

Abstract
Mice immunized against Mycobacterium tuberculosis (Mtb) infection by curing them of a primary lung infection were compared with naive mice in terms of the ability to generate a Th1 cell immune response and to control growth of an airborne Mtb challenge infection. Immunized mice generated and expressed Th1 cell immunity several days sooner than naive mice, as demonstrated by an earlier increase in the synthesis in the lungs of mRNA for Th1 cytokines and for inducible nitric oxide synthase, an indicator of macrophage activation. This Th1 cytokine/mRNA synthesis was accompanied by an earlier accumulation of Mtb-specific Th1 cells in the lungs and the presence of CD4 T cells in lesions. An earlier generation of immunity was associated with an earlier inhibition of Mtb growth when infection was at a 1-log lower level. However, inhibition of Mtb growth in immunized, as well as in naive, mice was not followed by resolution of the infection, but by stabilization of the infection at a stationary level. The results indicate that there is no reason to believe that the secondary response to an Mtb infection is quantitatively or qualitatively superior to the primary response.
Tuberculosis is a major world disease estimated to kill more than two million people annually (1). This is the case even though only 5–10% of immunocompetent humans are susceptible, presumably because of an inability to mount a successful immune response against Mycobacterium tuberculosis (Mtb), the causative agent of the disease. It is generally believed that this deficiency in susceptible humans can be overcome by vaccination. However, the only approved vaccine currently available, the attenuated bacillus Calmette-Guerin (BCG) strain of M. bovis, has proved unreliable for this purpose (2). Consequently, research in many laboratories is aimed at developing a vaccine that is more effective than BCG. It is known (3, 4) that immunity to tuberculosis is mediated predominantly by Th1 cells, with the aid of CD8 T cells, and is expressed by activated macrophages in which Mtb resides at infectious foci. We have recently argued (4) that mouse tuberculosis may be considered a model of tuberculosis in susceptible humans; in mice, as in >85% of immunocompetent susceptible humans (5), tuberculosis is a disease exclusively of the lungs. On the other hand, in immunocompromised mice and humans, the disease is systemic and can involve multiple organs. This is the situation in humans immunocompromised by HIV infection (6) or other means (7), and it is the situation in mice that have been functionally deleted of genes required for the production of Th1 cells or cytokines (8). Therefore, it is apparent that both host species respond to Mtb infections by acquiring a level of immunity that is capable of protecting all organs, except the lungs, from progressive disease.
In this regard, it is known (9) that Mtb-infected mice generate a Th1 cell immune response that is capable of inhibiting further Mtb growth in the lungs at about day 20 of infection and of holding the infection at an approximate stationary level from that time on. However, stationary lung infections in mice cause a progressive lung pathology that is eventually lethal (4). Therefore, in order to be successful in this host species, immunity would need to be capable of causing a lung infection to completely resolve, or resolve to a level incapable of inducing pathology. Susceptible humans with active diseases may also be considered capable of acquiring the capacity to restrict the growth of Mtb; otherwise, they would succumb to an infection as early as immunocompromised humans. In support of this proposition, it is known that humans with active pulmonary tuberculosis generate an Mtb-specific Th1 cell immune response, as evidenced by an acquired ability to express delayed-type sensitivity to intracutaneously injected Mtb antigens (10, 11) and by the acquisition of CD4 and CD8 T cells capable of responding to Mtb antigens in vitro (12–14). Presumably, the purpose of vaccination is to ensure that enough of these T cells are generated in response to an infection to cause it to resolve. It is apparent, however, that attempts to achieve this in mice with a variety of recently developed vaccines have been unsuccessful, so that the protection afforded by them has been no better, or only marginally so, than that afforded by BCG. It has been the general finding (15) that vaccination with recently designed vaccines, like vaccination with BCG, enables mice to reduce the level of an Mtb lung infection by ∼1 log. However, the immunologic significance of 1 log of protection appears not to have been fully investigated.
This paper will show that, according to the levels of Th1 cytokine gene transcription and the amount of CD4 Th1 cells generated, mice immunized by curing them of a primary airborne Mtb infection generate Th1 cell immunity to an airborne challenge infection sooner than naive mice, thereby enabling them to inhibit Mtb growth earlier, but not to resolve the infection. The results provide no reason to believe that the secondary response, although generated earlier, is qualitatively or quantitatively superior to the primary response.
Results
Earlier inhibition of Mtb growth in immunized mice
The ability of mice cured of a primary airborne Mtb infection to deal with a secondary airborne infection with the same strain of Mtb is shown in Fig. 1. It can be seen that Mtb grew at the same rate (doubling time = 28 h) in the lungs of immunized and naive mice for about the first 15 d of infection, after which further Mtb growth was inhibited in the lungs of immunized, but not of naive, mice. In the latter, Mtb growth was not inhibited until about day 20. Thus, immunized mice were able to shorten the period of progressive Mtb growth by the equivalent of approximately four Mtb doubling times, which corresponds to about a 1-log lower level of infection. In immunized and naive mice, the levels of infection remained approximately stable after Mtb growth was inhibited. This result was obtained in two separate experiments. Thus, according to Fig. 1, immunized, like naive, mice showed no ability to immediately reduce the number of Mtb bacilli that implanted in the lungs, to slow the initial rate of Mtb growth, or to cause an established Mtb infection to resolve.
Figure 1.

Course of Mtb infection in the lungs of immunized and naive mice infected with ∼2 × 102 CFUs of Mtb via the airborne route. Mtb grew log linearly for ∼20 d in the lungs of naive mice and for 15 d in the lungs of immunized mice before further growth was inhibited. Both in naive and immunized mice, inhibition of Mtb growth was followed by a stationary level infection that was 1 log lower in immunized mice. Means ± SD of five mice per group.
Earlier increases in Th1 cytokine gene transcription in immunized mice
A previous study (9) showed that control of Mtb growth in the lungs of naive mice occurs at about day 20 of infection and is dependent on the ability to generate Th1 cell immunity. It was anticipated, therefore, that an earlier control of lung infection by immunized mice would be associated with an earlier generation and expression of Th1 cell immunity. This was initially investigated by quantifying changes against time of infection in the levels of expression of Th1 cytokine genes with real-time RT-PCR. The results in Fig. 2 clearly show that an increased synthesis of mRNA for IL-2, IFN-γ, and TNF-α was initiated several days earlier and reached higher levels sooner in the lungs of immunized mice than in the lungs of naive mice. The same applied to an increased synthesis of mRNA for chemokines, IP-10, and MCP-1, and to an increased synthesis of mRNA for inducible nitric oxide synthase (NOS2), an indicator of IFN-γ–induced transcriptional activation of macrophages (16). According to the levels of IFN-γ and NOS2 gene transcription, the magnitude of the secondary response was somewhat lower, rather than higher, than that of the primary response at day 30. In both cases, the synthesis of Th1 cytokines and NOS2 mRNAs was sustained at high levels until the experiment was terminated at day 50. It will be noted in Fig. 2 that, although the levels of Th1 cytokine/mRNA synthesis were higher in the lungs of immunized mice before the challenge infection was given, an increased synthesis of these mRNAs did not take place in response to an infection in mice of either group until after an appreciable delay.
Figure 2.
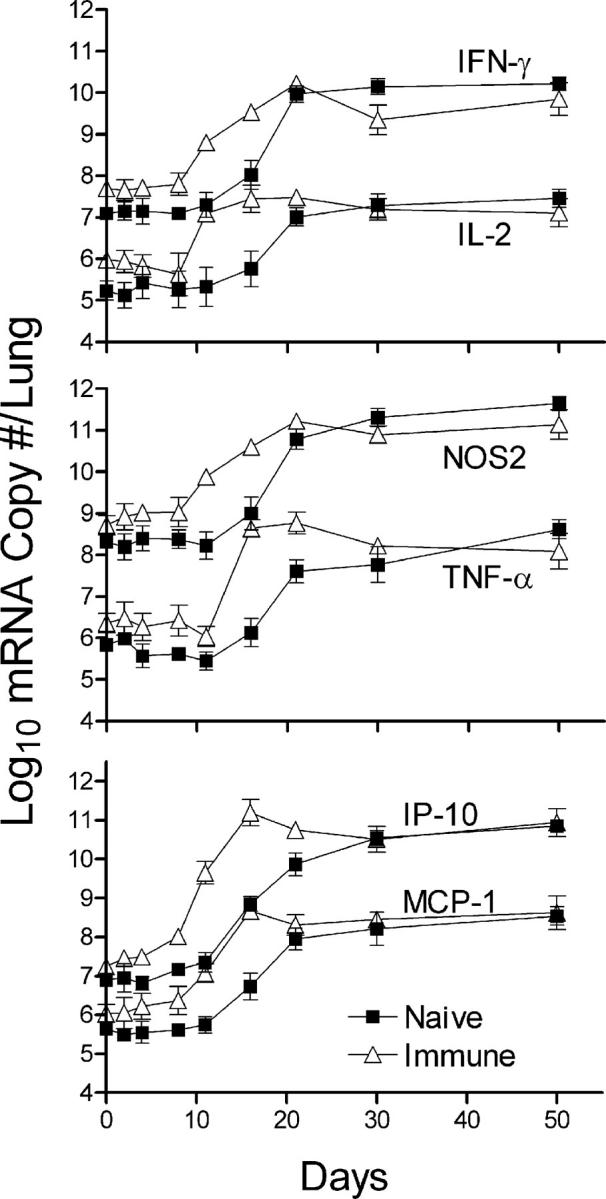
Real-time RT-PCR quantification of changes in Th1 cytokine gene transcription in the lungs of naive and immunized mice. Large increases in the synthesis of mRNA for IL-2, IFN-γ, and TNF-α were evident by day 12 of infection in immunized mice, 4 d earlier than in naive mice. The same applied to the increased synthesis of mRNA for IP-10 and MCP-10, and for NOS2. After day 12 in immunized and day 16 in naive mice, mRNA synthesis continued to increase to peak at 1–3 log higher levels, depending on the particular mRNA. Peak levels of synthesis were reached earlier in immunized mice, but were no higher than in naive mice. In all cases, mRNA synthesis was sustained at high levels until day 50, when the experiment was terminated. The results were obtained with the pooled lung RNA of four mice per time point per group. Shown are the means and SDs of four separate readings. The experiment was performed twice with essentially the same result.
Earlier increase in the number of IFN-γ–producing CD4 T cells in immunized mice according to flow cytometry
To determine whether earlier generation of immunity in immunized mice, as evidenced by earlier increases in the expression of Th1 cytokine genes in the lungs, is associated with the earlier increase in the number Th1 cells, flow cytometry was used to measure changes against time in the number of IFN-γ–producing CD4 cells. Fig. 3 shows that in both naive and immunized mice, an infection resulted in an increase in the total number of CD4 T cells in the lungs, and that these cells increased in number sooner in the lungs of immunized mice. Peak accumulation of CD4 T cells occurred on day 18 in the lungs of these mice and on day 30 in the lungs of naive mice, resulting in a higher peak number in the lungs of the latter. These numbers were approximately sustained until the experiment was terminated on day 85. The same results were obtained in two separate experiments.
Figure 3.

The results of a flow cytometry analysis of the number of CD4 T cells and IFN-γ–secreting CD4 T cells that accumulated in the lungs of naive and immunized mice against time of the challenge infection. CD4 T cells began accumulating earlier in immunized mice and reached peak numbers on day 20, after which the number remained relatively constant. In naive mice, the peak number was reached on day 30. The same kinetics applied to the accumulation of IFN-γ–producing CD4 T cells in the naive and immunized mice, except that only a small percentage of the CD4 cells were positive for IFN-γ. The results were obtained with pooled lung cells from four mice stained for surface CD4 and intracellular IFN-γ as described in Materials and methods. The same result was obtained in two separate experiments.
According to Fig. 3, IFN-γ–producing CD4 cells were not present in large numbers in the lungs of immunized or naive mice on day 9 of infection, but began accumulating in the lungs of the former between days 9 and 12 and in the latter between days 12 and 15. Peak accumulation occurred on day 20 in immunized mice and on day 30 in naive mice. It will be noted in Figs. 3 and 4 that <10% of the CD4 T cells were making IFN-γ at any one time of infection. According to Fig. 5, the majority of the gated IFN-γ–producing CD4 cells displayed the CD44hi CD62Llow activation phenotype. It is assumed that the majority of cells making IFN-γ, according to flow cytometry, were making this cytokine in response to an infection at the time of cell harvest. The presence of ESAT-6 peptide during the 5-h incubation period with brefeldin A had little or no effect on the number of cells synthesizing IFN-γ (unpublished data).
Figure 4.

Only a small percentage of CD4 T cells were making IFN-γ at peak responses. Only 10.5% of CD4 T cells in the lungs of naive mice (day 30), and 8.35% in the lungs of immunized mice (day 20), were making IFN-γ according to flow cytometry. The results were obtained with pooled lung cells of four mice stained for surface CD3 and CD4 and for intracellular IFN-γ as described in Materials and methods.
Figure 5.
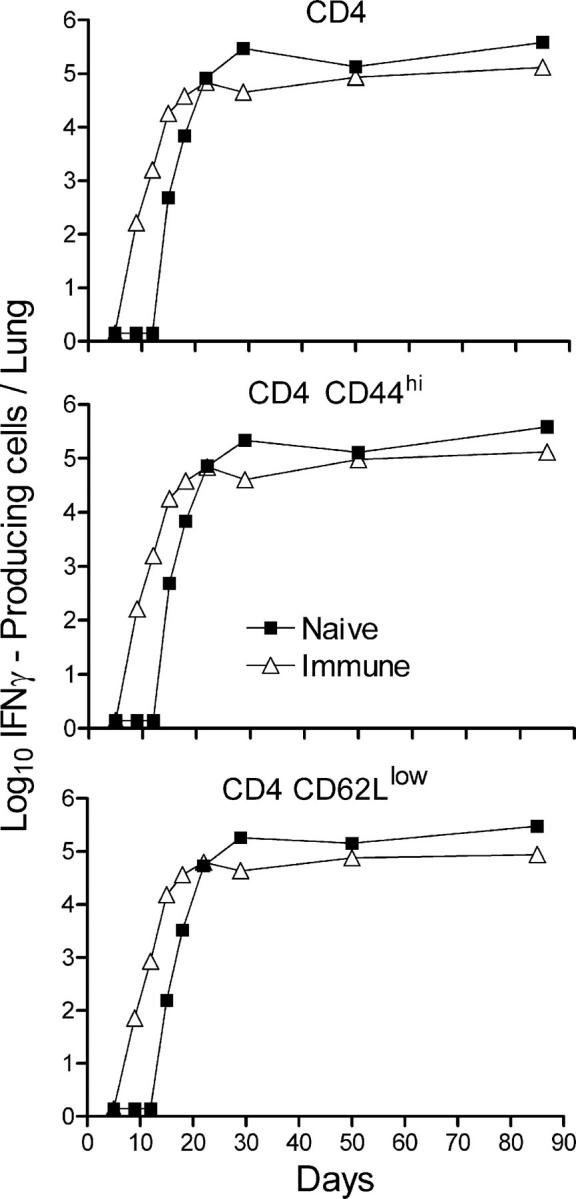
IFN-γ–producing CD4 T cells in the lungs displayed an activation phenotype. The majority of the CD4 T cells making IFN-γ in naive and vaccinated mice in response to a challenge infection displayed the CD44hiCD62Llow activation surface phenotype. Cells were stained for surface CD4, CD62L, and CD44, and intracellular IFN-γ as described in Materials and methods.
Earlier increase in the number of Mtb-specific CD4 T cells in the lungs of immunized mice as enumerated by Elispot
The generation kinetics of Th1 cells specific to Mtb were determined with the Elispot assay. Fig. 6 shows that cells capable of making IFN-γ in response to ESAT-61-20 peptide, to Ag85B240-254 peptide, or to antigens present in the Mtb sonicate accumulated and reached peak numbers sooner in the lungs of immunized mice. In these mice, cells capable of making IFN-γ in response to the previously mentioned antigen preparations were present in an increased number in the lungs by day 12 of infection and continued to increase in number until day 20, after which their number approximately stabilized. In contrast, in the lungs of naive mice, cells capable of making IFN-γ in response to these antigens were not present in increased numbers until day 18, after which they continued to increase until day 30 and then slowly declined. Consequently, the production of T cells capable of making IFN-γ in response to Mtb antigens reached greater numbers in the lungs of naive mice. The number of cells that responded to ESAT-6 in the lungs of naive and immunized mice was not much smaller than the number that responded to antigens in the Mtb sonicate. This result was obtained in two separate experiments. If one assumes that the Mtb sonicate contained enough ESAT-6 to stimulate all ESAT-6–specific T cells, it would follow that a large proportion of the Mtb-specific T cells present in the lungs were generated against this particular antigen. This finding would be in keeping with the demonstration of Brandt et al. (17) that ESAT-6 is a dominant Th1 cell antigen in mice.
Figure 6.
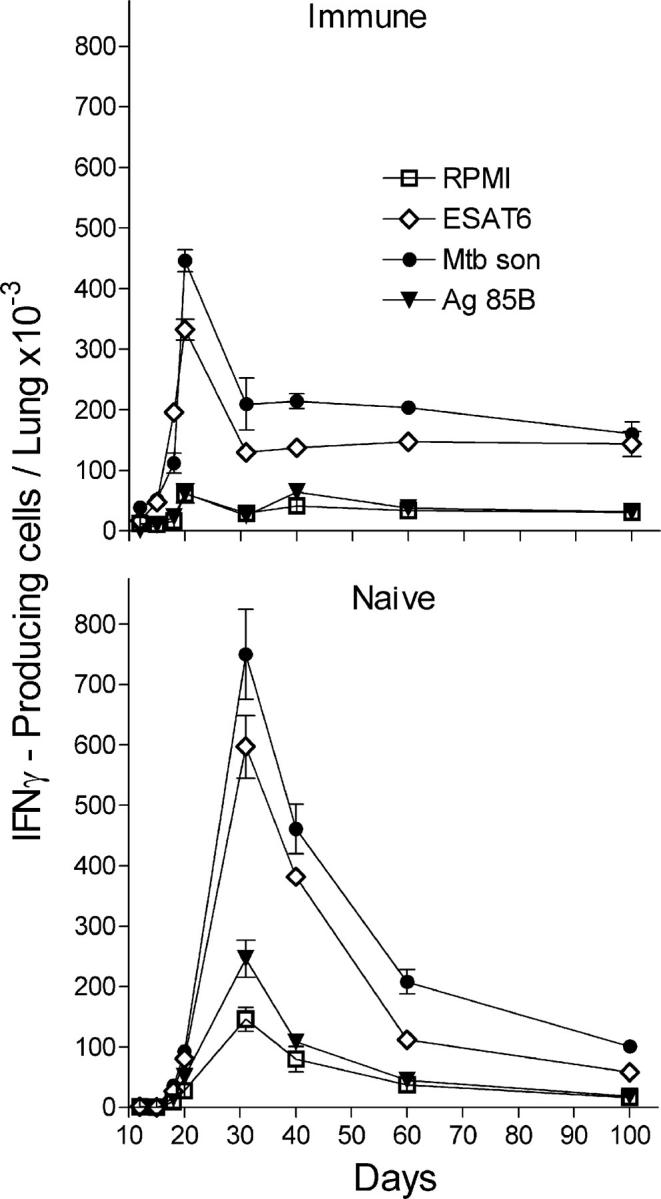
Kinetics of accumulation of Mtb-specific T cells in the lungs of naive and immunized mice. T cells capable of synthesizing IFN-γ in response to exposure to ESAT-61-20 peptide, Ag85240-254 peptide, and Mtb sonicate were enumerated with the Elispot assay. Increased numbers of Mtb-specific T cells were present in the lungs of immunized mice by day 12 of infection, but not until day 18 in the lungs of naive mice. Mtb-specific cells increased to peak on day 21 in the case of immunized mice and on day 30 in the case of naive mice. Consequently, Mtb-specific cells accumulated in higher numbers in the lungs of the latter mice. Results were obtained with pooled lung cells from four mice. Shown are the means ± SD of the number of spots in triplicate wells. The same result was obtained in two separate experiments.
Fig. 6 also shows that several lung cells made IFN-γ in the absence of added antigen, and that these cells accumulated with kinetics similar to those that were antigen specific. It seems reasonable to suspect that they were making IFN-γ in response to Mtb antigens at the time of cell harvest and that they continued to do so in the assay. It is likely that IFN-γ made in the absence of added antigen was in response to the presence of viable Mtb in the lung cell suspension. That viable intracellular Mtb was present is evidenced by the results of an experiment that determined the number of Mtb CFUs present in positively selected CD11b cells from pooled lung cells of four naive mice harvested on day 30 of infection, as described in Materials and methods. It was found that one lung equivalent of positively selected CD11b cells (2.3 × 106 cells) contained 106.19 CFUs of Mtb. In contrast, the lung cell suspension depleted of CD11b cells contained only 104.2 CFUs of Mtb. Thus, in the in vitro assays used, a potential antigen was present in the form of viable intracellular Mtb. It is assumed that this also applied to lung cells obtained from immunized mice. The result is not surprising given that no attempt was made to remove macrophages from the cell suspensions used for in vitro assays. This will be the subject of a future publication from this laboratory.
Because the ESAT-61-20 and Ag85B240-254 peptides are Class II (IAb) presented, it is reasonable to conclude that the cells that made IFN-γ in response to them were CD4 T cells. This was confirmed by the results of an Elispot assay performed with lung cell suspensions magnetically depleted of ≥90% CD4 cells. It can be seen in Fig. 7 that removing CD4 T cells from the lung cell suspension harvested on day 30 from naive mice and on day 20 from immunized mice removed most of the cells capable of making IFN-γ in response to ESAT-6, Ag85B, and the Mtb sonicate. The contribution of different T cell subsets to immunity is the subject of a study currently in progress in this laboratory.
Figure 7.
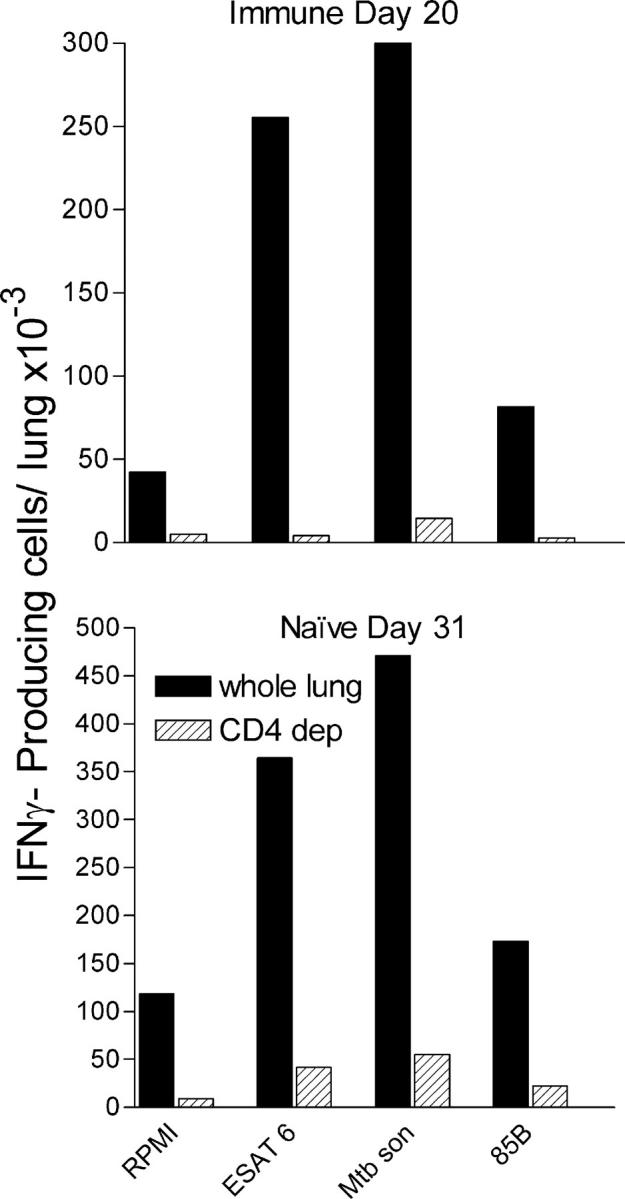
Evidence that the majority of cells that made IFN-γ in the Elispot assay were CD4 positive. Magnetic depletion of ≥90% CD4 T cells from the lung cell suspension resulted in the removal of the majority of cells capable of making IFN-γ in response to ESAT-61-20 peptide, to Ag85B240-254 peptide, or to the Mtb sonicate in the Elispot assay. The experiment was performed with pooled cells harvested from four immunized mice on day 20 and from four naive mice on day 31 of infection.
Location of CD4 cells in lesions
Our results show that the expression of Th1 cell–mediated immunity to an Mtb infection was associated with an appreciable increase in the number of CD4 T cells in the lungs. To mediate immunity, these cells would need to be located at sites of infection in the lungs where Mtb resides in macrophages. To determine the location of CD4 cells, immunocytochemistry was performed on lung sections using anti-CD4 mAb as a primary reagent. Fig. 8 shows that each site of infection (lesion) in naive mice on day 30 and in immunized mice on day 20 consisted of a region of alveolitis populated almost exclusively by mononuclear cells. Groups of air sacs scattered throughout each lesion contained Mtb-infected macrophages. Cells that stained for surface CD4 were present in relatively large numbers throughout each lesion in the interstitium and to a lesser extent in air sacs containing infected macrophages. CD4 cells were also present around veins and arteries in close proximity to lesions. Given that lesions did not exist before the infection was initiated, the relatively large number of lesion-based CD4 cells would account for a large proportion of the increased number of CD4 cells that accumulated in the lungs in response to infection.
Figure 8.
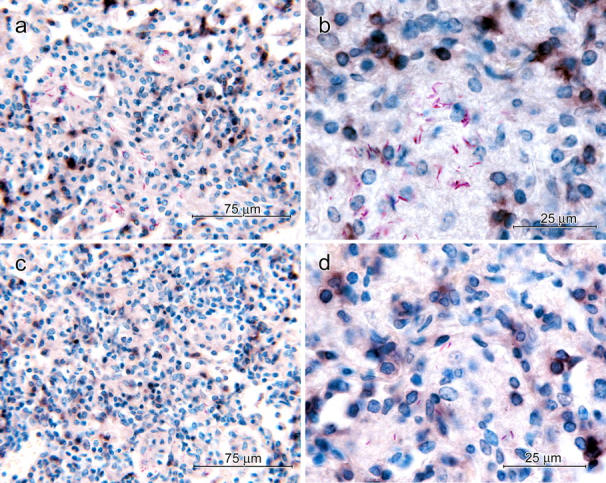
Immunocytochemical demonstration of CD4 cells in lung lesions of immunized and naive mice at the peak of CD4 cell accumulation. 40× micrographs of lung lesion of a 30-d-infected naive mouse (a) and a 21-d-infected immunized mouse (c) showing CD4-positive cells (dark brown) throughout the lesion. 100× micrographs of a lesion of a naive (b) and immunized mouse (d) showing some CD4-positive cells in proximity to macrophages containing acid-fast bacilli. Discrete black/brown smudges represent CD4-positive cells out of the plane of focus.
Discussion
This paper shows that mice immunized against an Mtb infection by curing them of a primary lung infection acquired a capacity to generate Th1 cell immunity to an airborne Mtb challenge infection sooner than unimmunized mice. Earlier generation of Th1 cell immunity was associated with an earlier inhibition of Mtb growth when the number of CFUs in the lungs was ∼1 log lower. On the other hand, immunized mice were unable to immediately kill Mtb bacilli that implanted in the lungs, cause Mtb infection to progress at a slower initial rate, or cause the infection to resolve. Instead, they were able to hold infection at a lower stationary level from day 15 on. Thus, the protection afforded by immunization was wholly attributable to an earlier inhibition of Mtb growth. In the apparent absence of any evidence to the contrary, it is reasonable to propose that the protection afforded mice by vaccinating them with BCG, or any one of several recently designed vaccines, will also prove to be wholly attributable to an ability to inhibit Mtb growth during an earlier stage of infection. It is predicted that this will also be found to apply to any marginal increase in the level of protection afforded by one vaccine over another, or by giving booster injections of a particular vaccine. It needs to be noted that shortening the period of progressive Mtb growth by the equivalent of one Mtb doubling time (28 h) would enable a vaccine to reduce the level of stationary infection by ∼50%. The finding that the pathogen grew at the same rate in immunized and naive mice for the first 12–15 d of infection is in accordance with results obtained with guinea pigs (18) and mice (19). The reason for the exceptionally long delay in immunized, as well as naive, mice before anti-Mtb immunity is expressed in the lungs remains unknown.
The earlier generation and expression of Th1 cell immunity by Mtb-immunized mice was measured here in terms of an earlier increase in the synthesis in the lungs of mRNA for the Th1 cytokines (IL-2, IFN-γ, and TNF-α) and in the synthesis of mRNA for NOS2, an inducible enzyme of macrophages that signifies a transcriptional response that results in up-regulation of their antimycobacterial function (16). It was also associated with earlier increases in the synthesis of mRNA for the chemokines (IP-10 and MCP-1), indicating that an earlier expression of secondary immunity was associated with an earlier inflammatory response. The earlier generation of Th1 cell immunity in immunized mice was also evidenced by the earlier appearance and accumulation of Mtb-specific T cells in the lungs. Thus, cells capable of making IFN-γ in response to Mtb antigens, as enumerated in the Elispot assay, increased in number between days 12 and 20 in the lungs of immunized mice and between days 18 and 30 in the lungs of naive mice. Immunocytochemistry revealed that many of these cells were present in lesions.
The finding that a large proportion of Mtb-specific cells generated in the primary and secondary responses were specific for the ESAT-61-20 peptide is in keeping with the conclusion of Brandt et al. (17) that this secreted protein is a major Mtb antigen in mice. ESAT-6 has also been shown to be a major Mtb antigen in humans (20) and cattle (21). Because the ESAT-61-20 and Ag85B240-254 peptides are MHC Class II (IAb) presented, it follows that the ESAT-6– and Ag85B-specific cells enumerated were CD4 T cells. This was confirmed by the additional finding that depleting CD4 T cells from the lung cell suspension removed the majority of cells capable of making IFN-γ in response to these peptides. In spite of the high frequency of ESAT-6–specific T cells made in the primary and secondary responses, however, there is no compelling reason to believe that T cells specific for this antigen were necessary for the anti-Mtb primary or secondary response to be protective. It is apparent from the literature (15) that the level of protection afforded mice by vaccinating them with an ESAT-6–expressing DNA vaccine is no better than that afforded by vaccinating them with DNA vaccines expressing Ag85A or Ag85B, or with BCG. Moreover, because it was shown by Schell et al. (19) some years ago, and more recently by others (22), that vaccination with BCG is no more protective than vaccinating with Mtb itself, it is reasonable to propose that the limited protective value of BCG is not a consequence of its inability to make ESAT-6 and other Mtb antigens encoded by genes known to be absent from BCG (23). Again, the level of protection afforded mice by immunizing them via the respiratory route with Mtb, as reported here, is similar to the level that has been reported by others for mice vaccinated with BCG via the i.v. route (15), indicating that vaccination via the respiratory route does not offer any protective advantage. This is in keeping with the early findings of Schell et al. (19) and the more recent findings of others (24, 25).
Moreover, an important point to make about vaccination via the airborne route with live BCG or Mtb is that in order to avoid the complexity of superimposing one infection on another, it is necessary to cure the immunizing lung infection before giving the challenge infection. This is not always done. It is known that a lung infection with BCG (19, 23), like lung infections with virulent (9) or avirulent Mtb (26), persists at an approximately stationary level for a protracted period of time. It is also known (26) that the maintenance of a stationary level of lung infection with virulent or avirulent Mtb is associated with an ongoing, active Th1 response, as evidenced by a sustained increase in the level of Th1 cytokine gene transcription. The influence of persistent BCG infection on the level of immunity to an Mtb challenge infection was recently shown to be considerable (27), presumably because it provides the antigenic stimulus for maintaining immunity in an active state. In this study, the immunizing Mtb lung infection was terminated by 100 d of chemotherapy before the secondary infection was initiated. Therefore, the response to the secondary infection may be considered an active secondary response, a conclusion supported by the finding that it was not expressed until after a delay of ∼15 d. We are aware, as shown many years ago (28), that in spite of the apparent absence of viable Mtb in mouse tissues after long-term chemotherapy, some viable bacilli remain present in a latent state. Initiating an Mtb infection via the respiratory route followed by curing the infection with chemotherapy almost certainly will not be used as a means to vaccinate humans against tuberculosis. Nevertheless, this method of vaccination is performed each time a susceptible human with tuberculosis is treated successfully with chemotherapy. According to recent studies (29, 30), however, humans cured of the active disease by chemotherapy can become exogenously reinfected with different strains of Mtb within weeks. This would indicate that, in the case of at least a proportion of susceptible humans, a successful vaccination against tuberculosis will be difficult to achieve.
It should be noted that this study deals with the response to infections in the lung, the only mouse organ in which an Mtb infection causes progressive disease (4). However, it is generally accepted that primary and secondary immune responses to infectious agents are initiated in draining lymph nodes. Presumably, therefore, the earlier increase in the number of Mtb-specific T cells in the lungs of immunized mice was a reflection of an earlier production of these cells in the draining nodes and their earlier release into the circulation from which they entered the lungs at sites of infection. This aspect of the anti-Mtb response is currently under study in this laboratory. Results obtained thus far (unpublished data) indicate that the number of Mtb-specific T cells in the draining node at any one time of infection is considerably smaller than the number in the lungs. Moreover, because it has been demonstrated (31) that Mtb-specific Th1 cells in the lungs belong to a replicating population, it is likely that most of them are generated locally in response to antigens presented by infected macrophages. The interpretation that the continuous expression of active Th1 cell immunity is responsible for holding an infection at a stationary level is supported by the knowledge (32) that depleting mice with a stationary lung infection of CD4 cells results in a resumption of Mtb growth.
This study does not show why the secondary and primary immune responses, although capable of stabilizing an infection at a stationary level, were not capable of causing the infection to resolve. However, because it shows that there is no reason to believe that the secondary response is quantitatively or qualitatively superior to the primary response, it seems obvious that the secondary response failed to resolve the infection for the same reason as the primary response. Because macrophages are the cells that express immunity, the failure to resolve the infection was presumably the result of the failure of macrophages to acquire an adequate antimicrobial function. On the other hand, it is possible that the inhibition of Mtb growth, as evidenced by a stationary level infection, is initiated by the pathogen itself as part of a counterresponse to the acquired antimycobacterial function of macrophages. It has been demonstrated (33), in this regard, that the ingestion of Mtb by activated macrophages causes a dramatic transcriptional response on the part of the pathogen that results in its acquiring a “dormancy transcriptome” that likely enables it to survive host adaptive defenses. Mtb has been shown (33) to undergo a similar transcriptional response in the lungs of mice at the time of expression of adaptive immunity. It is therefore possible that, in vaccinated mice, the activation of macrophages by an earlier generation of Th1 cells merely serves to trigger an earlier protective transcriptional response on the part of the pathogen. Until it is shown that the secondary anti-Mtb response is quantitatively and qualitatively superior to the primary response and causes macrophages to acquire a higher level of antimycobacterial function, there is no reason to believe that vaccination should enable mice to resolve an infection.
As for the survival consequences of infection levels ≥1 log lower in Mtb-immunized mice, it almost certainly would result in a longer time of survival. It was shown by Wiegeshaus et al. (34) that BCG vaccination enables guinea pigs to hold infection at a level ∼2 log lower than naive guinea pigs and to survive for about twice as long from a small airborne Mtb challenge infection. It also has been shown (35) that BCG vaccination of mice of the genetically susceptible DBA/2 strain enabled them to survive approximately twice as long as naive DBA/2 mice from an Mtb challenge given via the i.v. route. The same study showed that naive mice of the genetically resistant BALB/c strain all died from an i.v. Mtb challenge infection before any BCG-vaccinated BALB/c mice died. It is apparent that although vaccination provides guinea pigs and mice the ability to stabilize and hold an Mtb challenge infection at a lower stationary level, a lower level of infection nevertheless induces the development of progressive, lethal lung pathology.
Materials and Methods
Mice.
C57BL/6 male mice were purchased from The Jackson Laboratory and were 12 wk old when used in experiments. The experiments were performed and the mice were housed under barrier conditions in a Level III Biosafety Animal Facility in accordance with guidelines formulated by the Trudeau Institute Animal Care and Use Committee.
Immunization and infection
The H37Rv strain of Mtb (Trudeau Mycobacterial Culture Collection, no. 102) was grown in a suspension culture in Proskauer and Beck medium containing 0.01% Tween 80 and harvested during log-phase growth, as described previously (36). The culture was subjected to two 5-s bursts of ultrasound to break up clumps and appropriately diluted in PBS containing 0.01% Tween 80 for infection via the respiratory route in an aerosol infection chamber, as described previously (9). Mice were immunized by infecting them via the respiratory route with ∼102 CFUs of H37Rv, allowing the infection to proceed for 30 d, and then treating them for 100 d with 0.3 mg isoniazid and 0.1 mg rifampin (Sigma-Aldrich) per milliliter in drinking water. This study shows that the primary Th1 response to lung infection with H37Rv peaks on about day 30. At the end of chemotherapy, the lungs were considered free of viable Mtb as judged by the absence in lung homogenates of Mtb capable of forming colonies on nutrient agar. Immunized and naive mice were challenged at the same time in the same aerosol infection chamber with ∼2 × 102 CFUs of H37Rv. The lung infection was monitored by measuring changes against time in the total number of CFUs in the lungs. This involved killing five mice at the times indicated, homogenizing the lungs, plating serial dilutions of lung homogenates on enriched Middlebrook 7H11 agar, and counting colonies after incubating the plates for 21 d at 37°C.
Mtb antigens
The Mtb antigens used to stimulate IFN-γ production by lung cells were Esat-61-20 peptide (17), Ag85B240-254 peptide (37), and an ultrasound-disrupted H37Rv culture. Both peptides were purchased from New England Peptide. ESAT-6 is a secreted Mtb protein of unknown function (17), whereas Ag85B is a member of a family of mycolyl transferases involved in cell wall lipid biosynthesis (37). The Mtb sonicate was prepared by subjecting a culture of H37Rv (2 × 108 CFUs/ml in Proskauer and Beck medium) at 10°C to 5 min of ultrasound generated by a sonicator (model 1510; Braun-Sonic) set at 400 W. Because Proskauer and Beck medium is composed of inorganic salts and glycerol, all antigens present in the Mtb sonicate were derived from Mtb.
Lung cell preparation.
To harvest lung cells, mice were killed by cervical dislocation, and their lungs were perfused via the right ventricle with PBS containing 10 U/ml heparin to remove intravascular leukocytes. The lungs were then perfused with an enzyme cocktail consisting of 150 U/ml collagenase, 0.2 U/ml elastase (Roche), and 1 mg/ml DNase (Sigma-Aldrich) in RPMI 1640. The lungs were removed, placed in a dish, and diced into small fragments, and the fragments were incubated in the enzyme mixture at 37°C for 1 h. The preparation was passed through a 60-mesh/in2 stainless screen, and the resulting suspension was triturated with a pipette to break up aggregates. The cells were pelleted and resuspended in red cell lysis buffer (Sigma-Aldrich). They were then washed, passed through a 70-μm nylon cell strainer (BD Biosciences), pelleted, and resuspended in RPMI 1640 with 10% FCS (RPMI-FCS) for counting and analysis.
Cell depletion.
Lung cell suspensions were selectively depleted of CD4 or CD11b cells magnetically. This involved incubating the cell suspension with 10 μg/ml of biotin-conjugated anti-CD4 or anti-CD11b rat anti–mouse mAbs (BD Biosciences) for 40 min at 4°C. The cells were washed twice with buffered PBS containing 0.5% BSA, pelleted, and resuspended in 0.1 ml of buffered PBS containing streptavidin-coupled magnetic nanoparticles (IMag Streptavidin Particles; BD Biosciences) according to the manufacturer's instructions. The cells were placed in a 5-ml tube that was inserted into a close-fitting Perspex tube holder with a recess containing a 45 × 35-mm neodynium-iron-boron magnet (ForceField). After 6 min at 10°C, cells not held magnetically to the side of the tubes were removed with a pipette. The procedure was repeated three times, after which the depleted cells and the magnetically enriched cells were pelleted and resuspended for analysis by flow cytometry and Elispot. The procedure resulted in ≥90% depletion of CD4 T cells. Part of the enriched CD11b cell suspension was subjected to 5 s of ultrasound to cause cell disruption and was then 10-fold serially diluted in Tween 80–PBS. The dilutions were plated on nutrient agar and after an 18-d incubation at 37°C, CFUs of Mtb were counted with the aid of a dissecting microscope.
BM-derived DCs.
These were generated from C57BL/6 BM according to a published procedure (38). A GM-CSF–expressing J558L cell line (supplied by I. Mellman, Yale University School of Medicine, New Haven, CT) was used as a source of GM-CSF. The DCs were generated in large numbers and distributed in 1-ml lots each containing 2 × 106 cells. They were frozen and stored at −150°C in RPMI 1640 with 20% FCS and 10% DMSO. For experiments, the cells were thawed, washed, and resuspended in RPMI-FCS.
Elispot.
Changes in numbers of Mtb-specific cells in the lungs against time of infection were determined by enumerating changes in the numbers of cells capable of making IFN-γ in response to Mtb antigens in the Elispot assay. This was performed with a commercially available Elispot kit (Mouse IFN-γ ELISPOT Set; BD Biosciences) according to the manufacturer's instructions using pooled cells from four mice. It involved coating 96-well nitrocellulose plates with a rat anti–mouse IFN-γ mAb, washing with RPMI, and blocking with RPMI-FCS. To supplement APCs already present in the lung cell suspension, BM-derived DCs were added to the cell suspension at a ratio of 5:1 lung cells/DC. Twofold serial dilutions of the 100-μl admixture were added in triplicate to the wells starting at 105 lung cells/well. The wells then received 100 μl RPMI-FCS containing no antigen, 100 μg ESAT-61-20 peptide, 100 μg Ag85B240-254 peptide, or 100 μl of a 1:100 dilution of Mtb sonicate. After 18 h of incubation at 37°C, unattached cells were aspirated from the wells and the remaining cells were lysed with distilled water. The wells were washed again with PBS containing 0.05% Tween 20 and incubated with a second biotinylated rat anti–mouse IFN-γ mAb. The wells were then washed with PBS–Tween 20, incubated for 1 h with streptavidin-alkaline phosphatase, washed, and developed with 3-amino-9-ethyl-carbazol as substrate. After washing and drying, the nitrocellulose well bottoms were punched onto a transparent adhesive film, and the number of spots per well was counted with the aid of a microscope at 40×. The number of cells specific for each antigen preparation was calculated by subtracting the number of spots that formed in the absence of added antigen from the number that formed in its presence. Each experiment was repeated twice.
Flow cytometry.
Lung cells were prepared and admixed with BM-derived DCs at a ratio of 5:1 lung cells/DC, suspended in RPMI-FCS in two 5-ml tubes at 5 × 107 cells/ml, and incubated in the presence of brefeldin A (Epicenter Technologies) at a concentration of 10 μg/ml for 5 h at 37°C. The cells in one tube were washed and resuspended in medium containing FITC–anti-CD3, R-phycoerthrin–anti-CD4, and Peridinin chlorophyll protein–anti-CD8 mAbs. The cells in the other tube were washed and suspended in medium containing Peridinin chlorophyll protein–conjugated anti-CD4, R-phycoerthrin–anti-CD44, and FITC–anti-CD62L mAbs. Anti–mouse CD16/CD32 was added to block nonspecific binding, and the cells were incubated for 1 h at 4°C. They were then washed and fixed overnight in 0.5% paraformaldehyde, washed again, and stained for intracellular IFN-γ with allophycocyanin–anti–IFN-γ mAb. This involved treating the cells with 0.1% saponin (Sigma-Aldrich) in PBS, and then incubating them with allophycocyanin–anti–IFN-γ mAb. All monoclonal reagents were purchased from BD Biosciences. After washing, the cells were subjected to analysis with a flow cytometer (model FACSCalibur; BD Biosciences) using FloJo software (Tree Star).
Real-time RT-PCR quantification of mRNA synthesis for IFN-γ and NOS2.
The real-time RT-PCR procedure used has been described in detail elsewhere (26). Lungs were harvested at the times indicated in Results, and snap frozen in liquid nitrogen. RNA extraction, clean-up, and quantification were performed as previously described (26). In brief, to make standard curves for IFN-γ, NOS2, IL-2, TNF-α, IP-10, and MCP-1, a T7 transcript of each was generated (MEGAshortscript T7 Kit; Ambion), 10-fold serially diluted from 108 to 102 according to transcript copy number, and each dilution subjected to the real-time RT-PCR. The level of expression of these genes in the lungs of primary and secondary Mtb-infected mice was calculated according to the formula N = (Ct − b)/m, where N is the copy number, Ct is the threshold cycle, b is the y intercept, and m is the slope of the standard curve line obtained. Results were obtained with pooled lung RNA of four mice per group per time point. Shown are the means ± SDs of four separate readings per group per time point. The experiment was performed twice with essentially the same result.
Immunocytochemistry.
Lungs were fixed by an intratrachial infusion of zinc chloride/zinc acetate fixative (BD Biosciences) and an immersion in the fixative for 24 h at room temperature. The lungs were then dehydrated in 70 and 100% ethanol and embedded in wax according to standard procedures. 6-μm sections were cut with a rotary microtome and, after dewaxing, were treated with 5% BSA in PBS, washed in PBS, treated to block endogenous biotin (Avidin/Biotin Blocking Kit; Vector Laboratories), and treated with rat anti–mouse CD4 mAbs or rat isotype control IgG (BD Biosciences) for 18 h at 4°C. After washing in PBS, sections were treated with biotin-conjugated goat anti–rat IgG followed by avidin-biotinylated alkaline phosphatase (Vectastain ABC-AP Kit; Vector Laboratories), and then with the enzyme substrate (Vector Black; Vector Laboratories) to give a brownish/black reaction product. After washing, the cells were stained for acid-fast bacteria (39) and counterstained with methylene blue. Photomicrographs were taken with a microscope (model Microphot-Fx; Nikon) with a digital camera (model Spot RT; Diagnostic Instruments).
Acknowledgments
This research was supported by National Institutes of Health grants AI-37844 and HL-64565.
The authors have no conflicting financial interests.
Abbreviations used: BCG, bacillus Calmette-Guerin; Mtb, Mycobacterium tuberculosis; NOS2, inducible nitric oxide synthase.
References
- 1.Corbett, E.L., C.J. Watt, N. Walker, D. Maher, B.G. Williams, M.C. Raviglione, and C. Dye. 2003. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 163:1009–1021. [DOI] [PubMed] [Google Scholar]
- 2.Orme, I.M. 1999. Beyond BCG: the potential for a more effective TB vaccine. Mol. Med. Today. 5:487–492. [DOI] [PubMed] [Google Scholar]
- 3.Flynn, J.L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 19:93–129. [DOI] [PubMed] [Google Scholar]
- 4.North, R.J., and Y.J. Jung. 2004. Immunity to tuberculosis. Annu. Rev. Immunol. 22:599–623. [DOI] [PubMed] [Google Scholar]
- 5.Farer, L.S., A.M. Lowell, and W.B. Meador. 1979. Extrapulmonary tuberculosis in the United States. Am. J. Epidemiol. 109:205–217. [DOI] [PubMed] [Google Scholar]
- 6.Chaisson, R.E., G.F. Schecter, C.P. Theueer, G.W. Rutherford, D.F. Echenberg, and P.C. Hopewell. 1987. Tuberculosis in patients with the acquired immunodeficiency syndrome. Clinical features, response to therapy, and survival. Am. Rev. Respir. Dis. 136:570–574. [DOI] [PubMed] [Google Scholar]
- 7.Sinnott, J.T., and P.J. Emmanuel. 1990. Mycobacterial infections in the transplant patient. Semin. Respir. Infect. 5:65–73. [PubMed] [Google Scholar]
- 8.Cooper, A.M., B.M Saunders, C.D. D'Souza, A.A. Frank, and I.M Orme. 1997. Mycobacterium tuberculosis-driven processes in gene-disrupted mice. Bull. Inst. Pasteur. 95:85–95. [Google Scholar]
- 9.Mogues, T., M.E. Goodrich, L. Ryan, R. LaCourse, and R.J. North. 2001. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furcolow, M.L., B. Hewell, and W.E. Nelson. 1942. Quantitative studies of the tuberculin reaction: tuberculin sensitivity in relation to active tuberculosis. Am. Rev. Tuberc. 45:504–520. [Google Scholar]
- 11.Al Zahrani, K., H. Al Jahdali, and D. Menzies. 2000. Does size matter? Utility of tuberculin reactions for the diagnosis of mycobacterial disease. Am. J. Respir. Crit. Care Med. 162:1419–1422. [DOI] [PubMed] [Google Scholar]
- 12.Arend, S.M., P. Anderson, K.E. van Meijgaarden, R.L. Skjot, Y.W. Subronto, J.T. van Dissel, and T.H. Ottenhoff. 2000. Detection of active tuberculosis infection by T cell responses to early-secreted antigenic target 6-kDa protein and culture filtrate protein 10. J. Infect. Dis. 181:1850–1854. [DOI] [PubMed] [Google Scholar]
- 13.Ulrichs, T., P. Anding, S. Porcelli, S.E. Kaufmann, and M.E. Munk. 2000. Increased numbers of ESAT-6- and purified protein derivative-specific gamma interferon-producing cells in subclinical and active tuberculosis infection. Infect. Immun. 68:6073–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lalvani, A., A.A. Pathan, H. McShane, R.J. Wilkinson, M. Latif, C.P. Conlon, G. Pasvol, and A.V. Hill. 2001. Rapid detection of Mycobacterium tuberculosis infection by enumeration of antigen-specific T cells. Am. J. Respir. Crit. Care Med. 163:824–828. [DOI] [PubMed] [Google Scholar]
- 15.Britton, W.J., and U. Palendira. 2003. Improving vaccines against tuberculosis. Immunol. Cell Biol. 81:34–45. [DOI] [PubMed] [Google Scholar]
- 16.Ehrt, S., D. Schnappinger, S. Bekiranov, J. Drenkow, S. Shi, T.R. Gingeras, T. Gaasterland, G. Schoolnik, and C. Nathan. 2001. Reprogramming of the macrophage transcriptome in response to interferon-γ and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase–2 and phagocyte oxidase. J. Exp. Med. 194:1123–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandt, L., T. Oettinger, A. Holm, A.B. Andersen, and P. Andersen. 1996. Key epitopes on the ESAT-6 antigen recognized in mice during the recall of protective immunity to Mycobacterium tuberculosis. J. Immunol. 157:3527–3533. [PubMed] [Google Scholar]
- 18.Smith, D.W., D.N. McMurray, E.H. Wiegeshaus, A.A. Grover, and E.G. Harding. 1970. Host-parasite relationships in experimental airborne tuberculosis. IV. Early events in the course of infection in vaccinated and nonvaccinated guinea pigs. Am. Rev. Respir. Dis. 102:937–949. [DOI] [PubMed] [Google Scholar]
- 19.Schell, R.F., W.F. Ealey, G.E. Harding, and D.W. Smith. 1974. The influence of vaccination on the course of experimental airborne tuberculosis in mice. J. Reticuloendothel. Soc. 16:131–138. [PubMed] [Google Scholar]
- 20.Mustafa, A.S., H.A. Amoudy, H.G. Wiker, A.T. Abal, P. Ravn, F. Oftung, and P. Andersen. 1998. Comparison of antigen-specific T-cell responses of tuberculosis patients using complex or single antigens of Mycobacterium tuberculosis. Scand. J. Immunol. 48:535–543. [DOI] [PubMed] [Google Scholar]
- 21.Pollock, J.M., and P. Andersen. 1997. Predominant recognition of the ESAT-6 protein in the first phase of interferon with Mycobacterium bovis in cattle. Infect. Immun. 65:2587–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mollenkopf, H.J., M. Kursar, and S.H. Kaufmann. 2004. Immune response to postprimary tuberculosis in mice: Mycobacterium tuberculosis and Mycobacterium bovis bacille Calmette-Guerin induce equal protection. J. Infect. Dis. 190:588–597. [DOI] [PubMed] [Google Scholar]
- 23.Lewis, K.N., R. Liao, K.M. Guinn, M.J. Hickey, S. Smith, M.A. Behr, and D.R. Sherman. 2003. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J. Infect. Dis. 187:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orme, I.M., and F.M. Collins. 1986. Aerogenic vaccination of mice with Mycobacterium bovis BCG. Tubercle. 67:133–140. [DOI] [PubMed] [Google Scholar]
- 25.Paledira, U., A.G. Bean, C.G. Feng, and W.J. Britton. 2002. Lymphocyte recruitment and protective efficacy against pulmonary mycobacterial infection are independent of the route of prior Mycobacterium bovis BCG immunization. Infect. Immun. 70:1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung, Y.J., R. LaCourse, L. Ryan, and R.J. North. 2002. Virulent but not avirulent Mycobacterium tuberculosis can evade the growth inhibitory action of a T helper 1–dependent, nitric oxide synthase 2–independent defense in mice. J. Exp. Med. 196:991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen, A.W., L. Brant, E.M. Agger, L.A.H. van Pinxteren, and P. Andersen. 2004. The influence of remaining live BCG organisms in vaccinated mice on the maintenance of immunity to tuberculosis. Scand. J. Immunol. 60:273–277. [DOI] [PubMed] [Google Scholar]
- 28.McCune, R.M., F.M. Feldmann, H.P. Lambert, and W. McDermott. 1966. Microbial persistence: the capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 123:445–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Rie, A., R. Warren, M. Richardson, T.C. Victor, R.P. Gie, D.A. Enarson, N. Beyers, and P.D. van Helden. 1999. Exogenous reinfection as a cause of recurrent tuberculosis after curative treatment. N. Engl. J. Med. 341:1174–1179. [DOI] [PubMed] [Google Scholar]
- 30.Caminero, J.A., M.J. Pena, M.I. Campos-Herrero, J.C. Rodriguez, O. Afonso, C. Martin, J.M. Pavon, M.J. Torres, M. Burgos, P. Cabrera, et al. 2001. Exogenous reinfection with tuberculosis on a European island with a moderate incidence of disease. Am. J. Respir. Crit. Care Med. 163:717–720. [DOI] [PubMed] [Google Scholar]
- 31.Winslow, G.M., A.D. Roberts, M.A. Blackman, and D.L. Woodland. 2003. Persistence and turnover of antigen-specific CD4 T cells during chronic tuberculosis infection in the mouse. J. Immunol. 170:2046–2052. [DOI] [PubMed] [Google Scholar]
- 32.Scanga, C.A., V.P. Mohan, K. Yu, H. Joseoh, K. Tanaka, J. Chan, and J.L. Flynn. 2000. Depletion of CD4+ T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon-γ and nitric oxide synthase–2. J. Exp. Med. 192:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnappinger, D., S. Ehrt, M.I. Voskuil, Y. Liu, J.A. Mangan, I.M. Monahan, G. Dolganov, B. Efron, P.D. Butcher, C. Nathan, and G.K. Schoolnik. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiegeshaus, E.H., D.N. McMurray, A.A. Grover, G.E. Harding, and D.W. Smith. 1970. Host-parasite relationships in experimental airborne tuberculosis. III. Relevance of microbial enumeration to acquired resistance in guinea pigs. Am. Rev. Respir. Dis. 102:422–429. [DOI] [PubMed] [Google Scholar]
- 35.Medina, E., and R.J. North. 1999. Genetically susceptible mice remain proportionally more susceptible to tuberculosis after vaccination. Immunology. 96:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dunn, P.L., and R.J. North. 1995. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect. Immun. 63:3428–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Souza, S., V. Rosseels, M. Romano, A. Tanghe, O. Denis, F. Jurion, N. Costiglione, A. Vanonckelen, K. Palfiet, and K. Huygen. 2003. Mapping of murine Th1 helper T-cell epitopes of Mycolyl transferases Ag85A, Ag85B, and Ag85C from Mycobacterium tuberculosis. Infect. Immun. 71:483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inaba, K., M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu, and R.M. Steinman. 1992. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176:1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellis, R.C., and L.A. Zabrowarny. 1993. A safer staining method for acid fast bacilli. J. Clin. Pathol. 46:559–560. [DOI] [PMC free article] [PubMed] [Google Scholar]