs or B cells at a 4:1 ratio in round-bottom 96-well plates. Antigen-presenting capacity of purified APCs was tested by adding UV-irradiated reovirus (109 particles/well) to a second set of triplicate wells containing APCs and T cells. To control for the proliferative capacity of T cells, 20 μg/ml anti-CD3ɛ antibody (BD Biosciences) was added to a third set of triplicate wells. After incubation for 3 d, an aliquot of supernatant was removed from each well for IFN-γ ELISA and 1 μCi/ml [3H]thymidine was added to each well for the final 7 h of culture. Cells were harvested and [3H]thymidine incorporation was assessed by using a β emission scintillation counter (PerkinElmer). IFN-γ release from stimulated T cells was assayed by sandwich ELISA (OptEia; BD Biosciences).
Statistical Analysis.
Statistical significance of differences was determined by the Student's t test.
Results
Reovirus T1L Replicates within the FAE, and Viral Antigens Colocalize with CD11c+ DCs in the PP SED.
To identify viral antigen directly in murine tissues after peroral inoculation with a well-characterized reovirus strain, BALB/c mice were perorally inoculated with 108 PFUs of reovirus T1L and killed 8, 24, 48, and 72 h after infection. Cryosections were prepared from PPs of infected and uninfected control mice and stained using antibodies specific for CD11c and viral structural protein σ1. Confocal immunofluorescence microscopy demonstrated the presence of σ1 within the FAE and the SED at 8 h after infection, with higher levels at 24 h after infection, where it colocalized with CD11c+ DCs (Fig. 1, A and B). In contrast, σ1 was barely detectable at 48 and 72 h after infection (not depicted). To determine whether reovirus antigen staining represented active viral replication, we used an antibody specific for reovirus nonstructural protein σNS, which is expressed during reovirus infection but does not form part of the virion (22). Cells of the FAE were found to be the principal site of active viral replication, as it was only in this region that the nonstructural protein σNS was detectable (Fig. 1, C and D). These findings suggest that the staining for structural protein σ1 in the SED represented uptake of viral antigen and not active viral infection of DCs at that site.
Figure 1.
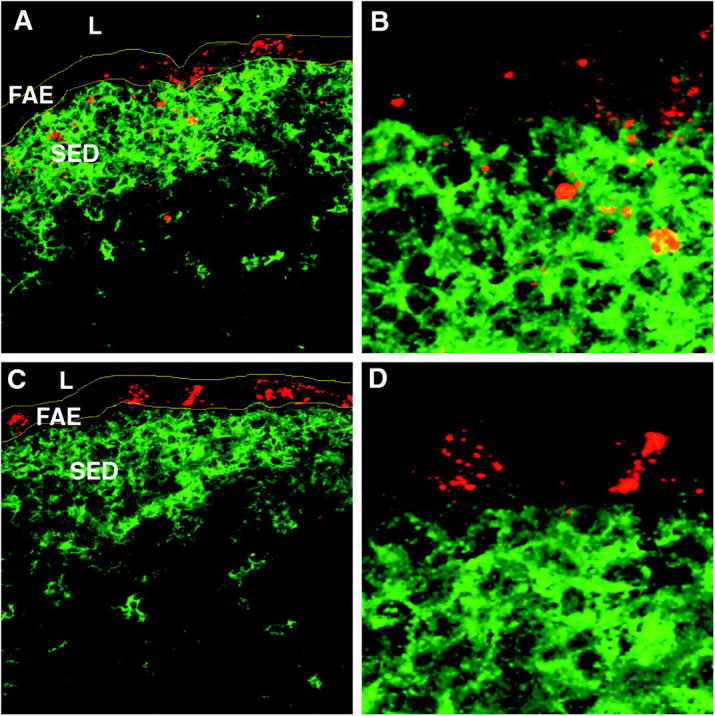
Reovirus T1L in the FAE and SED of PPs in association with CD11c+ DCs. (A) PP cryosections were prepared from mice 24 h after peroral inoculation with T1L. Sections were stained with antibodies specific for CD11c (green) and reovirus structural protein σ1 (red), and examined by confocal microscopy. (B) Higher magnification of A, showing σ1 in close association with CD11c+ DCs in the SED as indicated by the yellow color. (C) PP cryosections stained with antibodies specific for CD11c (green) and reovirus nonstructural protein σNS (red). (D) Higher magnification of C showing that σNS is detected only in cells of the FAE. L, lumen of small intestine; FAE, follicle-associated epithelium; SED, subepithelial dome. Borders of the FAE are indicated by yellow lines.
To confirm that viral antigen was ingested by PP DCs, CD11c+/CD19− cells were purified by flow cytometric sorting from PPs of mice 24 h after peroral inoculation with T1L and stained with antibodies specific for σ1 (Fig. 2, red) and σNS (Fig. 2, green). Staining for σ1 was clearly evident within CD11c+ DCs (Fig. 2 A), but staining for σNS was not detected (Fig. 2 B; see F for positive control staining). Furthermore, purified PP CD11c+ cells exposed to infectious T1L in vitro were not productively infected, but these cells were capable of acquiring viral antigen (Fig. 2, C and D). In contrast, infection of murine L cells with T1L resulted in productive viral infection with expression of both σ1 and σNS (Fig. 2, E and F). In addition, some of the σ1 in infected L cells demonstrated a diffuse staining pattern, whereas σ1 in DCs was distinctly punctate, suggesting antigen uptake into endosomes rather than active viral replication and assembly in the cytosol. Therefore, these data confirm the in situ studies and demonstrate that DCs capture T1L antigens but are not a primary site for viral replication.
Figure 2.
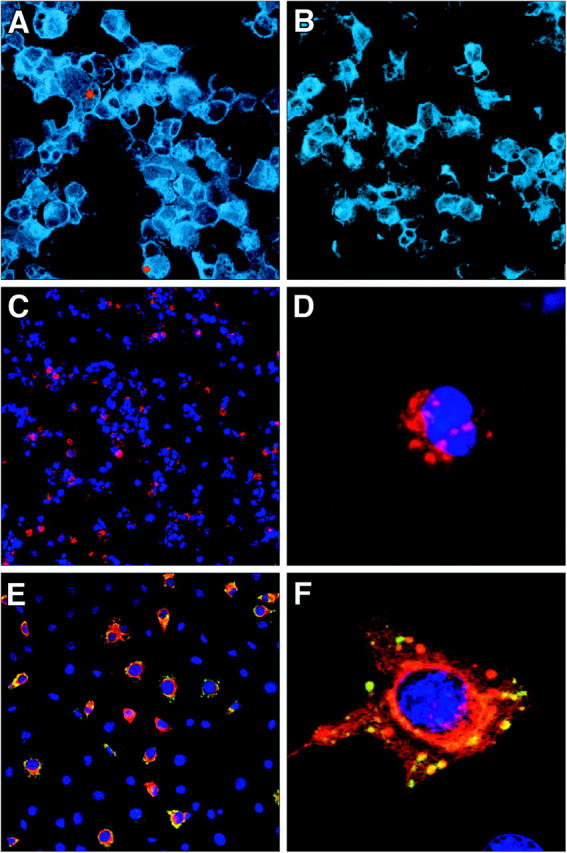
Replication of reovirus T1L within DCs in vivo and in vitro. (A) Cytospin preparation of CD11c+ DCs isolated from mice 24 h after peroral inoculation with T1L. Cells were stained with antibodies specific for CD11c (blue), reovirus σ1 (red), and reovirus σNS (green), and examined by confocal microscopy. (B) Cytospin preparation of CD11c+ DCs isolated from uninfected control mice and stained and imaged as in A. (C) Cytospin preparation of myeloid DCs (CD11c+/CD19−/CD8α−/CD11bhi) infected in vitro with reovirus T1L. Cells were stained with antibodies specific for reovirus σ1 (red) and reovirus σNS (green). Nuclei were labeled with Hoechst (blue). (D) Higher magnification of a single infected DC. Punctate staining of σ1 (red) can be seen. No evidence of σNS expression was detected. (E) Reovirus infection of L cells permissive for reovirus replication. Cells were stained as in C. (F) Higher magnification of a single infected L cell showing sites of reovirus replication as indicated by the yellow color (presence of both σ1 and σNS).
To identify the specific DC subset responsible for uptake of viral antigen, cytospin preparations of CD11c+ cells purified from infected mice were stained for CD11b, CD8α, and reovirus σ1. Viral antigen was detected only in CD8α− cells and confined to cells that displayed low levels of CD11b (Fig. 3, A and B). This result is also in concordance with in situ studies demonstrating that reovirus antigen is localized to CD8α−/CD11blo cells (not depicted). To exclude the possibility that antigen was contained within plasmacytoid DCs, we stained cytospin preparations for B220, which is highly expressed by both immature and mature/activated plasmacytoid DCs (23). B220-expressing cells were clearly demonstrable in cytospin samples; however, no viral antigen was contained within these cells (Fig. 3 C).
Figure 3.

Reovirus T1L in association with double negative DCs (CD11c+/CD8α−/CD11blo) in vivo. Cytospin preparations of CD11c+ DCs isolated from PPs of mice 24 h after peroral inoculation with T1L. Cells were stained with antibodies specific for reovirus σ1 protein and cell surface molecules, and examined by confocal microscopy. Nuclei were labeled with Hoechst (blue). (A) Anti-CD8α (green) and anti-σ1 (red). Reovirus is found in association with CD8α− cells. (B) Anti-CD11b (green) and anti-σ1 (red). Reovirus antigen is found in association with CD11blo cells. (C) Anti-B220 (red) and anti-σ1 (green). Reovirus antigen is found in B220− cells, indicating that it is not within plasmacytoid DCs.
To determine whether CD8α−/CD11blo DCs, in contrast to CD8α−/CD11bhi and CD8α+/CD11blo DC subsets, are capable of directly ingesting T1L antigen, the three CD11c+ DC types from PPs were purified by flow cytometric sorting and exposed for 24 h to equivalent inocula of T1L in vitro. Cytospin preparations were stained with antibodies specific for σ1 and σNS. We found that 70% of CD8α−/CD11bhi cells were positive for σ1 compared with 30% of CD8α−/CD11blo DCs and only 10% of CD8α+/CD11blo DCs. None of the purified PP DC subsets displayed positive staining for reovirus σNS. Thus, although all DCs are capable of taking up T1L in vitro, CD8α2/CD11blo DCs appear to be the primary cells that contain viral antigen in the SED after infection in vivo, despite the fact that CD8α−/CD11bhi DCs are also normally present in the SED.
PP DCs Can Acquire Reovirus Antigen from Infected Apoptotic Epithelial Cells.
Given that T1L does not appear to productively infect DCs, we reasoned that DCs likely acquire viral antigen from infected epithelial cells. Furthermore, because PP DCs ingest virus after exposure to T1L in vitro, it is likely that virus released from infected epithelial cells is directly sampled by underlying DCs. Reovirus is capable of inducing apoptosis of infected cells (24–26). Therefore, we hypothesized that reovirus-infected apoptotic epithelial cells are the source of the reovirus protein associated with the CD11c+ DCs in the SED. To test this hypothesis, we used the TUNEL technique to examine cells in reovirus-infected PP domes for evidence of apoptosis. TUNEL+ cells were detected in the SED of both uninfected and infected mice (Fig. 4, A–C). In infected mice, TUNEL staining in the SED colocalized with staining for σ1 (Fig. 4, B and C). It was not possible to perform double staining for TUNEL and CD11c, as the CD11c epitope is not recognized by the N418 antibody after the paraformaldehyde fixation required for TUNEL staining. Nonetheless, these results suggested that apoptotic cells in the SED contained σ1.
Figure 4.

Reovirus infection in association with apoptosis in the PP SED. (A) PP cryosection from an uninfected control mouse stained with TUNEL (green) and examined by confocal microscopy. (B) PP cryosection from a mouse 24 h after peroral inoculation with T1L stained with TUNEL (green) and an antibody specific for reovirus σ1 (red). (C) Higher magnification of B showing reovirus σ1 in association with TUNEL+ inclusions. (D) PP cryosection from an uninfected mouse stained with antibodies specific for CD11c (green), activated caspase-3 (red), and reovirus σ1 (blue). Note that apoptotic material can be detected in both the FAE and SED. (E) PP cryosections from a T1L-infected mouse stained as in D. Note two reovirus-infected cells in the FAE that are also positive for activated caspase-3 as indicated by the purple color. (F) Higher magnification of the SED of E showing association of reovirus σ1 and activated caspase-3 as indicated by the purple color. L, lumen of small intestine; FAE, follicle-associated epithelium; SED, subepithelial dome. Borders of the FAE are indicated by yellow lines.
To confirm the presence of apoptotic material in the DC region of the SED, we performed triple staining on PP cryosections using antibodies specific for CD11c, σ1, and the activated form of caspase-3, which is an early marker of apoptosis (Fig. 4, D–F; reference 27). Activated caspase-3 was detected in all domes of PPs infected with reovirus, often in close association with punctate σ1 staining. We also observed activated caspase-3 staining in PP domes from uninfected control mice (Fig. 4 D).
To determine whether the apoptotic cells in the SED were of epithelial origin, we stained tissues for CD11c, σ1, and cytokeratins, which are present in high concentration in epithelial cells of the FAE. Similar to the findings made using TUNEL and activated caspase-3 staining, punctate cytokeratin staining was detected in the SED of both uninfected (unpublished data) and infected mice (Fig. 5 A). In addition, σ1 and cytokeratin were observed to colocalize within the SED of infected animals (Fig. 5 B).
Figure 5.

Epithelial cell–derived cytokeratin in the FAE and SED of reovirus-infected mice. PP cryosections were prepared from mice 24 h after peroral inoculation with T1L. Sections were stained with antibodies specific for (A) CD11c (green) and cytokeratin (red), or (B) CD11c (green), cytokeratin (red), and reovirus σ1 (blue). Sections were examined by confocal microscopy. Cytokeratin can be detected in the DC region of the SED underlying the FAE. Reovirus σ1 shown in association with cytokeratin staining in the FAE and occasionally in the SED is indicated by the purple color. Borders of the FAE are indicated by yellow lines.
To test whether uptake of T1L leads to death of DCs, we counted viable DCs after trypan blue staining after incubation with T1L in vitro. In these experiments, we could not detect an effect of reovirus on DC viability. However, because a significant proportion of PP DCs die once they have been isolated from tissue in the absence of further stimulation, it might be difficult to exclude a more subtle effect. We also performed studies with an immature/inactivated murine DC cell line (D1) and found no evidence of cell death upon incubation with T1L.
To conclusively determine whether CD11c+ DCs in the SED acquire reovirus antigen from apoptotic epithelial cells, PP DCs were isolated from mice 24 h after peroral inoculation with T1L, and cytospins of these cells were stained with antibodies specific for CD11c, σ1, and either activated caspase-3 or cytokeratin. Confocal microscopic analysis indicated that both activated caspase-3 and cytokeratin colocalize with σ1 within CD11c+ DCs (Fig. 6). The vast majority of DCs containing viral antigen (85–95%) also contained active caspase-3. Analysis of intracellular staining by flow cytometry supported this finding (not depicted). Moreover, T1L antigen and activated caspase-3 were observed to colocalize into discrete vesicles within DCs (Fig. 6). Thus, these findings strongly suggest that SED DCs ingest apoptotic epithelial cells containing reovirus antigen for presentation to T cells.
Figure 6.

Reovirus T1L antigen in association with activated caspase-3 and epithelial cell–derived cytokeratin in CD11c+ DCs isolated from reovirus-infected PPs and purified by flow cytometry. Cytospin preparations were obtained from mice 24 h after peroral infection with T1L, stained, and examined by confocal microscopy. Nuclei were labeled with Hoechst (blue). Staining for (A) reovirus σ1 (green) and (B) activated caspase-3 (red) indicates that the majority of virus+ cells also express active caspase-3 within vesicular structures as indicated by the yellow color in the merged image (C). Expression of active caspase-3 in T1L-infected cells also is shown in D: reovirus σ1 (red), activated caspase-3 (green), and CD11c (gray). (E) Staining of cells for cytokeratin (green), reovirus σ1 (red), and CD11c (gray) demonstrates that reovirus σ1 also colocalizes with cytokeratin within vesicular structures as indicated by the yellow color. In this micrograph, a CD11c+ cell containing cytokeratin without reovirus is evident (large arrow), as is a cytokeratin-expressing contaminating epithelial cell (small arrow). Note that the cytokeratin in this cell stains diffusely and is not punctate.
PP DCs, But Not B Cells, from Reovirus-infected Mice Present Viral Antigens to Virus-primed CD4+ T Cells In Vitro.
To determine whether DCs isolated from PPs of T1L-infected mice present viral antigen to T cells, CD11c+ DCs were isolated from PPs of T1L-infected BALB/c mice by flow cytometric sorting and used to stimulate primed CD4+ T cells isolated from draining lymph nodes of mice immunized in the footpad with T1L in CFA. DCs from infected but not uninfected mice induced proliferation of, as well as IFN-γ production by, T1L-primed T cells (Fig. 7). In contrast, B cells isolated from infected mice did not present viral antigens to primed T cells, although they were capable of presenting antigen if reovirus was added to the cultures. These data provide strong evidence that PP CD11c+ DCs and not B cells present viral antigen to CD4+ T cells after peroral inoculation of mice with reovirus.
Figure 7.
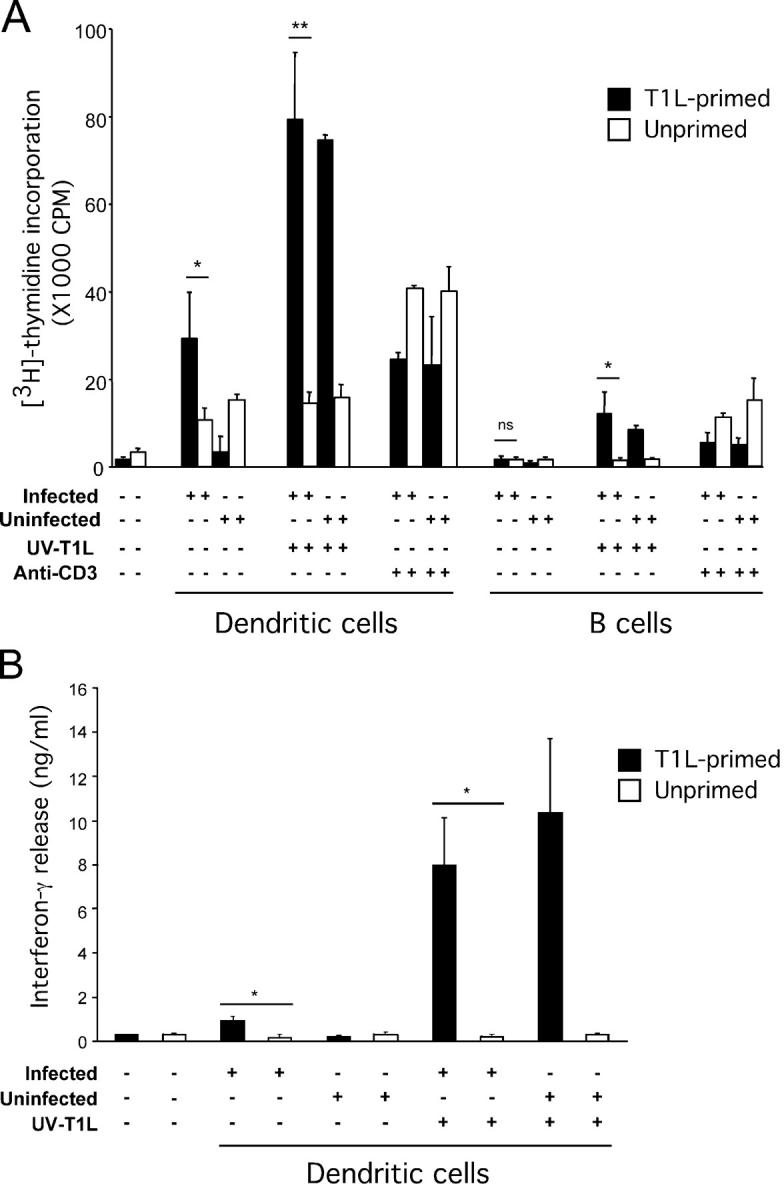
Activation of T1L-primed CD4+ T cells by PP DCs and B cells isolated from reovirus-infected mice. (A) Proliferation of CD4+ T cells. CD11c+/CD19− DCs or CD11c−/CD19+ B cells from either uninfected control mice or mice 24 h after peroral inoculation with T1L were purified by flow cytometry (>95% pure). Cells were cocultured with T1L-primed T cells (black bars) or unprimed T cells (white bars) for 72 h at an APC/T cell ratio of 1:4 and in the absence or presence of UV-inactivated T1L (UV-T1L). Cultures in the absence of UV-T1L were stimulated with anti-CD3 as a positive control. The first two bars represent baseline proliferation of T cells from T1L-primed and unprimed mice without DCs, B cells, or other stimuli. (B) IFN-γ release from CD4+ T cells. PP DCs isolated from uninfected or infected mice were cocultured with T1L-primed or control T cells in the absence or presence of UV-T1L as in A. The first two bars represent baseline IFN-γ release from T cells from T1L-primed and unprimed mice without DCs, or other stimuli. Results are presented as mean [3H]thymidine incorporation for A and mean IFN-γ release for B. Error bars represent standard error of the means for three independent experiments. *, P < 0.05; **, P < 0.0002; ns, not significant.
To identify the subpopulation of PP DCs capable of processing T1L in vivo, we isolated by flow cytometry the three major populations of CD11chi DCs, CD8α+/CD11blo, CD8α−/CD11blo, and CD8α−/CD11bhi DCs, 24 h after peroral inoculation with T1L and determined the capacity of these cells to activate T1L-primed T cells in vitro. Both CD8α−/CD11blo and CD8α+/CD11blo DCs, but not CD8α−/CD11bhi DCs, were capable of inducing the proliferation of T1L-primed T cells (Fig. 8). Thus, although CD8α−/CD11blo DCs located in SED appear to be the primary cells that process reovirus-infected apoptotic epithelial cells, reovirus antigen is also processed by CD8α+/CD11blo DCs for presentation to CD4+ T cells.
Figure 8.

Proliferation of T1L-primed CD4+ T cells induced by CD8α+, CD11bhi, and CD8α−/CD11blo subsets of DCs isolated from T1L-infected mice. DCs from uninfected (white bars) and infected (black bars) mice were purified by flow cytometry (>95% pure) and cocultured with T1L-primed T cells for 72 h at an APC/T cell ratio of 1:4 and in the absence or presence of UV-inactivated T1L (UV-T1L) or anti-CD3 as a positive control. Results are presented as mean [3H]thymidine incorporation. Error bars represent standard deviations for triplicate assays of one of two experiments performed with similar results. *, P < 0.05; ns, not significant.
Discussion
In this work, we examined early events that occur in murine PPs after infection with reovirus, which specifically targets the FAE and induces both mucosal and systemic immune responses. Previous studies have shown that reovirus strain T1L transcytoses through M cells and infects neighboring epithelial cells likely via receptors expressed on the basolateral epithelial cell surface (9). Electron microscopy studies demonstrate productive infection of epithelial cells along with viral particles in a small number of mononuclear cells in the SED (10). These observations prompted us to examine whether DCs in the SED are capable of taking up reovirus antigen for presentation to T cells.
We first tested whether reovirus proteins colocalize with CD11c+ cells in situ after viral infection. We detected reovirus structural protein σ1 not only in the epithelium, but also in discrete aggregates within the SED of infected PPs. In contrast, we did not detect reovirus nonstructural protein σNS, which is expressed only in infected cells during active viral replication (22). These findings indicate that active replication of T1L occurs only within the FAE and not within DCs of the SED. Consistent with these results, we found that purified DCs are not productively infected with T1L in vitro, although they are capable of avidly engulfing T1L antigen. Thus, unlike many viruses that directly infect DCs, such as influenza virus (28), reovirus T1L does not infect SED DCs, but instead these cells capture viral antigen from the FAE. Virus ingested by SED DCs might be input virus transported to the PP via M cells or progeny virus released from infected epithelial cells.
We also thought it possible that DCs in the SED might acquire viral antigen from infected epithelial cells in the FAE. This process could occur by a mechanism termed “nibbling,” in which antigen is taken up from living cells by contact with DCs (29). However, we considered it more likely that DCs process antigen from apoptotic epithelial cells nearby. This hypothesis is supported by two lines of evidence. First, reovirus T1L induces apoptosis of infected cells, although to a lesser extent than type 3 reovirus strains (24–26). Second, after infection of mice with reovirus, aggregates of viral antigen in the SED are observed, and these aggregates are much larger than those found in DCs loaded with virus in vitro (Figs. 1 and 2). These observations suggest that the viral antigen is contained within apoptotic bodies. Indeed, we found that a substantial proportion of reovirus antigen is associated with apoptotic bodies in the SED as judged by colocalization of reovirus σ1 protein with both TUNEL staining and activated caspase-3, the processed form of pro–caspase-3 detected during the early stages of apoptosis (27). Moreover, tissue staining for epithelial cell–expressed cytokeratins demonstrated that at least a portion of the apoptotic cells are epithelial cells. The fact that there is no T1L σNS protein detectable within these apoptotic bodies is likely because viral replication is normally complete before apoptosis of virally infected cells occurs (24, 25).
To distinguish between DCs in the SED that were undergoing apoptosis from DCs that had ingested apoptotic epithelial cells, we isolated DCs from infected mice and examined whether these cells contained reovirus antigen that colocalized with activated caspase-3 or cytokeratins. We detected DCs that contained each marker in the same inclusions as the reovirus protein. These data strongly suggest that a major pathway by which reovirus antigen is processed by DCs in vivo is via the sampling of apoptotic bodies from virus-infected epithelial cells.
This interpretation is consistent with extensive previous studies demonstrating the capacity of DCs to process viral antigens from apoptotic and dying cells (for review see reference 30). After the report by Bevan (31) demonstrating the phenomenon of cross-presentation and cross-priming of CTLs, Albert et al. (32) found that human DCs can use influenza virus–infected cells as sources of antigen. Since that time, in vitro studies have shown that DCs cross-present antigens from dead cells infected with a variety of viruses, including human immunodeficiency virus (33), measles virus (34), human cytomegalovirus (35–37), Epstein-Barr virus (38–40), canarypox virus (41, 42), and vaccinia virus (43). In contrast, in vivo studies, although implicating DCs in cross-priming to soluble or cell-associated nonviral proteins (for review see references 30, 44, and 45), have provided less direct evidence that DCs mediate cross-presentation of virus-infected cells. For example, although bone marrow–derived APCs mediate cross-priming against poliovirus in vivo in a transporter associated with antigen-processing–dependent manner (46), and infection of mice with adenovirus expressing influenza A virus nucleoprotein under tissue-specific promoters resulted in CTL activity against nucleoprotein (47), DCs were not specifically implicated in these studies. More direct evidence for DC presentation of viral antigens in vivo comes from studies of HSV. HSV-1 and -2 antigens are presented in the absence of HSV virions or DNA in the draining lymph nodes of infected mice, and presentation of these antigens is mediated primarily by CD8α+ and CD11bhi DCs, respectively (48, 49). Although these studies demonstrate cross-presentation of viral antigens by DCs in vivo, they do not demonstrate the source of viral antigen. The results presented here provide direct evidence that DCs process viral antigen from virus-infected apoptotic cells in vivo.
An additional important finding of our work is the demonstration of significant numbers of TUNEL+ and activated caspase-3+ cells in the SED of uninfected mice. Although it is our impression that there might be a higher number of TUNEL+ cells in the SED of reovirus-infected PPs, it is difficult to discern whether the absolute frequency of apoptotic cells differs from that of uninfected animals because of problems with orientation during the preparation of tissue for cryosectioning. Therefore, we are hesitant to draw firm conclusions about these observations. However, it is apparent that infection is not required for the presence of TUNEL+ cells in the PP dome. This result suggests that epithelial cells in the SED undergo apoptosis continually under normal physiologic conditions.
Two previous studies provide additional support for physiologic apoptosis of SED epithelial cells. In the first, Huang et al. (50) demonstrated in uninfected rats the presence of CD4−/OX41− DCs containing apoptotic bodies and cytokeratin+ inclusions in the lamina propria and T cell zones of mesenteric lymph nodes. They also showed that DCs contain epithelial cell–derived, nonspecific esterase in the basal layer and T cell zones of PPs. The OX41− DC subset is a weak inducer of T cell proliferation and constitutively migrates from the intestine to mesenteric lymph nodes, as evidenced by the presence of large numbers of these cells in the thoracic duct lymph after mesenteric lymph node adenectomy (51). In the second, Vezys et al. (52) demonstrated that expression of a nonsecreted form of OVA in intestinal epithelial cells results in OVA-specific CD8+ T cell unresponsiveness. OVA-specific, OT-1 T cell receptor–transgenic T cells transferred into OVA-expressing mice expanded first in PPs and mesenteric lymph nodes, and then became unresponsive to challenge with OVA-expressing vesicular stomatitis virus. OVA-specific CD8+ unresponsiveness can also be prevented by treatment of these mice with cholera toxin, activating CD40 antibodies, or vesicular stomatitis virus that does not express OVA (53). Taken together with the current studies, these data are consistent with the hypothesis originally offered by Huang et al. (50) that under steady-state conditions, antigens either expressed by epithelial cells or absorbed from the intestinal lumen into epithelial cells are taken up by DCs after epithelial cell apoptosis. The possibility that these DCs may then induce T cell unresponsiveness is supported by studies showing that apoptotic cells do not induce maturation of immunostimulatory DCs, which allows captured antigens to be presented in a fashion that induces tolerance (for review see reference 54).
The situation may differ in the case of reovirus infection. DCs loaded with virus-infected apoptotic epithelial cells are most likely induced to mature into fully immunostimulatory DCs resulting in the strong T cell immune responses seen to this virus. This process may occur by exposure of resident DCs to products of necrotic epithelial cells after virus-induced cell lysis, cytokines such as IFN-α, IL-1, or TNF-α, viral particles, or viral double stranded RNA released from infected epithelial cells. Studies are currently underway to distinguish between these possibilities. Regardless of the precise mechanism, reovirus infection may convert a normal tolerogenic response to epithelial cell–associated antigens into an effective immunogenic response to the virus. This model is consistent with the idea that tolerogenic DCs continuously induce T cell tolerance to self-antigens under steady-state conditions. After infection, when immunogenic DCs are induced (e.g., by cytokines, pathogen-associated molecular patterns, or factors released from necrotic cells) and present both self- and environmental proteins together with viral antigens, preexisting T cell tolerance (in this case to noninfectious epithelial cell–derived antigens) would allow the development of an adaptive immune response tightly focused on antigens derived from the invading pathogen (55). This scenario would certainly be the case if the mechanism of tolerance induced by steady-state DCs is T cell deletion or unresponsiveness, as has been shown in many systems (see reference 44).
Our work also addressed the role of specific DC subsets in the uptake of epithelial cell–associated antigen in the PP SED. Although previous studies have shown that both CD8α+ and CD8α− DCs can present soluble antigen in the context of MHC class II molecules after in vivo loading, CD8α+ DCs might be more efficient in the presentation of soluble antigens to CD8+ T cells. In addition, CD8α+ DCs are clearly specialized for the uptake and presentation of cell-associated antigen to both CD4+ and CD8+ T cells (56–60). We detected inclusions containing reovirus, cytokeratin, and activated caspase-3 in CD8α− DCs in the PP SED. However, the SED region of CD8α− DCs that contained apoptotic epithelial cells differ from the majority of CD8α− DCs in other tissues in that they do not express significant levels of CD11b.
In previous studies, we showed that CD8α−/CD11blo DCs are uniquely overrepresented in mucosal lymphoid tissues, concentrated in the SED and interfollicular T cell regions of the PP, and produce high levels of IL-12 upon stimulation with CD40L or Staphylococcus aureus and IFN-γ (2). These DCs are not plasmacytoid DCs because they do not express B220 or Ly6C, and express high levels of CD11c. In addition, these CD8α−/CD11blo DCs induce Th1 cell differentiation in vitro in a manner similar to CD8α+ PP or spleen DCs (2). However, despite this similarity, CD8α−/CD11blo PP DCs are not likely to be an immature or mature form of CD8α+ DCs. Freshly isolated or in vitro–stimulated CD8α−/CD11blo, CD8α−/CD11bhi, and CD8α+/CD11blo PP DCs express similar levels of MHC class II and costimulatory molecules (2, 3). In addition, in vitro maturation of CD8α−/CD11blo DCs does not result in an increase in CD8α expression (2). The unique lineage of these cells is also supported by a study demonstrating that CD8α−/CD4−/DEC-205lo DCs, which are likely the same population as the CD8α−/CD11blo DCs, appear in the mesenteric lymph nodes with kinetics similar to other DC subsets after BrDU labeling in vivo (61). We now have demonstrated that CD8α−/CD11blo DCs likely play a key role in the development of an antiviral immune response to reovirus. Because CD8α−/CD11blo DCs produce high levels of IL-12 and induce strong Th1 cell responses in vitro (2), these cells may play a role in driving Th1 cell responses to reovirus in vivo (13).
In a final series of experiments, we demonstrated that DCs isolated from PPs of reovirus-infected mice can present viral antigen to reovirus-primed CD4+ T cells. In addition, we found that both CD8α−/CD11blo and CD8α+/CD11blo DCs, but not CD8α−/CD11bhi DCs, from T1L-infected animals were capable of inducing proliferation (Fig. 8) and IFN-γ production (not depicted) by T1L-primed CD4+ T cells. These data demonstrate that although apoptotic bodies are associated with CD8α−/CD11blo DCs, T1L antigen is also processed by CD8α+/CD11blo DCs in vivo. Interestingly, these findings are similar to those of Scheinecker et al. (62), who found that a gastric self-antigen, H(+)/K(+)-ATPase, thought to be released from dying cells, is contained within vesicular compartments of CD8α− DCs in draining lymph nodes in situ. However, both CD8α− and CD8α+ DCs could stimulate antigen-specific T cells directly ex vivo. Thus, the presentation of T1L by CD8α+ DCs could be the result of direct viral antigen uptake or the cross-presentation of antigen transported into the T cell zone by CD8α−/CD11blo DCs or other types of cells.
Studies reported here demonstrate a direct role for PP DCs in the presentation of viral antigen to CD4+ T cells after reovirus infection of mice. We show that an important pathway by which PP DCs process reovirus antigen is by uptake of virus-infected, follicle-associated epithelial cells that have undergone apoptotic cell death. This function is mediated principally by the CD8α−/CD11blo subset of PP DCs. Although not demonstrated here, it is likely that T1L antigens are also cross-presented to CD8 T cells in a similar manner. These studies provide mechanistic insight into the process by which an intestinal viral pathogen induces both local and systemic immune responses.
Acknowledgments
We thank members of our laboratories for many useful discussions. We also acknowledge Warren Strober for his review of the manuscript, and Owen Scwartz for help with confocal microscopy.
This research was supported by Public Health Service award AI50080, and the Elizabeth B. Lamb Center for Pediatric Research. Additional support was provided by Public Health Service awards CA68485 for the Vanderbilt-Ingram Cancer Center and DK20593 for the Vanderbilt Diabetes Research and Training Center.
M.N. Fleeton's present address is Virology Unit, Trinity College, Dublin 2, Ireland.
Abbreviations used in this paper: FAE, follicle-associated epithelium; PP, Peyer's patch; SED, subepithelial dome; T1L, reovirus strain type 1 Lang; TUNEL, terminal deoxy-UTP nick-end labeling.
References
- 1.Kelsall, B.L., and W. Strober. 1996. Distinct populations of dendritic cells are present in the subepithelial dome and T cell regions of the murine Peyer's patch. J. Exp. Med. 183:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iwasaki, A., and B.L. Kelsall. 2001. Unique functions of CD11b+, CD8α+, and double-negative Peyer's patch dendritic cells. J. Immunol. 166:4884–4890. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki, A., and B.L. Kelsall. 2000. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3α, MIP-3β, and secondary lymphoid organ chemokine. J. Exp. Med. 191:1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki, A., and B.L. Kelsall. 1999. Mucosal immunity and inflammation. I. Mucosal dendritic cells: their specialized role in initiating T cell responses. Am. J. Physiol. 276:G1074–G1078. [DOI] [PubMed] [Google Scholar]
- 5.Virgin, H.W., K.L. Tyler, and T.S. Dermody. 1997. Reovirus. Viral Pathogenesis. N. Nathanson, editor. Lippincott-Raven, New York. 669–699.
- 6.Bodkin, D.K., M.L. Nibert, and B.N. Fields. 1989. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J. Virol. 63:4676–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bass, D.M., D. Bodkin, R. Dambrauskas, J.S. Trier, B.N. Fields, and J.L. Wolf. 1990. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J. Virol. 64:1830–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf, J.L., D.H. Rubin, R.S. Finberg, R.S. Kauffman, A.H. Sharpe, J.S. Trier, and B.N. Fields. 1981. Intestinal M cells: a pathway for entry of reovirus into the host. Science. 212:471–472. [DOI] [PubMed] [Google Scholar]
- 9.Rubin, D.H. 1987. Reovirus serotype 1 binds to the basolateral membrane of intestinal epithelial cells. Microb. Pathog. 3:215–219. [DOI] [PubMed] [Google Scholar]
- 10.Bass, D.M., J.S. Trier, R. Dambrauskas, and J.L. Wolf. 1988. Reovirus type I infection of small intestinal epithelium in suckling mice and its effect on M cells. Lab. Invest. 55:226–235. [PubMed] [Google Scholar]
- 11.Kauffman, R.S., J.L. Wolf, R. Finberg, J.S. Trier, and B.N. Fields. 1983. The sigma 1 protein determines the extent of spread of reovirus from the gastrointestinal tract of mice. Virology. 124:403–410. [DOI] [PubMed] [Google Scholar]
- 12.Fan, J.-Y., C.S. Boyce, and C.F. Cuff. 1998. T-helper 1 and T-helper 2 cytokine responses in gut-associated lymphoid tissue following enteric reovirus infection. Cell. Immunol. 188:55–63. [DOI] [PubMed] [Google Scholar]
- 13.Major, A.S., and C.F. Cuff. 1997. Enhanced mucosal and systemic immune responses to intestinal reovirus infection in β2-microglobulin-deficient mice. J. Virol. 71:5782–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.London, S.D., D.H. Rubin, and J.J. Cebra. 1987. Gut mucosal immunization with reovirus serotype 1/L stimulates virus-specific cytotoxic T cell precursors as well as IgA memory cells in Peyer's patches. J. Exp. Med. 165:830–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hopkins, S.A., F. Niedergang, I.E. Corthesy-Theulaz, and J.-P. Kraehenbuhl. 2000. A recombinant Salmonella typhimurium vaccine strain is taken up and survives within murine Peyer's patch dendritic cells. Cell. Microbiol. 2:59–68. [DOI] [PubMed] [Google Scholar]
- 16.Pron, B., C. Boumalia, F. Jaubert, P. Berche, G. Milon, F. Geissman, and J.L. Gaillard. 2001. Dendritic cells are early cellular targets of Listeria monocytogenes after intestinal delivery and are involved in bacterial spread in the host. Cell. Microbiol. 3:331–340. [DOI] [PubMed] [Google Scholar]
- 17.Shreedar, V.K., B.L. Kelsall, and M.R. Neutra. 2003. Cholera toxin induces migration of dendritic cells from the subepithelial dome region to T- and B-cell areas of Peyer's patches. Infect. Immun. 71:504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furlong, D.B., M.L. Nibert, and B.N. Fields. 1988. Sigma 1 protein of mammalian reoviruses extends from the surface of viral particles. J. Virol. 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith, R.E., H.J. Zweerink, and W.K. Joklik. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology. 39:791–810. [DOI] [PubMed] [Google Scholar]
- 20.Debiasi, R.L., M.K. Squier, B. Pike, M. Wynes, T.S. Dermody, J.J. Cohen, and K.L. Tyler. 1999. Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J. Virol. 73:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virgin, H.W., M.A. Mann, B.N. Fields, and K.L. Tyler. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 65:6772–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becker, M.M., M.I. Goral, P.R. Hazelton, G.S. Baer, S.E. Rodgers, E.G. Brown, K.M. Coombs, and T.S. Dermody. 2001. Reovirus sigmaNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J. Virol. 75:1459–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, et al. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 24.Tyler, K.L., M.K.T. Squier, S.E. Rodgers, B.E. Schneider, S.M. Oberhaus, T.A. Grdina, J.J. Cohen, and T.S. Dermody. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein σ1. J. Virol. 69:6972–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodgers, S.E., E.S. Barton, S.M. Oberhaus, B. Pike, C.A. Gibson, K.L. Tyler, and T.S. Dermody. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J. Virol. 71:2540–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connolly, J.L., E.S. Barton, and T.S. Dermody. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J. Virol. 75:4029–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter, A.G., and R.U. Janicke. 1999. Emerging roles of caspase 3 in apoptosis. Cell Death Differ. 6:99–104. [DOI] [PubMed] [Google Scholar]
- 28.Norbury, C.C., D. Malide, J.S. Gibbs, J.R. Bennink, and J.W. Yewdell. 2002. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat. Immunol. 3:265–271. [DOI] [PubMed] [Google Scholar]
- 29.Harshyne, L.A., M.I. Zimmer, S.C. Watkins, and S.M. Barratt-Boyes. 2003. A role for class A scavenger receptor in dendritic cell nibbling from live cells. J. Immunol. 170:2302–2309. [DOI] [PubMed] [Google Scholar]
- 30.Fonteneau, J.F., M. Larsson, and N. Bhardwaj. 2002. Interactions between dead cells and dendritic cells in the induction of antiviral CTL responses. Curr. Opin. Immunol. 14:471–477. [DOI] [PubMed] [Google Scholar]
- 31.Bevan, M.J. 1976. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143:1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class-I restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 33.Larsson, M., J.F. Fonteneau, M. Lirvall, P. Haslett, J.D. Lifson, and N. Bhardwaj. 2002. Activation of HIV-1 specific CD4 and CD8 T cells by human dendritic cells: roles for cross-presentation and non-infectious HIV-1 virus. AIDS. 16:1319–1329. [DOI] [PubMed] [Google Scholar]
- 34.Servet-Delprat, C., P.O. Vidalain, O. Azocar, F. Le Deist, A. Fischer, and C. Rabourdin-Combe. 2000. Consequences of Fas-mediated human dendritic cell apoptosis induced by measles virus. J. Virol. 74:4387–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tabi, Z., M. Moutaftsi, and L.K. Borysiewicz. 2001. Human cytomegalovirus pp65- and immediate early 1 antigen-specific HLA class I-restricted cytotoxic T cell responses induced by cross-presentation of viral antigens. J. Immunol. 166:5695–5703. [DOI] [PubMed] [Google Scholar]
- 36.Arrode, G., C. Boccaccio, J. Lule, S. Allart, N. Moinard, J.P. Abastado, A. Alam, and C. Davrinche. 2000. Incoming human cytomegalovirus pp65 (UL83) contained in apoptotic infected fibroblasts is cross-presented to CD8(+) T cells by dendritic cells. J. Virol. 74:10018–10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arrode, G., C. Boccaccio, J.P. Abastado, and C. Davrinche. 2002. Cross-presentation of human cytomegalovirus pp65 (UL83) to CD8+ T cells is regulated by virus-induced, soluble-mediator-dependent maturation of dendritic cells. J. Virol. 76:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subklewe, M., C. Paludan, M.L. Tsang, K. Mahnke, R.M. Steinman, and C. Munz. 2001. Dendritic cells cross-present latency gene products from Epstein-Barr virus–transformed B cells and expand tumor-reactive CD8+ killer T cells. J. Exp. Med. 193:405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maecker, H.T., S.A. Ghanekar, M.A. Suni, X.S. He, L.J. Picker, and V.C. Maino. 2001. Factors affecting the efficiency of CD8+ T cell cross-priming with exogenous antigens. J. Immunol. 166:7268–7275. [DOI] [PubMed] [Google Scholar]
- 40.Herr, W., E. Ranieri, W. Olson, H. Zarour, L. Gesualdo, and W.J. Storkus. 2000. Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4(+) and CD8(+) T lymphocyte responses. Blood. 96:1857–1864. [PubMed] [Google Scholar]
- 41.Motta, I., F. Andre, A. Lim, J. Tartaglia, W.I. Cox, L. Zitvogel, E. Angevin, and P. Kourilsky. 2001. Cross-presentation by dendritic cells of tumor antigen expressed in apoptotic recombinant canarypox virus-infected dendritic cells. J. Immunol. 167:1795–1802. [DOI] [PubMed] [Google Scholar]
- 42.Ignatius, R., M. Marovich, E. Mehlhop, L. Villamide, K. Mahnke, W.I. Cox, F. Isdell, S.S. Frankel, J.R. Mascola, R.M. Steinman, et al. 2000. Canarypox virus-induced maturation of dendritic cells is mediated by apoptotic cell death and tumor necrosis factor alpha secretion. J. Virol. 74:11329–11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsson, M., J.F. Fonteneau, S. Somersan, C. Sanders, K. Bickham, E.K. Thomas, K. Mahnke, and N. Bhardwaj. 2001. Efficiency of cross presentation of vaccinia virus-derived antigens by human dendritic cells. Eur. J. Immunol. 31:3432–3442. [DOI] [PubMed] [Google Scholar]
- 44.Mougneau, E., S. Hugues, and N. Glaichenhaus. 2002. Antigen presentation by dendritic cells in vivo. J. Exp. Med. 196:1013–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norbury, C.C., and L.J. Sigal. 2003. Cross priming or direct priming: is that really the question? Curr. Opin. Immunol. 15:82–88. [DOI] [PubMed] [Google Scholar]
- 46.Sigal, L.J., S. Crotty, R. Andino, and K.L. Rock. 1999. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 398:77–80. [DOI] [PubMed] [Google Scholar]
- 47.Prasad, S.A., C.C. Norbury, W. Chen, J.R. Bennink, and J.W. Yewdell. 2001. Cutting edge: recombinant adenoviruses induce CD8 T cell responses to an inserted protein whose expression is limited to nonimmune cells. J. Immunol. 166:4809–4812. [DOI] [PubMed] [Google Scholar]
- 48.Mueller, S.N., C.M. Jones, C.M. Smith, W.R. Heath, and F.R. Carbone. 2002. Rapid cytotoxic T lymphocyte activation occurs in the draining lymph nodes after cutaneous herpes simplex virus infection as a result of early antigen presentation and not the presence of virus. J. Exp. Med. 195:651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao, X., E. Deak, K. Soderberg, M. Linehan, D. Spezzano, J. Zhu, D.M. Knipe, and A. Iwasaki. 2003. Vaginal submucosal dendritic cells, but not Langerhans cells, induce protective Th1 responses to herpes simplex virus-2. J. Exp. Med. 197:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang, F.P., N. Platt, M. Wykes, J.R. Major, T.J. Powell, C.D. Jenkins, and G.G. MacPherson. 2000. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 191:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu, L., M. Zhang, C. Jenkins, and G.G. MacPherson. 1998. Dendritic cell heterogeneity in vivo: two functionally different dendritic cell populations in rat intestinal lymph can be distinguished by CD4 expression. J. Immunol. 161:1146–1155. [PubMed] [Google Scholar]
- 52.Vezys, V., S. Olson, and L. Lefrancois. 2000. Expression of intestine-specific antigen reveals novel pathways of CD8 T cell tolerance induction. Immunity. 12:505–514. [DOI] [PubMed] [Google Scholar]
- 53.Vezys, V., and L. Lefrancois. 2002. Cutting edge: inflammatory signals drive organ-specific autoimmunity to normally cross-tolerizing endogenous antigen. J. Immunol. 169:6677–6680. [DOI] [PubMed] [Google Scholar]
- 54.Steinman, R.M., D. Hawiger, and M.C. Nussenzweig. 2003. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21:685–711. [DOI] [PubMed] [Google Scholar]
- 55.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pooley, J.L., W.R. Heath, and K. Shortman. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8− dendritic cells, but cross presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 57.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iyoda, T., S. Shimoyama, K. Liu, Y. Omatsu, Y. Akiyama, Y. Maeda, K. Takahara, R.M. Steinman, and K. Inaba. 2002. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belz, G., C. Behrens, C. Smith, J. Miller, C. Jones, K. Lejon, C. Fathman, S. Mueller, and K. Shortman. 2002. The CD8α+ dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 196:1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu, K., T. Iyoda, M. Saternus, Y. Kimura, K. Inaba, and R.M. Steinman. 2002. Immune tolerance after delivery of dying cells to dendritic cells in situ. J. Exp. Med. 196:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamath, A.T., S. Henri, F. Battye, D.F. Tough, and K. Shortman. 2002. Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood. 100:1734–1741. [PubMed] [Google Scholar]
- 62.Scheinecker, C., R. McHugh, E.M. Shevach, and R.N. Germain. 2002. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J. Exp. Med. 196:1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]