

Abstract
After cell death, via apoptosis or necrosis, the uptake of dead cells by neighboring cells or phagocytes prevents the release of intracellular content. An array of molecules, including initiation molecules of the complement system, are involved in marking dead cells for uptake. After binding of these molecules, complement activation takes place, which when uncontrolled might result in a proinflammatory state. In the current study we demonstrate that complement inhibitor, C4b-binding protein (C4BP), binds strongly to necrotic cells, irrespective of the cell type used or the method of induction. After binding of the C4BP–protein S (PS) complex to necrotic cells via PS-phosphatidylserine and C4BP-DNA interactions, C4BP-PS inhibits complement activation on these cells. C4BP binds DNA via a patch of positively charged amino acids, mainly on the second complement control domain of the C4BP α-chain (affinity constant: 190 nM). Furthermore, C4BP limits DNA release from necrotic cells and inhibits DNA-mediated complement activation in solution. The C4BP–necrotic cell interaction also occurs in vivo as necrotic areas of arteriosclerotic plaques and of various cancers stain strongly positive for C4BP. This study describes a novel mechanism in which C4BP limits the inflammatory potential of necrotic cells.
Under physiologic conditions cell death is a tightly regulated process in which apoptotic cells are cleared rapidly by neighboring cells or professional phagocytes (1–3). Nonphysiologic cell death, necrosis, can be the result of a primary stress signal or occur secondary to apoptosis (1). Dead cells need to be taken up rapidly to prevent the release of intracellular content and the onset of autoimmune diseases, such as systemic lupus erythematosus (4). Fast removal of dead cells is dictated by a vast array of molecules that are derived from the dying cell or from plasma, and collectively, often are referred to as “eat me” signals (5). Several of these proteins that mark cells for uptake are complement components, such as C1q and mannose-binding lectin (MBL) (6). In addition to serving as prophagocytic factors they also activate the complement system (7).
Complement activation via C1q and MBL bound to dead cells may be beneficial up to the level of C3b because it allows interaction with complement receptors that are present on phagocytes, and thereby enhance phagocytosis. However, complement activation should not proceed beyond C3b because this would generate the anaphylatoxin, C5a, that evokes a local inflammatory response, and leads to the formation of the membrane attack complex (C5b-9) that induces lysis and releases intracellular contents that could serve as autoantigens. Only late apoptotic cells, and especially necrotic cells, bind C1q and MBL (8–10); this led us to hypothesize that a strong complement inhibitor should be present on necrotic cells to prevent further complement activation.
DNA is one of the autoantigens that is released from apoptotic cells, and in larger quantities from necrotic cells (11). In addition, DNA release from granulocytes (12) is a recently described mechanism that contributes to the plasma DNA pool and even is present in healthy individuals. High concentrations of extracellular DNA can be found in several clinical situations, such as the inflamed synovium in rheumatoid arthritis (13), and in bronchial lavage fluid of patients who have cystic fibrosis (14). Initially, it was reported that autologous DNA may not be immunogenic and dangerous as such (15); however, recent studies suggest that free DNA, also mammalian (16), and especially DNA–protein complexes are able to activate cells (17, 18). DNA is potentially dangerous because it can activate complement (19, 20), activate cells (21), and serve as a target of autoantibodies (22); however, because DNA is present in plasma of healthy individuals, some molecules of the immune system apparently limit its proinflammatory potential.
We have hypothesized that C4b-binding protein (C4BP) may be one of those molecules. C4BP is a key complement inhibitory molecule for the classic and lectin pathways of complement (23), and is present in plasma at a concentration of ∼200 μg/ml. This multidomain molecule consists mostly of six or seven α-chains and one β-chain to which vitamin K dependent anticoagulant protein S (PS) is bound with high affinity (23). The C4BP α-chain is built up almost exclusively of eight complement control protein (CCP) domains, and the C4BP β-chain consists of three such CCP modules. PS has the capacity to interact with negatively charged phospholipids via its γ-carboxyglutamic acid domain. We showed previously that C4BP binds to the surface of apoptotic cells via PS (24, 25).
Serum amyloid–P component (SAP) initially was reported to be the only serum component to bind to DNA under physiologic conditions (26). However, in the same period, one report described the use of DNA-cellulose columns to purify several proteins from serum; one of these proteins was reported to be C4BP (27). Since then, several complement components have been identified to bind to DNA, such as C1q (28) and MBL (29), but some functional importance only has been described for C1q (30).
We have argued that complement recognition of DNA by C1q and MBL would be beneficial but that release of proinflammatory mediators should be prevented; therefore, we tested the complement inhibitory role of C4BP binding to DNA in detail. Because necrotic cells are a major source of DNA in vivo, we tested the binding of C4BP to necrotic cells in relation to DNA release, complement activation, and TNF-α release by macrophages.
Here we demonstrate that complement inhibitor, C4BP, binds to necrotic cells via DNA and phosphatidylserine, and that C4BP regulates complement activation on those cells. In addition, C4BP limits the release of DNA from necrotic cells by binding to DNA via a positively charged patch, mainly on CCP2 of its α-chain.
RESULTS
The C4BP–PS complex binds strongly to necrotic cells
Because necrotic cells are a possible source of autoantigens and have the potential to activate the complement system, we tested the effect of fluid-phase complement regulator C4BP on these processes. First we analyzed the binding of C4BP to live, apoptotic, and necrotic cells. C4BP-PS does not bind to live Jurkat T cells but binds strongly to apoptotic (24) and necrotic Jurkat T cells (Fig. 1 A). The distribution of C4BP-PS binding to apoptotic cells is more variable than that on necrotic cells, which show strong uniform binding. To rule out that factors specific for the cell-type or the method of inducing necrosis influenced this analysis, we tested C4BP-PS binding to three different cell lines using three different methods of inducing necrosis. We observed no significant binding of C4BP-PS to live Jurkat T cells, Ramos B cells, or monocytic THP-1 cells. Upon induction of necrosis with all three methods (heat, freeze-thawing, ethanol), we observed a strong binding of C4BP-PS to all three cell types (Fig. 1 B).
Figure 1.

C4BP binds to necrotic cells. (A) Live, apoptotic, and necrotic Jurkat T cells were analyzed by flow cytometry. Shown are forward scatter (FSC) and side scatter (SSC) as well as stainings to discriminate live, apoptotic, and necrotic cells using Annexin V and ViaProbe. For live cells double-negative cells were gated; for apoptotic cells Annexin V–positive, ViaProbe-negative cells were gated; and for necrotic cells double-positive cells were gated. Specific binding of C4BP-FITC to these cell populations was compared with the binding of the negative control FITC conjugated mAb 104 (anti-C4BP). (B) The binding of C4BP to necrotic cells is independent of the cell type used and the method to induce necrosis. Jurkat T cells were compared with Ramos B cells and monocytic THP-1 cells. Live cells were compared with cells induced into necrosis by heat, freeze/thaw cycles, or ethanol. The experiment was performed in triplicate and repeated twice; means and standard deviations of the mean fluorescence intensity are shown. (C) C4BP binding to necrotic cells in the presence of blocking mAb against the C4BP α-chain or PS. (D) C4BP binding to necrotic cells in the presence of an increasing concentration of DNA. (E) C4BP localizes to the outer membrane of necrotic cells. Live and necrotic Jurkat T cells were incubated with C4BP FITC and ViaProbe. Using different filters for FITC, ViaProbe, and phase contrast, pictures were obtained from cells loaded onto object slides. Original magnification, 400.
Next we wanted to determine the mode of interaction between necrotic cells and the C4BP-PS complex, which is composed of several subunits. C4BP-PS-FITC was preincubated with blocking antibodies against the C4BP α-chain or against PS before administration to the necrotic cells. We observed that the binding to necrotic cells was inhibited partially by the mAb against the α-chain, but was inhibited strongly by the mAb against PS, which indicated that it binds mostly via PS–phospholipids interactions (Fig. 1 C). Necrotic cells have been reported to be a source of DNA; therefore, we tested if the binding of C4BP to necrotic cells could be influenced by preincubating C4BP with exogenous DNA. Increasing concentrations of exogenous DNA inhibited the binding of C4BP to necrotic cells only partially (Fig. 1 D), and to a similar extent as the blocking mAb against the α-chain (Fig. 1 C).
To have a visual impression of how C4BP binds to necrotic cells, we stained live and necrotic Jurkat T cells with ViaProbe—to stain nuclei of dead cells that permit access of this nuclear dye—and with C4BP-PS-FITC. Fig. 1 E shows that C4BP binds strongly and evenly to the outer membrane of dead cells only, and does not enter the cell or reach the nucleus.
C4BP-PS inhibits complement activation on necrotic cells
We wondered if this strong C4BP deposition has physiologic consequences; therefore, we tested if bound C4BP-PS still regulates complement activation. For this purpose we analyzed complement activation on necrotic cells using normal human serum (NHS), C4BP-deficient serum, or C4BP-deficient serum reconstituted with physiologic concentrations of C4BP-PS. It is known that apoptotic cells activate some complement, but it is mostly the necrotic cells that bind C1q and MBL and activate complement strongly. Comparing NHS with C4BP-deficient serum on necrotic cells shows that although both sera deposit equal quantities of the classic pathway initiation molecule, C1q, the C4BP-deficient serum induces much more C3b deposition (Fig. 2), which indicates that C4BP regulates the amount of C3b that is deposited on necrotic cells. Reconstituting with C4BP-PS in the fluid-phase fully reversed this effect. To ascertain that this actually is due to C4BP-PS bound to the necrotic cells, we preincubated necrotic cells at physiologic concentration of C4BP-PS and subsequently washed and incubated with C4BP-deficient serum. C3b deposition was similar to the NHS condition, which indicates that C4BP-PS on the necrotic cell regulates complement activation on these cells. By regulating the amount of C3b, it also regulates the amount of C5b-9 on the cells and C5a release (Fig. 2), and by doing so, C4BP dampens the formation and release of these proinflammatory proteins.
Figure 2.

C4BP regulates complement activation on necrotic cells. Necrotic cells were incubated with NHS or C4BP-deficient serum (C4BP df) or C4BP-deficient serum reconstituted with C4BP in the fluid-phase (C4BP df + C4BP), or the necrotic cells were preincubated with purified C4BP, washed, and then added to the C4BP-deficient serum. Deposition of complement components on the necrotic cells was analyzed by flow cytometry; staining for C1q, C3b, and C5b-9 is depicted as mean fluorescence intensity (MFI). The release of the anaphylatoxin, C5a, in the fluid phase was detected by ELISA and is depicted as μg/l. Mann-Whitney tests were performed to test for significant differences between the groups. N.S., not significant. *P < 0.05.
C4BP is present on necrotic cells in clinically relevant necrotic lesions
To verify that the binding of C4BP to necrotic cells is (patho) physiologically relevant, we tested several clinically important conditions for the presence of C4BP on necrotic cells. For this purpose we selected sections of two different types of cancer and sections of human atherosclerotic plaques that were known, from pathology reports, to contain necrotic areas. We stained sections of five cases of renal cell carcinoma, five cases of ductal carcinoma in situ of the breast, and tissue from two patients who had carotid atherosclerotic plaques. All patients displayed a strong deposition of C4BP in the necrotic areas but not in nonnecrotic tissue around these lesions. There was some variation in the intensity of staining for C4BP; representative pictures are shown in Fig. 3. These data indicate that C4BP binding to necrotic cells occurs in several clinically relevant conditions.
Figure 3.

C4BP binds to necrotic cells in vivo. Tissue sections from two clinically important conditions—atherosclerosis and cancer—were stained for the presence of C4BP on necrotic tissue. Samples from patients who suffered from atherosclerotic plaques, renal cell carcinoma, or ductal carcinoma in situ of the breast were stained with polyclonal antibodies against C4BP, or as a negative control by omitting the first antibody, and with H/E. *Location of the necrotic area in the hematoxylin-eosin–stained sections. Original magnification for the atherosclerotic sections was 10, and for the cancer samples, 100 and 400, respectively.
C4BP binds to DNA via its α-chains
Our blocking experiments showed that binding of C4BP-PS to necrotic cells is influenced by DNA. Therefore, we tested the ability of complement inhibitor, C4BP, to bind to DNA using band shift assays with Eco RI digested pcDNA3 vector (DNA) as a target. After incubating an increasing concentration of C4BP-PS with linearized DNA, we observed a dose-dependent retention of DNA as a high molecular weight complex, which indicated that C4BP-PS had bound DNA (Fig. 4 A). As a negative control we used prothrombin, another plasma protein, which like C4BP-PS is known to bind to apoptotic cells (31), and incubated a similar concentration range with the DNA but did not observe any high molecular retention (Fig. 4 B); this indicated that prothrombin does not bind DNA and that the binding of C4BP-PS in this assay was specific. C4BP-PS is composed of several subunits (see Fig. 6 A); therefore, we tested what part of this molecule is responsible for the DNA binding. Using blocking antibodies against the different domains of C4BP we observed that antibodies against α-chain blocked the interaction with DNA completely, whereas antibodies against β-chain or PS had no effect (Fig. 4 C). To prove that the α-chains are responsible for binding, we compared plasma-derived C4BP-PS—which consists of C4BP α-chains, one β-chain, and PS—with recombinant C4BP, which consists only of α-chains and free PS. We observed equally strong DNA binding by plasma-derived and recombinant C4BP, and no DNA binding by PS (Fig. 4 D). These results confirm that C4BP binds DNA through its α-chains.
Figure 4.

C4BP binds to DNA via the α-chains. Linearized pcDNA3 vector DNA was incubated with increasing concentrations of C4BP or prothrombin and separated on a regular agarose gel. As a DNA–protein complex the DNA migrates at high molecular weight, whereas free DNA migrates at low molecular weight. (A) Increasing concentrations of C4BP ranging from 0.2 μM to 1.6 μM result in C4BP-DNA binding. (B) Prothrombin, even incubated at a 1:7 μM ratio, to compensate for seven potential binding sites per C4BP molecule versus one in prothrombin, does not bind to DNA. (C) C4BP-PS was incubated with blocking antibodies to the α-chain, β-chain, and PS. (D) Plasma-derived C4BP-PS, recombinant C4BP, and free PS were compared for their ability to bind to DNA.
Figure 6.
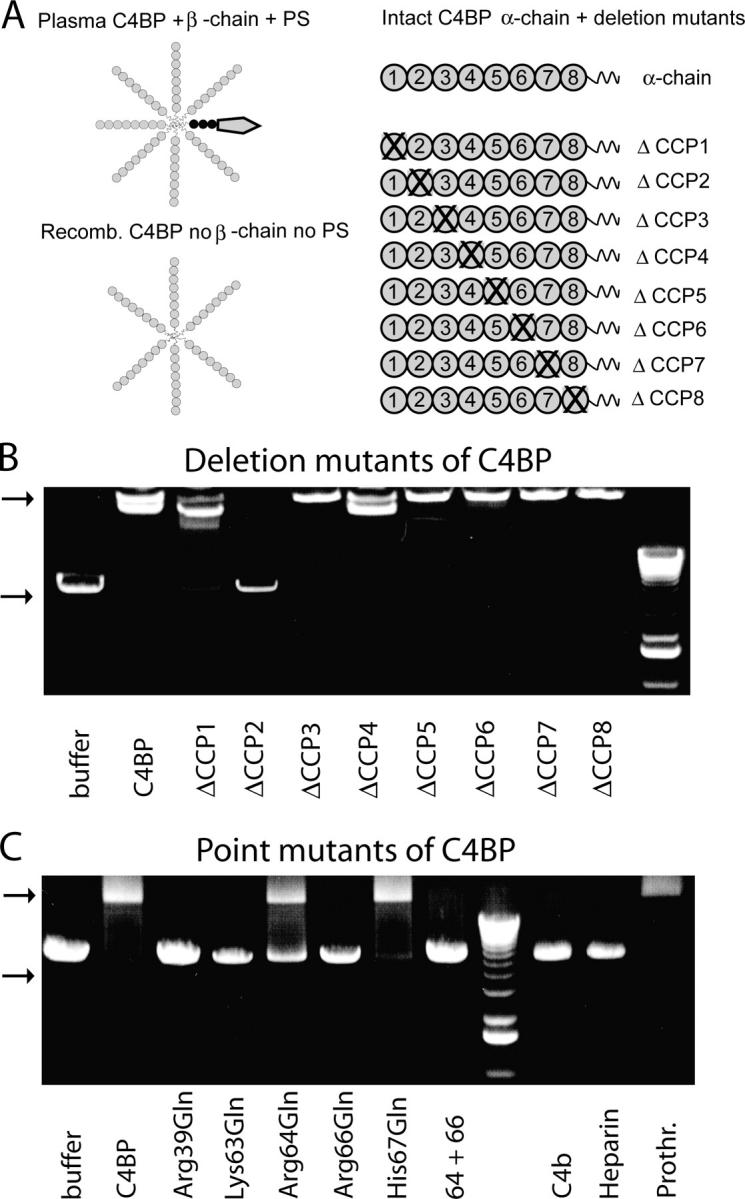
C4BP binds to DNA via positively charged amino acids on the interface between CCP-1 and CCP-2. (A) Schematic overview of different C4BP forms used in this study. Plasma-derived C4BP mostly consists of seven α-chains, one β-chain, and one molecule of PS bound with high affinity to the β-chain. Also, PS is built up of several domains but for simplicity is depicted as one unit. Recombinantly produced C4BP consists of six α-chains and lacks the β-chain and PS. α-chains are built up of eight CCP domains followed by short COOH-terminal extensions involved in polymerization of the whole molecule. Deletion mutants lacking individual CCP domains are depicted as ΔCCP. (B) Deletion mutants lacking individual CCP domains were tested for their ability to bind to DNA. (C) Single amino acid mutants in which the positively charged amino acids have been replaced by a neutral amino acid were tested for their ability to bind DNA. Excess C4b, heparin, and prothrombin were used to compete for the binding site of DNA on C4BP. Top arrow in B and C indicates DNA–protein complexes; bottom arrow indicates free DNA.
The C4BP-DNA interaction is ionic in nature and can be inhibited by free nucleotides
To identify the type of interaction between C4BP and DNA, C4BP-PS was incubated with DNA in the presence of an increasing concentration of NaCl. Increasing the ionic strength of the buffer resulted in inhibition of the DNA-C4BP interaction, and indicated that the interaction is ionic in nature (Fig. 5 A). The interaction was inhibited by 50% at 600 mM NaCl. In addition, we tested if the interaction between C4BP and DNA was inhibited by free nucleotides. The C4BP-DNA mixture was incubated in the presence of an increasing concentration of deoxyribonucleoside triphosphate (dNTP) and an equimolar mixture of free nucleotides; a dose-dependent inhibition of the interaction between linear pcDNA3 vector DNA and C4BP-PS was observed (Fig. 5 B). To test which of the nucleotides in the mixture was responsible for this inhibition, we added increasing concentrations of each of the four nucleotides—A, T, C, and G—separately. Each nucleotide had a similar capacity to inhibit the interaction of C4BP-PS with DNA (Fig. 5 C).
Figure 5.

The C4BP–DNA interaction is ionic in nature and can be inhibited by free nucleotides. (A) C4BP-DNA interactions are inhibited by increasing the ionic strength of the buffer as indicated. (B) Inhibition of C4BP-DNA interactions by increasing concentrations of dNTPs from 4 to 32 mM. (C) Dose-dependent inhibition of the C4BP-DNA interaction by individual nucleotides A, T, C, G each tested at 4 and 32 mM.
C4BP binds DNA via a positively charged cluster of amino acids on the interface between CCP1 and CCP2
Within the multidomain molecule of C4BP-PS, the α-chains are responsible for the interaction. By testing a set of recombinant deletion mutants in which individual domains had been deleted (Fig. 6 A), we were able to determine that the binding site for DNA is located on CCP2 with a small contribution by CCP1 (Fig. 6 B). It is known that there is a patch of positively charged amino acids on this interface; because such a positively charged patch may be responsible for the observed ionic interaction with negatively charged DNA, we analyzed point mutants in which the individual positively charged amino acids were substituted by the uncharged amino acid, Gln. The positively charged amino acids—Arg-39 on CCP1 and Lys-63 and Arg-66 on CCP2—are essential, whereas Arg-64 is intermediate and His-67 does not seem to be essential (Fig. 6 C). By testing two ligands that are known to interact with this cluster on the C4BP α-chain, C4b and heparin (32), we observed that these two ligands could compete with DNA for binding, which indicated that they had similar/overlapping binding sites on C4BP (Fig. 6 C). As a negative control, we performed the same with prothrombin, which could not compete. From this part of the study we conclude that C4BP-PS binds DNA, and that this interaction is ionic in nature and can be inhibited by individual nucleotides. The binding site on C4BP is located on the interface between CCP1 and CCP2 and involves positively charged amino acids at positions 39, 63, 64, and 66, a similar binding site as used by C4b and heparin.
C4BP binds to several forms of DNA and RNA
Given the resemblance between DNA and RNA, we also tested C4BP-RNA interaction and observed that RNA migrates at high molecular weight after incubation with C4BP (Fig. 7 A). However, we observed that only the higher band of the RNA preparation was interacting with C4BP. Therefore, we tested if there are size constraints for RNA/DNA for the C4BP interaction by incubating a commercial DNA ladder with buffer only or with 0.8, 1.6, or 3.2 μM C4BP. This analysis shows that C4BP binds preferably to high molecular weight DNA; however, increasing the concentration of C4BP resulted in high molecular retention of all DNA fragments and indicated no real size cut-off for the interaction between C4BP-PS and DNA (Fig. 7 B).
Figure 7.
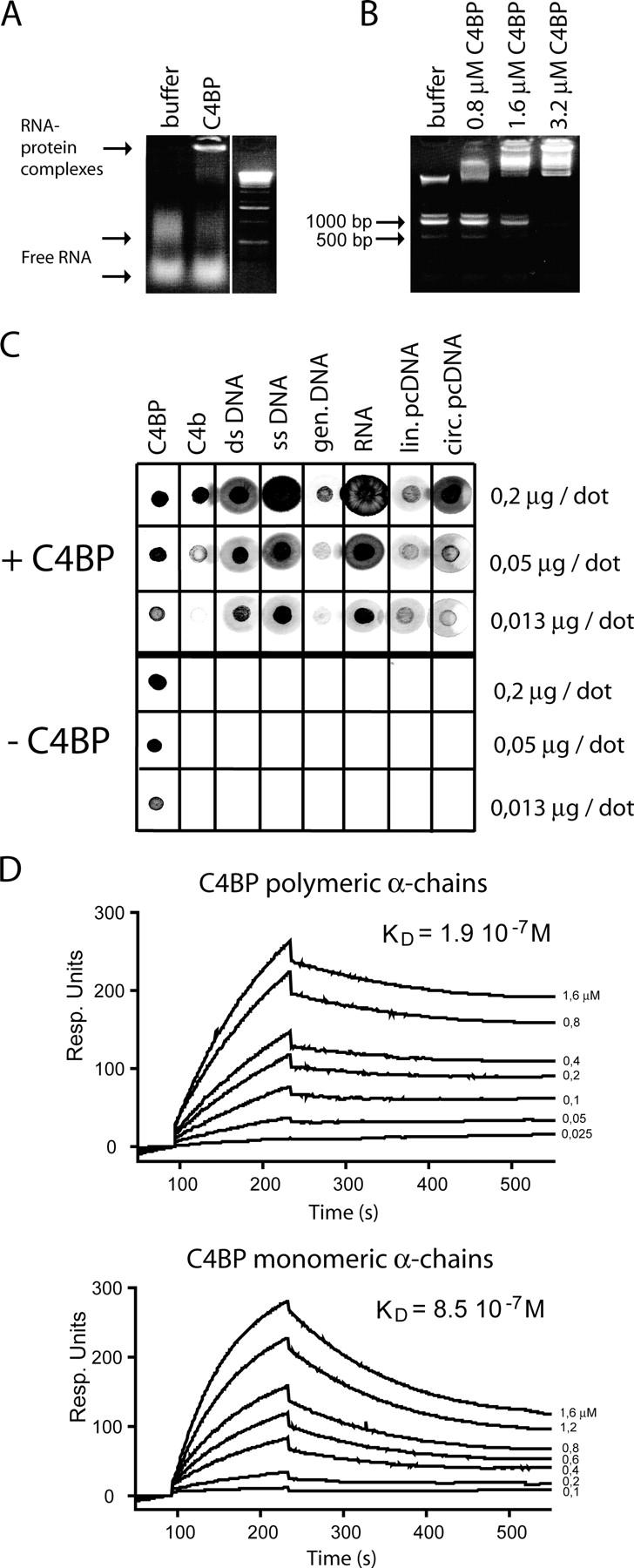
C4BP binds to various forms of DNA as well as to RNA. (A) RNA was incubated in buffer only or with C4BP and run on an agarose gel. (B) A DNA ladder was incubated with buffer only or with 0.8, 1.6, or 3.2 μM C4BP to analyze the preferential sizes of DNA for C4BP binding at these C4BP concentrations. Band sizes are indicated in bp. (C) Dot-blot analysis of C4BP binding to different forms of DNA and RNA. C4BP and C4b were dotted as positive controls. One blot was incubated with C4BP and one blot was incubated without C4BP, the developing procedure was similar. (D) Biacore analysis of the interaction of polymeric versus monomeric α-chains of C4BP with immobilized DNA. The concentrations of the analytes are indicated on the right side. The y-axis displays response units.
In addition to analyzing the binding site for DNA on C4BP, we also studied the molecular requirements of DNA for this interaction to occur. For this purpose we used dot-blots onto which we dotted increasing concentrations of different forms of DNA and RNA. We generated two identically dotted blots, on which we dotted C4BP-PS as a positive control and C4b, another natural ligand for C4BP-PS, as an additional control. One blot was incubated with C4BP and the other with buffer only; after incubation both blots were treated equally and stained for the presence of C4BP. C4BP-PS binds strongly to dsDNA, ssDNA, genomic DNA, RNA, linear, and circular pcDNA3 vector (Fig. 7 C). The observed preference of C4BP-PS to bind to higher molecular weight DNA may relate to the polymeric nature of C4BP. Therefore, we tested the affinity of polymeric C4BP versus monomeric C4BP α-chain using Biacore. Biotinylated DNA was loaded onto streptavidin chips, and the two forms of C4BP were injected at several concentrations. Intact, multivalent C4BP binds DNA more strongly, but monovalent α-chains also have the capacity to bind DNA (Fig. 7 D). The affinity constants for binding DNA are 8.5 10−7 M (ka = 6.7 10−3 1/Ms; kd = 5.7 10−3 1/s) for monomeric α-chains and 1.9 10−7 M (ka = 1.4 10−4 1/Ms; kd = 2.6 10−3 1/s) for polymeric C4BP α-chains; this is in the same affinity range as the affinity between C4BP and C4b (33). As can be observed from the sensograms, the association and the dissociation rate are affected by the multivalency of C4BP.
C4BP inhibits complement activation on DNA
DNA was shown to bind several complement initiation molecules and to activate the classic pathway of the complement system (19, 20). Knowing now that C4BP binds DNA, we wondered if C4BP is involved in the regulation of complement activation on DNA in solution. We incubated increasing concentrations of DNA with NHS or C4BP-deficient serum. As a readout for this complement activation we used the residual capacity of these sera to activate complement in a classic pathway complement activation assay. The C4BP-deficient serum had equal complement activity as did NHS in the absence of DNA. Upon incubation with DNA, both sera became dose-dependently depleted of classic pathway activity, but the C4BP-deficient serum already was depleted at lower DNA concentrations (Fig. 8) which indicated that C4BP limits complement activation on DNA in the fluid-phase.
Figure 8.

C4BP-PS inhibits complement activation by DNA. Complement activation on fluid-phase DNA was analyzed by incubating DNA with NHS or C4BP-deficient serum. Residual classic pathway activity was determined using lysis of immunoglobulin sensitized sheep erythrocytes as a read out. The experiment was performed in triplicate and means ± SD are shown.
C4BP bound to necrotic cells limits DNA release
To test if C4BP on the necrotic cell also may be involved in the regulation of DNA release, we analyzed the amount of DNA released in the supernatant after induction of necrosis in the presence or absence of C4BP-PS. We generated live and necrotic Jurkat, Ramos, and THP-1 cells in medium without FCS or in medium containing 100 μg/ml C4BP-PS, and cultured these cells for an additional 8 h. Just after the induction of necrosis and at the moment of collecting the supernatant, we verified by FACS that similar amounts of necrotic cells were present in the various conditions. We observed a highly reproducible amount of necrotic cells using heat, which was not affected by the presence of C4BP (unpublished data). The DNA concentration was determined in cell-free supernatant. Live cells release some DNA during culture, which is not inhibited by C4BP (Fig. 9 A). However, necrotic cells release more DNA and this release is inhibited by the presence of C4BP-PS (Fig. 9 A) which indicates that C4BP-PS on the outer membrane of necrotic cells captures DNA and retains it on the necrotic cell. We observed that C4BP-PS preferentially binds high molecular weight DNA; therefore, we analyzed what sizes of DNA are released from cells rendered necrotic in the presence or absence of C4BP-PS. We ran necrotic cells on a regular agarose gel and observed that in the presence of C4BP-PS there was only the low molecular weight DNA band, whereas in the absence of C4BP-PS there also is a high molecular weight band (Fig. 9 B). C4BP-PS captures the high molecular weight DNA that is being released from necrotic cells, and the residual DNA, as measured in the supernatant, most likely is low molecular weight DNA. We did not observe a clear inhibition of the release of intact nucleosomes by incubation with C4BP and we also did not observe significant binding of C4BP to intact chromosomes on cell smears (unpublished data); collectively, these data indicated that C4BP may only bind DNA when it is sufficiently free of histones.
Figure 9.

C4BP limits DNA release from necrotic cells. (A) DNA release by live and necrotic cells generated and cultured in medium only or medium containing 100 μg/ml C4BP as determined using a PicoGreen assay on the supernatant. The experiment was performed in triplicate four times, means and standard deviations are shown. Student's t test was performed to test for significant differences between the groups. NS, not significant. * P < 0.05, ** P < 0.001, ***P < 0.0001. (B) Necrotic cells were incubated with either buffer only or with C4BP and then loaded onto a regular agarose gel stained with ethidium bromide. Necrotic cells release DNA, and when these cells are generated in the presence of C4BP, only the low molecular weight band is visible.
C4BP limits the proinflammatory potential of necrotic cells
We generated THP-1–derived macrophages and analyzed the effect of coincubating necrotic cells in the presence of heat-inactivated NHS, NHS, C4BP-deficient serum, or reconstituted deficient serum, and as a readout we analyzed the production of the proinflammatory cytokine, TNF-α. Comparing heat-inactivated NHS with NHS, we observed a strong increase in TNF-α production which indicated that when complement activation can take place, more TNF-α is released (Fig. 10). Using C4BP-deficient serum we observed increased amounts of TNF-α; this is reversed by reconstituting with purified C4BP (Fig. 10) and indicates that C4BP limits the proinflammatory effects of necrotic cells and serum.
Figure 10.

C4BP limits the pro-inflammatory potential of necrotic cells and complement. THP-1 derived macrophages were incubated with necrotic cells in the presence of either heat inactivated NHS (HI NHS), NHS, C4BP-deficient serum (C4BP df) or reconstituted serum. After incubation, TNF-α release by the macrophages was analyzed by ELISA. The experiment was performed four times in triplicate; (means and SEM) are shown. Analysis of variance with the Tukey posttest was performed to test for significant differences between the groups. *P < 0.05; ***P < 0.001.
DISCUSSION
We describe the binding of a key complement inhibitor, C4BP, to necrotic cells and DNA. By binding to necrotic cells C4BP inhibits complement activation on those cells and limits DNA release.
Although deficiencies have been reported for virtually all complement proteins, including inhibitors (34–37), no complete deficiency for C4BP has been described, although it has been studied extensively (38, 39). Whether this means that C4BP also has other functions is still a matter of debate. No genetic deficiencies have been described for the DNA binding proteins, SAP and C-reactive protein, either (40). SAP was reported to bind to DNA (26) and to form complexes with C4BP (41); in the current paper we show also that C4BP in purified form binds DNA which strengthens the idea that SAP and C4BP have roles in regulating DNA processing.
Noncell-associated DNA is present in plasma of healthy individuals and in highly increased concentrations in plasma of patients who have cancer (42), systemic lupus erythematosus (43, 44), or rheumatoid arthritis (13). Such DNA can be derived from degranulating neutrophils as a physiologic process in innate defense (12) or from dying cells (45). Because in many cancers the DNA that is present in plasma can be used to identify the type of tumor by PCR technology, it must be derived from dying tumor cells (46). All of the noncell-associated DNA in plasma was reported to be complexed to proteins (47), and we are studying the relative contribution of C4BP to the formation of such complexes. DNA can stimulate cells via Toll-like receptors; this is well-known for CpG motif–bearing nonmammalian DNA, but also is true for mammalian DNA (16–18), especially via the simultaneous triggering of different sets of receptors by DNA–protein complexes (16–18). Because of its polymeric nature, C4BP may be involved in cross-linking such cellular receptors. One of the direct practical implications of our findings relates to gene therapy. DNA delivery for gene therapy is of limited success (48), in part because of complement activation on either the DNA itself or the poly-l-lysine that serves as a carrier molecule (19). Preincubation of such complexes with C4BP may block binding sites for complement-activating molecules—and at the same time—inhibit complement activation, and thereby, increase the therapeutic success.
Binding of C4BP to necrotic cells is a broad phenomenon. We showed that C4BP-PS binds strongly and specifically to necrotic cells of three different cell lines, after induction of necrosis using three different methods, and to necrotic cells that are present in normal in vitro cultures. In addition, we observed C4BP binding to necrotic cells in clinically important conditions, such as vascular disease and cancer, in all samples tested. Therefore, we propose that C4BP binding to apoptotic (24) and necrotic cells is a fundamental property that may be the main function of the C4BP-PS complex.
We showed previously that C4BP-PS binds to apoptotic cells via PS–phosphatidylserine interactions (24). Phosphatidylserine also is available on necrotic cells and C4BP-PS binds to them mainly via PS, but also via DNA on the outer membrane of necrotic cells, not in the nucleus. Because C4BP-PS is polymeric, several α-chains will be available to inhibit complement activation and to capture free DNA that is released from such cells.
Generating a C4BP-deficient mouse would seem to be an attractive approach to confirm these data in vivo; however, murine C4BP is different from human C4BP because it lacks the β-chain and PS (49). This may make mouse C4BP behave differently; it probably will not bind to apoptotic cells directly but should be able to interact with DNA. This does not mean that the C4BP-PS system cannot be important in man, but it indicates that a murine system may not be optimal to study the role of C4BP-PS in vivo.
Binding between C4BP and DNA is established via positively charged amino acids at the interface between CCP1 and CCP2. A similar site also is used for binding to heparin (32), streptococcal M-proteins (50), and C3b and C4b (23). The estimated affinity constant for the C4BP–DNA interactions of the polymeric molecule versus the monomeric α-chains were 1.9 10−7 M versus 8.5 10−7 M. These numbers are in the same range as the affinities for C4b (51). We are studying if high concentrations of DNA can affect C4BP function.
Although necrosis generally is considered to be a more proinflammatory condition than apoptosis, in our opinion it is essential to realize that several factors are involved in dampening the proinflammatory potential of necrotic cells just as is the case with apoptotic cells. The current paper shows that C4BP limits C3b and C5b9 deposition on necrotic cells and the release of C5a and DNA. C4BP also limits the proinflammatory response of macrophages induced by necrotic cells and serum. Although necrotic cell death is uncontrolled in its death process at the level of the individual cell, it is not unregulated by the host.
MATERIALS AND METHODS
Cells and induction of necrosis.
Jurkat T cells; Ramos B cells; and the monocytic cell line, THP-1, (all purchased from ATCC) were grown in RPMI 1640, supplemented with glutamin, penicillin, streptomycin, and 10% heat-inactivated FCS (all from Invitrogen). Washed cells were induced into apoptosis or necrosis. Apoptotic cells were generated by addition of 1 μM staurosporine (Sigma-Aldrich) to Jurkat T cells for 3 h. Necrosis was induced by using three different methods: (a) heat: cells were brought to a concentration of 106 per ml and incubated at 56°C for 30 min, (b) repeated freeze-thaw cycles: five times freeze-thaw from −80°C to +56°C in 60 min, or (c) by incubation in 10% ethanol for 60 min at 37°C.
THP-1 cells were differentiated into macrophages at 25,000 cells per well in a 96-well plate using 100 nM PMA (Sigma-Aldrich) in complete RPMI 1640 for 72 h in total with a change to fresh medium after 48 h. Necrotic Jurkat cells (heat) were used at 250,000 per well in RPMI 1640, without FCS, containing 0.5 μg/ml LPS (Escherichia coli 026:B6, Sigma-Aldrich) supplemented with NHS, heat-inactivated NHS, C4BP-deficient serum, or deficient serum reconstituted with C4BP all at a final concentration of 20%. Cells were coincubated for 6 h and cell-free supernatant was tested in TNF-α ELISA (R&D Systems) following the instructions of the manufacturer.
Proteins and sera.
The C4BP-PS complex was purified from human plasma as described (52). Recombinant wild-type C4BP and different mutants lacking individual CCP domains or with introduced point mutations Arg39Gln, Lys63Gln, Arg64Gln, Arg66Gln, His67Gln, and Arg64Gln-Arg66Gln were generated and purified as described (53, 54). PS was purified as described (52), prothrombin was purified using BaCl2 precipitation and DEAE-Sephacel chromatography, C4 was purified, and C4b was generated as described (51). Heparin was obtained from ICN Biomedicals. mAb 104 against the α-chain of C4BP, mAb 2B against the β-chain of C4BP, and mAb 21 against PS were a gift of B. Dahlbäck, Lund University, Sweden. NHS was obtained from freshly drawn blood from healthy volunteers that was allowed to clot for 30 min at room temperature and all further steps on ice; individual sera were pooled and stored in aliquots at –70°C or used to generate depleted serum. Human serum deficient in C4BP-PS was prepared by passing fresh serum through a HiTrap column (Amersham Biosciences) coupled with mAb 104 (55). The flow through was collected and the depleted serum was stored in aliquots at –70°C. The C4BP-PS preparations did not contain any DNase I activity.
Nucleic acids.
pcDNA3 plasmid DNA (Invitrogen) was linearized using EcoRI (Fermentas). Human genomic DNA was isolated using QIAGEN Blood Maxi kit following instructions of the manufacturer and whole blood from a healthy volunteer. dsDNA and ssDNA from calf thymus were obtained from Sigma-Aldrich. RNA was isolated using RNA-easy kit from QIAGEN using mouse spleen as a source. The dNTPs were from Amersham Biosciences.
Flow cytometry.
Cell populations were analyzed using flow cytometry. To discriminate live, apoptotic, and necrotic populations, cells were stained with annexin V–PE (Molecular Probes) and ViaProbe (7-AAD, Molecular Probes). C4BP binding was analyzed by incubating cells with 2 μg of C4BP-FITC or as a control with 2 μg of mAb 104-FITC in 50 μl binding buffer (10 mM Hepes, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2) for 30 min at room temperature. Blocking experiments were performed by preincubating C4BP-FITC with anti–α-chain mAb 104 or with anti-PS mAb 21 at a 1 μg mAb:1 μg C4BP-FITC ratio or with 0.25, 1.25, or 2.5 μg dsDNA per μg C4BP-FITC at room temperature for 30 min. Complement deposition was analyzed using rabbit anti-C1q FITC (DakoCytomation), rabbit anti-C3 FITC (DakoCytomation), goat–anti-C9 (Advanced Research Technologies), and rabbit anti–goat FITC (DakoCytomation).
Cell staining.
Live and necrotic Jurkat T cells were stained for C4BP binding and cell viability using C4BP-FITC and ViaProbe. Cells were incubated with a combination of 1 μg C4BP-FITC and 5 μl ViaProbe in 25 μl binding buffer. Cells were mounted on glass slides and visualized using a regular Olympus microscope equipped with the appropriate filters. Original magnification was 400.
Immunohistochemistry.
Sections for three conditions of clinically observed necrosis were stained and permission for their use was obtained from the relevant local ethical committees. The sections were selected by a pathologist with the presence of one or more necrotic lesions as the only inclusion criterion, based on histologic features of necrosis. We stained sections from five cases of renal cell carcinoma and five cases of ductal carcinoma in situ of the breast from archived formalin-fixed, paraffin-embedded, surgical tumor specimens. In addition, we stained frozen tissue from two patients who had carotid atherosclerosis. Sections were deparaffinized following standard procedures or stained directly for frozen sections. Polyclonal rabbit anti–human C4BP was used as primary antibody at 10 μg/ml, washed and followed by goat anti–rabbit HRP, substrate and hematoxylin counter stain. As a negative control we stained the same sections by omitting primary antibody.
Gel shift analyses.
Binding of C4BP to DNA was evaluated using gel shift analysis. DNA and proteins were mixed using 0.2 μg linearized pcDNA3 vector DNA with 1 to 8 μg protein in a 10-μl reaction volume containing Tris EDTA buffer (10 mM Tris-HCl, 5 mM EDTA, pH 7.6) and incubated for 30 min at 37°C. DNA–protein complexes were separated by 0.8% agarose gel electrophoresis in 40 mM Tris acetate, pH 8, buffer and 0.5 μg/ml ethidium bromide and visualized by UV transluminator. For most consecutive studies, 0.2 μg DNA and 2 μg protein were incubated in a 10-μl reaction volume containing competitors in TE-buffer for 30 min at 37°C and analyzed as described. Blocking antibodies were incubated at 1 μg mAb per μg of C4BP. mAb 104 was used to block the α-chain of C4BP-PS, mAb 2B was used to block the α-chain of C4BP-PS, and mAb 21 was used to block PS. Plasma-derived C4BP-PS and recombinant C4BP were compared for binding to DNA at 0.8 μM; free PS was analyzed at 5 μM. To test for ionic interactions, C4BP-PS and DNA were incubated at increasing concentrations of NaCl (150 mM, 300 mM, 600 mM, and 1,200 mM). To test if the interaction between C4BP-PS and DNA was dependent on a particular nucleotide, mixtures of all four dNTPs or individual nucleotides were added at 4 mM, 8 mM, 16 mM, or 32 mM. Both deletion mutants lacking individual CCP domains or point mutants as described at proteins and sera were analyzed at 0.8 μM. Blocking the interaction between C4BP-PS and DNA was performed with 4 μg C4b, 4 μg heparin, and 4 μg prothrombin per 1 μg C4BP-PS. RNA–C4BP–PS interactions were visualized by performing gel-shift analysis using 0.1 μg RNA and 0.8 μM C4BP-PS. To determine size dependency of the C4BP–DNA interaction, 4 μg of a 1-kb DNA-ladder (Fermentas) was incubated with buffer only or with 0.8 or 3.2 μM C4BP-PS and analyzed in the gel-shift assay.
Dot blot.
Several forms of DNA and RNA, as described in the “nucleic acids” section, were dotted in decreasing concentration onto Hybond-C extra nitrocellulose membrane (Amersham Biosciences). Two identical blots were produced dotting C4BP-PS and C4b as positive controls at 1, 0.25, and 0.06 μg per dot, and DNA/RNA samples at 0.2, 0.05 and 0.013 μg per dot in a 2-μl volume. Blots were washed using wash buffer (50 mM Tris HCl, 0.15 M NaCl, 0.1% Tween) and blocked using Quench (wash buffer supplemented with 3% fish gelatin) for 1 h at room temperature. One blot was incubated with 5 μg/ml of C4BP-PS for 1 h at room temperature, the other blot was incubated with buffer only. Both blots were washed three times with washing buffer and stained for C4BP-PS using our anti-C4BP mAb 104 and goat anti–mouse alkaline phosphatase (DakoCytomation). Blots were developed using color substrate reaction.
Surface plasmon resonance.
To measure the kinetics of DNA-C4BP interaction we coupled biotinylated DNA to a streptavidin chip (Biacore). Double-stranded 25-mer oligonucleotides (G-C)25 and (A-T)25 were produced using equimolar amounts of single-stranded 5′ biotinylated 25-mer oligonucleotides (MWG) as described (56). Equal concentrations of G–C and A–T were coupled to the streptavidin sensor chip surface to a level of 300 response units. All experiments were performed at a continuous flow rate of 30 μl/min using Biacore buffer (150 mM NaCl, 10 mM Hepes, 2.5 mM CaCl2, 0.002% Tween-20, pH 7.4). Analytes, C4BP intact molecule, and C4BP monovalent α-chains were run over the chip in a concentration gradient. The chip was regenerated using pulse injection of 3 M guanidium chloride followed by 1 M NaCl. The obtained sensograms were analyzed using Bio-evaluation software 3.0 using 1:1 model of interaction with drifting baseline and Rmax local.
Complement activation assays.
Complement activation on fluid-phase dsDNA was analyzed by incubating increasing concentrations of dsDNA with NHS or C4BP-deficient human serum. The mixtures of DNA and serum were incubated at 37°C for 1 h. Under these conditions DNA will activate the complement system and consume it partially. As a readout of complement consumption, residual classic pathway activity was determined using a hemolytic assay as described previously (55). In brief, sheep erythrocytes coated with antibodies were incubated with controls or serial dilutions of sera that had been incubated previously with the DNA preparations. Release of hemoglobulin in the fluid-phase was used as a readout of complement-mediated lysis.
Analysis of complement activation and deposition on necrotic cells was performed by flow cytometry. Cells were rendered necrotic by heat as described in the section entitled “Cells and induction of necrosis” and washed with DGVB++ (2.5 mM veronal buffer, pH 7.3, containing 70 mM NaCl, 140 mM glucose, 0.1% gelatin, 1 mM MgCl2 and 0.15 mM CaCl2) and transferred to 96-well plates (Greiner) at 105 cells in a 50-μl volume containing DGVB++ and 10% NHS, C4BP-deficient human serum, or C4BP-deficient serum reconstituted with physiologic concentrations of C4BP-PS. Samples were incubated on a shaker for 30 min at 37°C. Deposition of C1q, C3, and C5b-9 was analyzed as described in the section entitled “Flow cytometry.” C5a-release in the fluid phase was determined in the supernatant using a C5a detection kit (IBL) according to the instructions of the manufacturer. Assays were performed twice in triplicate.
DNA-release assay.
Jurkat T cells, Ramos B cells, and THP-1 cells were kept alive or made necrotic as described in the Cells and induction of necrosis section in RPMI 1640 without FCS in the presence or absence of 100 μg/ml C4BP-PS. Cells were plated at 105 cells in 100 μl and cultured for an additional 8 h. Cells were analyzed just before and after the experiment by flow cytometry for the percentage of live, apoptotic, and necrotic cells by staining with Annexin V and ViaProbe as described in the Flow cytometry section. Cells and supernatant were separated by centrifugation in V-bottom plates and the DNA content of the supernatant was analyzed using a PicoGreen DNA quantification kit (Molecular Probes) as described by the manufacturer. Experiments were performed in triplicate and repeated four times. Averages and standard deviations are shown.
Statistical analysis.
We used Mann-Whitney, Student's t and ANOVA with Tukey posttest, to test for significant differences between groups.
Acknowledgments
We greatly appreciate the excellent technical support of E. Nilsson regarding immunohistology, Prof. Dr. E. Slota (Institute of Zootechnics, Krakow, Poland) for providing cell smears for chromosome staining, Prof. Dr. B. Dahlbäck (Lund University, Malmö, Sweden) for providing the mAb against C4BP and PS, and Dr. K. Gelderman (Lund University, Malmö, Sweden) for providing mouse RNA and critical comments on the text.
The authors acknowledge the financial support of Foundations of Tore Nilsson and the Royal Physiographic Society in Lund (L.A. Trouw) and Cancerfonden, Swedish Research Council, Swedish Foundation for Strategic Research (INGVAR) and Foundations of Kock, Påhlsson, Österlund, Bergvalls, Hain, Crafoord, King Gustav V's 80th anniversary, Svartz, Zoegas, and a research grant from the University Hospital in Malmö (A.M. Blom). L.A. Trouw is a recipient of postdoctoral stipends from foundations of Wenner-Gren and Anna-Greta Crafoord.
The authors have no conflicting financial interests.
Abbreviations used: C4BP, C4b-binding protein; C4BP-PS, C4b-binding protein–protein S complex; CCP, complement control protein (domain); dsDNA, double-stranded DNA; dNTP, deoxyribonucleoside triphosphate; MBL, mannose-binding lectin; NHS, normal human serum; PS, protein S; SAP, serum amyloid–P component; ssDNA, single-stranded DNA.
References
- 1.Savill, J., I. Dransfield, C. Gregory, and C. Haslett. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2:965–975. [DOI] [PubMed] [Google Scholar]
- 2.Savill, J., and V. Fadok. 2000. Corpse clearance defines the meaning of cell death. Nature. 407:784–788. [DOI] [PubMed] [Google Scholar]
- 3.Fadok, V.A., and G. Chimini. 2001. The phagocytosis of apoptotic cells. Semin. Immunol. 13:365–372. [DOI] [PubMed] [Google Scholar]
- 4.Manderson, A.P., M. Botto, and M.J. Walport. 2004. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 22:431–456. [DOI] [PubMed] [Google Scholar]
- 5.Ren, Y., and J. Savill. 1998. Apoptosis: the importance of being eaten. Cell Death Differ. 5:563–568. [DOI] [PubMed] [Google Scholar]
- 6.Ogden, C.A., A. deCathelineau, P.R. Hoffmann, D. Bratton, B. Ghebrehiwet, V.A. Fadok, and P.M. Henson. 2001. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J. Exp. Med. 194:781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fishelson, Z., G. Attali, and D. Mevorach. 2001. Complement and apoptosis. Mol. Immunol. 38:207–219. [DOI] [PubMed] [Google Scholar]
- 8.Gaipl, U.S., S. Kuenkele, R.E. Voll, T.D. Beyer, W. Kolowos, P. Heyder, J.R. Kalden, and M. Herrmann. 2001. Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ. 8:327–334. [DOI] [PubMed] [Google Scholar]
- 9.Nauta, A.J., N. Raaschou-Jensen, A. Roos, M.R. Daha, H.O. Madsen, M.C. Borrias-Essers, L.P. Ryder, C. Koch, and P. Garred. 2003. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur. J. Immunol. 33:2853–2863. [DOI] [PubMed] [Google Scholar]
- 10.Zwart, B., C. Ciurana, I. Rensink, R. Manoe, C.E. Hack, and L.A. Aarden. 2004. Complement activation by apoptotic cells occurs predominantly via IgM and is limited to late apoptotic (secondary necrotic) cells. Autoimmunity. 37:95–102. [DOI] [PubMed] [Google Scholar]
- 11.Jahr, S., H. Hentze, S. Englisch, D. Hardt, F.O. Fackelmayer, R.D. Hesch, and R. Knippers. 2001. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 61:1659–1665. [PubMed] [Google Scholar]
- 12.Brinkmann, V., U. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D.S. Weiss, Y. Weinrauch, and A. Zychlinsky. 2004. Neutrophil extracellular traps kill bacteria. Science. 303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 13.Yu, D., P.M. Rumore, Q. Liu, and C.R. Steinman. 1997. Soluble oligonucleosomal complexes in synovial fluid from inflamed joints. Arthritis Rheum. 40:648–654. [DOI] [PubMed] [Google Scholar]
- 14.Whitchurch, C.B., T. Tolker-Nielsen, P.C. Ragas, and J.S. Mattick. 2002. Extracellular DNA required for bacterial biofilm formation. Science. 295:1487. [DOI] [PubMed] [Google Scholar]
- 15.Krieg, A.M., A.K. Yi, S. Matson, T.J. Waldschmidt, G.A. Bishop, R. Teasdale, G.A. Koretzky, and D.M. Klinman. 1995. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 374:546–549. [DOI] [PubMed] [Google Scholar]
- 16.Goeckeritz, B.E., M. Flora, K. Witherspoon, Q. Vos, A. Lees, G.J. Dennis, D.S. Pisetsky, D.M. Klinman, C.M. Snapper, and J.J. Mond. 1999. Multivalent cross-linking of membrane Ig sensitizes murine B cells to a broader spectrum of CpG-containing oligodeoxynucleotide motifs, including their methylated counterparts, for stimulation of proliferation and Ig secretion. Int. Immunol. 11:1693–1700. [DOI] [PubMed] [Google Scholar]
- 17.Leadbetter, E.A., I.R. Rifkin, A.M. Hohlbaum, B.C. Beaudette, M.J. Shlomchik, and A. Marshak-Rothstein. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 416:603–607. [DOI] [PubMed] [Google Scholar]
- 18.Viglianti, G.A., C.M. Lau, T.M. Hanley, B.A. Miko, M.J. Shlomchik, and A. Marshak-Rothstein. 2003. Activation of autoreactive B cells by CpG dsDNA. Immunity. 19:837–847. [DOI] [PubMed] [Google Scholar]
- 19.Plank, C., K. Mechtler, F.C. Szoka Jr., and E. Wagner. 1996. Activation of the complement system by synthetic DNA complexes: a potential barrier for intravenous gene delivery. Hum. Gene Ther. 7:1437–1446. [DOI] [PubMed] [Google Scholar]
- 20.Jiang, H., B. Cooper, F.A. Robey, and H. Gewurz. 1992. DNA binds and activates complement via residues 14-26 of the human C1q A chain. J. Biol. Chem. 267:25597–25601. [PubMed] [Google Scholar]
- 21.Kirschning, C.J., and S. Bauer. 2001. Toll-like receptors: cellular signal transducers for exogenous molecular patterns causing immune responses. Int. J. Med. Microbiol. 291:251–260. [DOI] [PubMed] [Google Scholar]
- 22.Rahman, A. 2004. Autoantibodies, lupus and the science of sabotage. Rheumatology (Oxford). 43:1326–1336. [DOI] [PubMed] [Google Scholar]
- 23.Blom, A.M., B.O. Villoutreix, and B. Dahlback. 2004. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol. Immunol. 40:1333–1346. [DOI] [PubMed] [Google Scholar]
- 24.Webb, J.H., A.M. Blom, and B. Dahlbäck. 2002. Vitamin K–dependent protein S localizing complement regulator C4b-binding protein to the surface of apoptotic cells. J. Immunol. 169:2580–2586. [DOI] [PubMed] [Google Scholar]
- 25.Webb, J.H., A.M. Blom, and B. Dahlbäck. 2003. The binding of protein S and the protein S-C4BP complex to neutrophils is apoptosis-dependent. Blood Coagul. Fibrinolysis. 14:355–359. [DOI] [PubMed] [Google Scholar]
- 26.Pepys, M.B., and P.J. Butler. 1987. Serum amyloid P component is the major calcium-dependent specific DNA binding protein of the serum. Biochem. Biophys. Res. Commun. 148:308–313. [DOI] [PubMed] [Google Scholar]
- 27.Abdullah, M., R.J. Davies, and J.A. Hill. 1985. The application of DNA-cellulose chromatography in the isolation of immunoglobulin M and complement component C4b-binding protein from human serum. J. Chromatogr. 347:129–136. [DOI] [PubMed] [Google Scholar]
- 28.Van Schravendijk, M.R., and R.A. Dwek. 1982. Interaction of C1q with DNA. Mol. Immunol. 19:1179–1187. [DOI] [PubMed] [Google Scholar]
- 29.Palaniyar, N., J. Nadesalingam, H. Clark, M.J. Shih, A.W. Dodds, and K.B. Reid. 2004. Nucleic acid is a novel ligand for innate, immune pattern recognition collectins surfactant proteins A and D and mannose-binding lectin. J. Biol. Chem. 279:32728–32736. [DOI] [PubMed] [Google Scholar]
- 30.Gaipl, U.S., T.D. Beyer, P. Heyder, S. Kuenkele, A. Bottcher, R.E. Voll, J.R. Kalden, and M. Herrmann. 2004. Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum. 50:640–649. [DOI] [PubMed] [Google Scholar]
- 31.D'Agnillo, P., J.S. Levine, R. Subang, and J. Rauch. 2003. Prothrombin binds to the surface of apoptotic, but not viable, cells and serves as a target of lupus anticoagulant autoantibodies. J. Immunol. 170:3408–3422. [DOI] [PubMed] [Google Scholar]
- 32.Blom, A.M., J. Webb, B.O. Villoutreix, and B. Dahlbäck. 1999. A cluster of positively charged amino acids in the N-terminal modules of the C4BP α-chain is crucial for C4b binding and factor I cofactor function. J. Biol. Chem. 274:19237–19245. [DOI] [PubMed] [Google Scholar]
- 33.Blom, A.M., L. Kask, B. Ramesh, and A. Hillarp. 2003. Effects of zinc on factor I cofactor activity of C4b-binding protein and factor H. Arch. Biochem. Biophys. 418:108–118. [DOI] [PubMed] [Google Scholar]
- 34.Morgan, B.P., and M.J. Walport. 1991. Complement deficiency and disease. Immunol. Today. 12:301–306. [DOI] [PubMed] [Google Scholar]
- 35.Pappalardo, E., L.C. Zingale, A. Terlizzi, A. Zanichelli, A. Folcioni, and M. Cicardi. 2002. Mechanisms of C1-inhibitor deficiency. Immunobiology. 205:542–551. [DOI] [PubMed] [Google Scholar]
- 36.Shichishima, T., and H. Noji. 2002. A new aspect of the molecular pathogenesis of paroxysmal nocturnal hemoglobinuria. Hematology (Am Soc Hematol Educ Program). 7:211–227. [DOI] [PubMed] [Google Scholar]
- 37.Riley-Vargas, R.C., D.B. Gill, C. Kemper, M.K. Liszewski, and J.P. Atkinson. 2004. CD46: expanding beyond complement regulation. Trends Immunol. 25:496–503. [DOI] [PubMed] [Google Scholar]
- 38.Malm, J., M. Laurell, and B. Dahlbäck. 1988. Changes in the plasma levels of vitamin K–dependent proteins C and S and of C4b-binding protein during pregnancy and oral contraception. Br. J. Haematol. 68:437–443. [DOI] [PubMed] [Google Scholar]
- 39.Marcovina, S.M., A. Zoppo, S. Vigano-D'Angelo, G. Di Cola, and A. D'Angelo. 1991. Determination of serum levels of complement component C4b-binding protein: influence of age and inflammation. Int. J. Clin. Lab. Res. 21:171–175. [DOI] [PubMed] [Google Scholar]
- 40.Butler, P.J., G.A. Tennent, and M.B. Pepys. 1990. Pentraxin-chromatin interactions: serum amyloid P component specifically displaces H1-type histones and solubilizes native long chromatin. J. Exp. Med. 172:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Beer, F.C., M.L. Baltz, S. Holford, A. Feinstein, and M.B. Pepys. 1981. Fibronectin and C4-binding protein are selectively bound by aggregated amyloid P component. J. Exp. Med. 154:1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu, T.L., D. Zhang, J.H. Chia, K.H. Tsao, C.F. Sun, and J.T. Wu. 2002. Cell-free DNA: measurement in various carcinomas and establishment of normal reference range. Clin. Chim. Acta. 321:77–87. [DOI] [PubMed] [Google Scholar]
- 43.Rumore, P.M., and C.R. Steinman. 1990. Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J. Clin. Invest. 86:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bengtsson, A., R. Nezlin, Y. Shoenfeld, and G. Sturfelt. 1999. DNA levels in circulating immune complexes decrease at severe SLE flares-correlation with complement component C1q. J. Autoimmun. 13:111–119. [DOI] [PubMed] [Google Scholar]
- 45.Choi, J.J., C.F. Reich III, and D.S. Pisetsky. 2004. Release of DNA from dead and dying lymphocyte and monocyte cell lines in vitro. Scand. J. Immunol. 60:159–166. [DOI] [PubMed] [Google Scholar]
- 46.Taback, B., S.J. O'Day, and D.S. Hoon. 2004. Quantification of circulating DNA in the plasma and serum of cancer patients. Ann. N. Y. Acad. Sci. 1022:17–24. [DOI] [PubMed] [Google Scholar]
- 47.Rieber, M., C.E. Contreras, M.S. Rieber, and N.E. Bianco. 1986. Novel DNA-protein complex and a large DNA in SLE cryoprecipitates. Clin. Exp. Immunol. 66:61–67. [PMC free article] [PubMed] [Google Scholar]
- 48.Ward, C.M., M.L. Read, and L.W. Seymour. 2001. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 97:2221–2229. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez de Cordoba, S., M. Perez-Blas, R. Ramos-Ruiz, P. Sanchez-Corral, F. Pardo-Manuel de Villena, and J. Rey-Campos. 1994. The gene coding for the beta-chain of C4b-binding protein (C4BPB) has become a pseudogene in the mouse. Genomics. 21:501–509. [DOI] [PubMed] [Google Scholar]
- 50.Blom, A.M., K. Berggård, J.H. Webb, G. Lindahl, B.O. Villoutreix, and B. Dahlbäck. 2000. Human C4b-binding protein has overlapping but not identical binding sites for C4b and streptococcal M proteins. J. Immunol. 164:5328–5336. [DOI] [PubMed] [Google Scholar]
- 51.Blom, A.M., L. Kask, B. Ramesh, and A. Hillarp. 2003. Effects of zinc on factor I cofactor activity of C4b-binding protein and factor H. Arch. Biochem. Biophys. 418:108–118. [DOI] [PubMed] [Google Scholar]
- 52.Dahlbäck, B. 1983. Purification of human C4b-binding protein and formation of its complex with vitamin K–dependent protein S. Biochem. J. 209:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blom, A.M. 2000. A cluster of positively charged amino acids in the alpha-chain of C4b-binding protein (C4BP) is pivotal for the regulation of the complement system and the interaction with bacteria. Scand. J. Clin. Lab. Invest. Suppl. 233:37–49. [PubMed] [Google Scholar]
- 54.Blom, A.M., L. Kask, and B. Dahlbäck. 2001. Structural requirements for the complement regulatory activities of C4BP. J. Biol. Chem. 276:27136–27144. [DOI] [PubMed] [Google Scholar]
- 55.Kask, L., L.A. Trouw, B. Dahlback, and A.M. Blom. 2004. The C4b-binding protein-protein S complex inhibits the phagocytosis of apoptotic cells. J. Biol. Chem. 279:23869–23873. [DOI] [PubMed] [Google Scholar]
- 56.Eivazova, E.R., J.M. McDonnell, B.J. Sutton, and N.A. Staines. 2000. Specificity and binding kinetics of murine lupus anti-DNA monoclonal antibodies implicate different stimuli for their production. Immunology. 101:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]