

Abstract
The generation of high-affinity antibodies requires somatic hypermutation (SHM) and class switch recombination (CSR) at the immunoglobulin (Ig) locus. Both processes are triggered by activation-induced cytidine deaminase (AID) and require UNG-encoded uracil-DNA glycosylase. AID has been suggested to function as an mRNA editing deaminase or as a single-strand DNA deaminase. In the latter model, SHM may result from replicative incorporation of dAMP opposite U or from error-prone repair of U, whereas CSR may be triggered by strand breaks at abasic sites. Here, we demonstrate that extracts of UNG-proficient human B cell lines efficiently remove U from single-stranded DNA. In B cell lines from hyper-IgM patients carrying UNG mutations, the single-strand–specific uracil-DNA glycosylase, SMUG1, cannot complement this function. Moreover, the UNG mutations lead to increased accumulation of genomic uracil. One mutation results in an F251S substitution in the UNG catalytic domain. Although this UNG form was fully active and stable when expressed in Escherichia coli, it was mistargeted to mitochondria and degraded in mammalian cells. Our results may explain why SMUG1 cannot compensate the UNG2 deficiency in human B cells, and are fully consistent with the DNA deamination model that requires active nuclear UNG2. Based on our findings and recent information in the literature, we present an integrated model for the initiating steps in CSR.
Recent research revealed a surprising molecular link between the ancient DNA repair mechanisms and the vertebrate adaptive immune system apparently mediated through the nucleobase, uracil. Generally, uracil in DNA is the result of spontaneous or chemical deamination of cytosine, or misincorporation of dUMP instead of dTMP during replication (for review see reference 1). Such uracil is removed by cellular uracil-DNA glycosylases (UDGs), which initiate a multistep base-excision repair pathway eventually restoring the correct DNA sequence. In B cells, uracil in genomic DNA also may result from enzymatic deamination of cytosine by activation-induced cytidine deaminase (AID). AID is required for somatic hypermutation (SHM) and class switch recombination (CSR) of Ig genes (2, 3), but its precise role in antibody diversification has been the focus of extensive debate (for review see reference 4). Based on its homology with APOBEC-1, AID initially was suggested to be involved in RNA editing (5). This view was supported by apparent dispensability for active uracil-DNA glycosylase in CSR (6). In the alternative model, AID specifically catalyses deamination of cytosine to uracil in single-stranded DNA in Ig variable and switch regions of activated B cells, thereby facilitating SHM and CSR, respectively (7, 8). The latter model is supported by a substantial body of evidence, including results from Ung-deficient mice (8, 9) as well as human patients (10). Based on the altered SHM pattern in the variable region of Ig genes and partially defective CSR in Ung −/− mice, it was suggested that uracil-DNA glycosylase from the UNG gene is central downstream of AID (8). Furthermore, recent data indicate that the major route for CSR involves uracil excision by the uracil-DNA glycosylase, UNG2, with recognition of U:G mismatches by MutS homologue (MSH)2 providing a backup, at least in mice (9). In contrast, the second step in hypermutation, generating mutations at A:T base pairs, apparently is triggered by mismatch recognition, with uracil excision providing a backup (9). The involvement of UNG in CSR and SHM was demonstrated in three unrelated hyper-IgM (HIGM) patients (10). The characteristics of these patients were similar to those associated with AID deficiency, including susceptibility to bacterial infections and lymphoid hyperplasia. However, in contrast with the partial defect CSR observed in the Ung −/− mice, these patients were characterized by a much more severe lack of serum IgG and IgA; this suggested that the mismatch recognition system plays a less important role in CSR in humans compared with mice. Surprisingly, other uracil-DNA glycosylases, such as the single-strand–specific monofunctional uracil-DNA glycosylase (SMUG1), are not able to compensate for UNG deficiency in mice (9), although it was reported to be the major uracil-DNA glycosylase for U:G mismatches in mice (11).
Uracil in DNA may be excised by at least four mammalian uracil-DNA glycosylases in addition to UNG (SMUG1, methyl-CpG binding domain 4 glycosylase [MBD4], thymine-DNA glycosylase [TDG], and Nei-like glycosylase [NEIL]1). Of these, UNG2 seems to be quantitatively dominating in nuclei, with SMUG1 acting as a backup (12). The substrate preference of SMUG1 in vitro is similar to that of UNG2, and it remains unknown why SMUG1 cannot complement the function of UNG2 in UNG-deficient patients (and mice). To investigate this we analyzed the major uracil-DNA glycosylase activities in EBV-immortalized lymphoid cell lines (LCLs) from UNG-proficient individuals and from UNG-deficient HIGM patients (P1, P2, and P3; reference 10). Two of these patients (P1 and P3) carry biallelic germline mutations leading to premature translational stop and catalytically dead proteins. The third patient (P2) carries a homozygous missense mutation leading to a phenylalanine to serine substitution (F251S) in UNG2. The molecular basis of the HIGM phenotype in patient P2 was studied by analyzing the enzymatic properties and the intracellular behavior of the UNG2-F251 mutant. Finally, we analyzed the total genomic uracil content in the LCLs to investigate whether SMUG1 or other uracil-DNA glycosylases could have different backup functions in the overall genome in contrast with at Ig loci. Based on the results from the present study and previous work from our laboratory and others, we hypothesize that removal of deaminated cytosine from single-stranded Ig switch regions by UNG2 is essential for CSR that is mediated via a MutSα-independent pathway and also important for MutSα-dependent pathways.
RESULTS
UNG2 is the major nuclear uracil-DNA glycosylase in human lymphoid cells and SMUG1 contributes insignificantly to removal of uracil in single-stranded DNA
Here we have focused on the capacity of WT and UNG mutant human LCLs to process uracil in different DNA contexts. We first investigated the capacity of nuclear extracts from an UNG-proficient LCL to remove uracil from single-stranded compared with double-stranded [3H]dUMP-labeled calf thymus DNA. To ensure that the putative major UDGs, UNG2 and SMUG1, were maintained during preparation of the extracts, immunoprecipitates from nuclear extracts and total cell lysates were subjected to Western blot analysis. The results indicated that both proteins were readily extractable and maintained to a similar extent during preparation of the nuclear extracts. Furthermore, no bands representing potential degradation products could be observed; this indicated that UNG2 and SMUG1 remained intact during extraction (Fig. 1 A). The UDG activity against the ssDNA substrate was ∼2.5-fold higher than with double-stranded DNA (Fig. 1 B). UNG-neutralizing antibodies inhibited >99% of the activity against both substrates, whereas no effect was observed with SMUG1-neutralizing antibodies; this indicated that UNG2 is the major nuclear uracil-DNA glycosylase in the cells, with a preference for ssDNA. When using [3H]dUMP-labeled calf thymus DNA as substrate, the relatively high substrate concentration used (3.6 μM) would favor UDGs having high catalytic turnover, such as UNG. Moreover, U:G-specific UDGs would not be detected using this substrate. To enhance detection of UDGs with lower catalytic turnover and high substrate affinity (e.g., SMUG1) as well as U:G-specific enzymes (e.g., TDG and MBD4), the extracts were analyzed using a low concentration (0.02 μM) of a 19-mer [33P]-labeled oligodeoxynucleotide (containing uracil opposite A or G, or in ssDNA) as substrate and combined with long incubation time (Fig. 1 C). In the presence of anti-UNG antibodies (lane 2) and Ugi (lane 5), uracil excision was detectable from the U:A and U:G substrates. This was attributed mainly to SMUG1, because both activities were abolished (U:A) or reduced (U:G) in the presence of anti-UNG and anti-SMUG1 antibodies (lane 4). Although total uracil excision was most efficient from the single-stranded DNA (Uss) substrate, almost no activity against the Uss substrate was detected when UNG was inhibited by antibodies (lane 2) or Ugi (lane 5). SMUG1 originally was described (and named) as a single-strand selective monofunctional uracil-DNA glycosylase (13). However, the substrate preference of SMUG1 is influenced strongly by apurinic/apyrimidinic endonuclease (APE)1 and the concentration of salt (12, 14). Therefore, we analyzed and compared UNG2 and SMUG1 activity with purified enzymes using the oligonucleotide substrate described in Fig. 1 C (U:A, U:G, and Uss) and in the presence of APE1. Under these conditions, purified human SMUG1 shows a more than 100-fold preference for U:G substrate compared with Uss. Furthermore UNG2 is ∼1,000-fold more efficient than SMUG1 on Uss (Fig. 1 D). These substrate activity profiles of UNG2 and SMUG1 (in the presence of APE1) resemble those seen in the nuclear extracts, and further support that UNG2 is the major, if not sole, nuclear glycosylase releasing uracil in Uss in B cells. This is likely to be important in the UNG-deficient HIGM phenotype, because cytosine deamination by AID introduces uracil only in ssDNA (15, 16). In conclusion, UNG2, but not SMUG1, in LCL nuclear extracts excises uracil from ssDNA very efficiently. Our findings likely explain the inability of SMUG1 to compensate for UNG2 in B cells of UNG-deficient HIGM patients and mice.
Figure 1.

UNG2 is the major uracil-DNA glycosylase in B cell nuclei. (A) Immunoprecipitation and Western blot analysis of UNG and SMUG1 in total and nuclear extracts prepared from the UNG proficient control LCL. (B) Total specific UDG activity in UNG-proficient LCL nuclear extracts measured by high turnover activity assays using 36 pmol (1.8 μM) [3H]-dUMP labeled calf thymus DNA substrates (gray bars: double-stranded substrate; black bars: single-stranded substrate). Extracts (2 μg protein) were pretreated with 0.5 μg preimmune IgGs (0-Ab), neutralizing anti-hUNG (PU101), or neutralizing anti-hSMUG1 (PSM1) IgGs, or both neutralizing IgGs. (C) UDG activities in UNG-proficient LCL nuclear extract measured by low turnover activity assays using 0.2 pmol (0.02 μM) [33P]-labeled 19-mer deoxyoligonucleotide with uracil in U:A, U:G, or Uss contexts. The samples in lane 1–4 were pretreated as in (A). Lane 5: nuclear extract was preincubated with 10 ng Ugi. Lane 6: samples were treated as in lane 4. In addition, 200 ng purified UNG2 was included in the reaction. Lane 7: samples were treated as in lane 4. In addition, 200 ng purified SMUG1 was added to the reaction. Note that two product bands appear to be formed consistently. The lower of these bands represents the 9-bp product formed by cleavage of the AP site by hot piperidine. The upper band represents a 9-bp product in which the AP site has been cleaved by AP endonuclease (present in the extracts), before the addition of hot piperidine. Such cleavage results in loss of the 3′-phosphate and slower migration during PAGE. (D) UDG activity of purified, recombinant UNG2 and SMUG1 in the presence of 0.2 pmol purified, recombinant APE1 was measured using 0.2 pmol [33P]-labeled 19-mer deoxyoligonucleotide with uracil in U:A, U:G, or Uss as substrate. Lane 1: 0 pmol UNG2/SMUG1; lane 2: 0.2 fmol UNG2/SMUG1; lane 3: 2 fmol UNG2/SMUG1; lane 4: 20 fmol UNG2/SMUG1; lane 5: 0.2 pmol UNG2/SMUG1; and lane 6: 2.0 pmol UNG2/SMUG1.
UNG2-F251S is fully active and stable in vitro, but the mutation profoundly impairs the expression of active protein in B cells
The homozygous UNG mutations carried by patients P1 and P3 encode COOH-terminally truncated, and thus, catalytically dead proteins (10). Thus, the impaired ability to process AID-generated uracil in Ig loci may explain their HIGM phenotype. However, this is not obvious for the UNG2-F251S substitution mutation in patient P2 (Fig. 2 A). Begum et al. (6) recently reported that the mouse counterpart of this mutant (denoted F242S) removed uracil and restored CSR in transfected mouse Ung −/− B-cells. To study this apparent paradox in more detail, we analyzed nuclear extract from the B cell line derived from patient P2 using the uracil-containing 19-mer [33P]-labeled DNA-oligo as substrate. No UNG activity was detected in this assay, thus no inhibitory effect was achieved with addition of UNG-Ab or Ugi. However, the extract clearly revealed SMUG1 activity against U:A and U:G substrates, but not against Uss (Fig. 2 B). To analyze the effect of the F251S mutation at the protein level, we expressed UNG2-F251S in Escherichia coli. Although the expression level of the mutant protein in the bacteria was considerably lower than that of the WT, the specific activities as well as the temperature optima and thermal stabilities of the purified enzymes essentially were identical (Table I, Fig. 2 C). However, this could not rule out the possibility that the F251S mutation leads to truncated forms of UNG in the B cells. We previously showed that NH2-terminal truncations may alter the catalytic properties and protein:protein interaction capabilities of UNG (12, 17, 18). Intact or processed forms of UNG could not be detected by immunoprecipitation or Western blot analysis in P2 LCL total cell lysates when using a polyclonal antibody directed against the entire common catalytic domain of the mitochondrial UNG1 and nuclear UNG2 proteins (Fig. 2 D). However, more sensitive and UNG-selective enzymatic assays (using high extract concentration, increased incubation temperature and time, and the high turnover substrate) revealed that the P2 LCL contained trace levels of UNG activity as demonstrated by the inhibitory effect of neutralizing UNG antibodies (Fig. 2 E). This residual UNG activity amounted to ∼0.4% compared with that of the WT control (Fig. 1 B).
Figure 2.

UNG2-F251S is fully active and stable in vitro, but the mutation impairs expression of active protein in B cells. (A) Overall structure of the WT UNG complexed with a uracil-containing DNA duplex, with F251 highlighted. The close-ups show that F251 is surrounded by several hydrophobic side chains, and that the F251S substitution results in a “hole” in the central hydrophobic region of UNG. (B) P2 nuclear extract analyzed using 0.2 pmol (0.02 μM) [33P]- labeled 19-mer deoxyoligonucleotide with uracil in U:A, U:G, or Uss contexts. The samples were pretreated as in Fig 1 B. (C) Temperature optimum (left panel) and thermal stability (right panel) of purified, recombinant UNG2-WT and UNG2-F251S measured by high turnover activity assays using 36 pmol (1.8 μM) [3H]-dUMP labeled calf thymus DNA substrates. (D, left) Immunoprecipitation and Western blot analysis of purified, recombinant UNG2-WT and UNG2-F251S, demonstrating that the PU101 antibody precipitates the WT and mutant proteins with equal efficiency. (right) IP and Western blot analysis of UNG (using PU101) and SMUG1 (using PSM1) in whole cell extracts from P2 (UNG2-F251S) and C1 (control 1). (E) Trace levels of UNG activity in P2 nuclear (gray bars) and cytosolic + mitochondrial (black bars) fractions measured by high turnover activity assays using 36 pmol [3H]-dUMP [3H]-labeled heat-denatured calf thymus DNA substrate under high sensitivity assay conditions (4 μg protein extracts, 30 min incubation at 37°C). Note the higher resolution of the y-axis compared with Fig. 1 B.
Table I.
Specific activities and relative substrate preferences of purified, recombinant UNG2 and UNG2-F251S measured using single-stranded and double-stranded [3H]UMP-labeled DNA substrates
| Substrate | Specific activity |
WT | single-stranded/ double-stranded |
|
|---|---|---|---|---|
| U/mg | % | |||
| UNG2 | double-stranded | 1,779 ± 71 | 100 | 8.5 ± 0.2 |
| UNG2-F251S | double-stranded | 1,836 ± 209 | 103 ± 11 | 8.2 ± 0.3 |
| UNG2 | single-stranded | 15,231 ± 325 | 100 | |
| UNG2-F251S | single-stranded | 15,107 ± 496 | 99 ± 3 |
The F251S mutation impairs nuclear accumulation of UNG2 and causes abnormal translocation to mitochondria via interaction with UNG1
We previously used human HeLa cells to study intracellular localization of UNG1- and UNG2-WT and mutants in great detail (12, 18–21). To study the intracellular behavior of UNG2-F251S, mutant and WT proteins were expressed as fluorescent fusion proteins and analyzed in live HeLa cells. When the cultures were followed for 2 d, an increasingly aberrant sorting of the mutant UNG protein was observed, with a shift toward cytoplasmic accumulation (Fig. 3 A). Moreover, the nuclear fraction of the mutant did not accumulate in replication foci (Fig. 3 B; reference 18). Coexpression of UNG2-F251S-EYFP with mitochondrial UNG1-ECFP (Fig. 3 C; reference 21), as well as visualizing the mitochondria with a monoclonal anti–human mitochondria antibody (Fig. 3 D), demonstrated that the mutant unambiguously colocalized with mitochondria, whereas WT UNG2-EYFP did not. Moreover, coexpression of the two proteins completely abolished nuclear accumulation of UNG2-F251S-EYFP (Fig. 3 C). The most likely explanation for this is that UNG2-F251S physically interacts with UNG1, thus redirecting its translocation from nuclei to mitochondria. Sequestering of UNG2 to UNG1 likely also promotes proteolytic degradation of the mutant. Thus, even if significant amounts of enzymatically active mutant UNG2-F251S are expressed in vivo in some cells, its normal nuclear accumulation will be perturbed.
Figure 3.
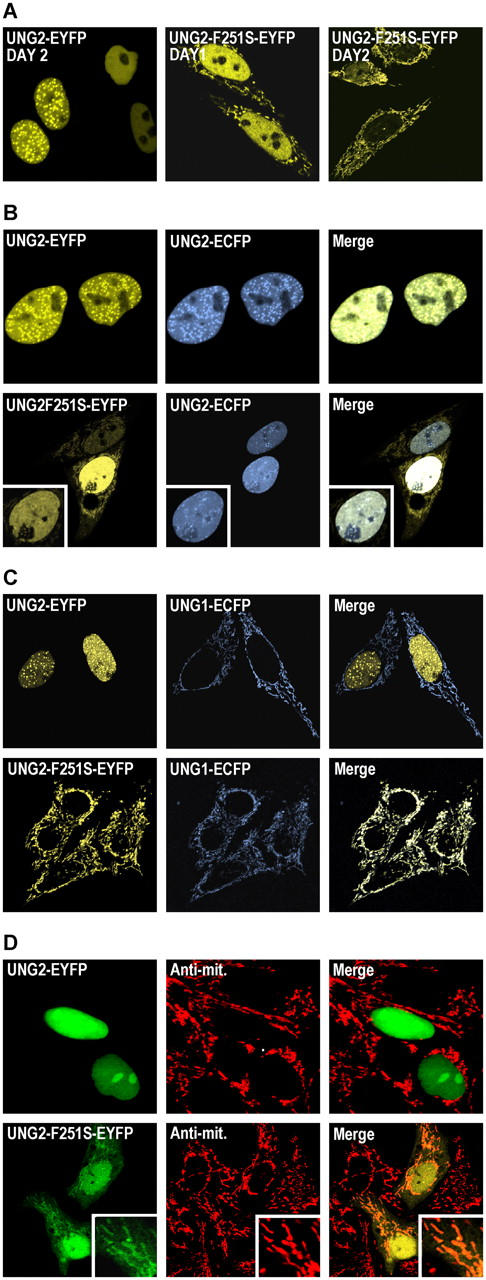
UNG2-F251S is abnormally translocated to mitochondria. (A) WT UNG2-EYFP or UNG2-F251S-EYFP transfected in HeLa cells and followed in live cells for 2 d. (B) Control coexpression of WT UNG2 with different fluorescent tags (upper row). UNG2-F251S and WT UNG2 are localized different in human cells, demonstrated by coexpression of UNG2-F251S-EYFP and WT UNG2-ECFP in the same cell (lower row). (C) Coexpression of WT UNG1-ECFP and UNG2-EYFP demonstrates distinct sorting to mitochondria and nuclei, respectively (upper row), whereas coexpression of mutant UNG2-F251S-EYFP and UNG1-ECFP abolishes translocation of the mutant to nuclei, while increasing its accumulation at mitochondria. (D) UNG2-F251S-EYFP but not WT UNG2-EYFP colocalizes with mitochondria in fixed cells as demonstrated using an antibody against human mitochondria.
Impaired nuclear UNG2 leads to profoundly increased overall genomic uracil content
To investigate if the inability of SMUG1 or other glycosylases to complement UNG2 in uracil removal was restricted to Ig loci, we analyzed the overall genomic uracil content in control and patient LCLs by the comet assay. Although genomic uracil was not detectable in the controls, the UNG mutant P1, P2, and P3 cell lines contained substantial levels of uracil (Fig. 4 A). Thus, SMUG1 cannot perform efficient sanitation of genomic uracil in any of the LCLs lacking UNG expression (Fig. 4 A, lower panel). Furthermore, because clearly visible comets were observed with virtually all cells in the patient LCLs after UNG treatment (Fig. 4 B), SMUG1 apparently is unable to compensate for the lack of overall genomic uracil removal by UNG2 at any stage of the cell cycle. To investigate whether up-regulation of SMUG1 or other potential backup UDG activities in UNG-deficient LCLs could be detected at the enzyme activity level, the total residual UDG activity in the presence of the UNG-specific inhibitor, Ugi (Fig. 4 C; reference 22), was analyzed in parallel in LCLs from patients P1–P3 and the controls. In accordance with the results in Figs. 1 C and 2 B, the removal of uracil from Uss by SMUG1 was close to the detection limit in the patient LCLs and in the controls, whereas significant activity against U:A and U:G could be observed. Furthermore, the enzymatic activity of SMUG1 and other (U:G-specific) UDGs in the LCLs apparently was not correlated to the UNG status and the overall genomic uracil content in the cells.
Figure 4.

Steady-state genomic uracil levels are increased in LCLs from UNG mutant patients; thus, SMUG1 or other UDGs cannot compensate for UNG deficiency. (A) Single cell gel electrophoresis (comet assay) of alkali-treated cells. Each of the comets was assigned a score for the level of apparent DNA damage (given as arbitrary units, see Materials and methods). Cells were treated with recombinant UNGΔ84 before electrophoresis (black bars) or with buffer alone (open bars). Error bars represent the standard deviation calculated from four independent experiments (each in duplicate). Immunoprecipitation and Western blot analysis of UNG and SMUG1 from the whole cell LCL lysates are shown below. (B) Photomicrographs illustrating results from the comet assay. C1 and UNG mutant P3 cells are representative for all cell lines in the respective groups. (C) SMUG1 or other UDG activities are not up-regulated in UNG-deficient LCLs. Nuclear extracts (2 μg protein) from UNG-proficient (C1, C2) and UNG-deficient (P1, P2, P3) LCLs were preincubated with 10 ng Ugi and subjected to analysis using low turnover activity assays using 0.2 pmol (0.02 μM) [33P]-labeled19-mer deoxyoligonucleotide with uracil in U:A, U:G, or Uss contexts. In addition, either 0.5 μg preimmune IgGs (0-Ab) or neutralizing anti-hSMUG1 IgGs (SMUG1-Ab) were added to the extracts.
Given the marked preference of SMUG1 for uracil in dsDNA, the elevated genomic uracil content in B cells from UNG-deficient patients may divert SMUG1 away from Uss in Ig loci. Thus, even if SMUG1, or very few molecules of active UNG2 (as in P2), theoretically could be sufficient to fulfill a specific function in CSR and SHM, the competition by a large overall DNA uracil pool in the patients may contribute to the HIGM phenotype.
DISCUSSION
Recently, it was demonstrated that overexpression of human SMUG1 in Ung-deficient mice did not restore CSR (9). This is in full agreement with our results with human cells; this indicates that the efficient removal of uracil from DNA cannot be compensated for by SMUG1 or other DNA glycosylases. The original naming of SMUG1 as a single-strand selective monofunctional uracil-DNA glycosylase (13) was based upon the substrate preference of purified, recombinant Xenopus SMUG1. As demonstrated in our laboratory and by others, the activity profile of hSMUG1 is different from that originally reported for xSMUG1, and the activity of the human enzyme strongly depends on the salt concentrations that are used in the assays and the presence of APE1 (12, 14). Thus, at physiologically relevant salt concentrations, the activity of recombinant hSMUG1 against Uss is reduced severely; this is confirmed by the present analyses of the LCL extracts. However, compensatory mechanisms may play different roles in humans and mice because CSR is less affected in Ung-deficient mice (8) than in UNG − / − patients (10). However, mouse Msh2 − / − Ung−/ − double knockouts proved to be completely defective in CSR (9) which underscores the importance of mismatch recognition in CSR in mice. Contribution by mismatch recognition also may explain recent findings by Begum et al. (6) which indicated that the precise step at which CSR is blocked differs in murine and human UNG-deficient B cells.
CSR in UNG mutant human B cells is blocked before formation of DNA double strand breaks (DSBs; reference 10). In contrast, CSR in murine CH12F3-2 B cells that overexpressed Ugi was affected at a step downstream of strand cleavage; this suggested that the catalytic activity of UNG was dispensable for the DNA-cleavage step in CSR (6). In addition, several catalytically deficient UNG mutants were able to rescue CSR in murine Ung −/− cells (6). However, these results should be interpreted in light of the exceptional catalytic turnover of UNG (kcat up to 5,000 uracils × min−1 from ssDNA; reference 23). Previous work from our laboratory demonstrated that the UNG mutants that are able to support CSR (D145N, N204V, H268L) used by Begum et al. (6) all exhibit residual catalytic activity between 0.04% and 0.52% (24). This is within the normal range of activity observed for most other DNA glycosylases (23), and when overexpressed they still may support CSR in mouse Ung −/− B cells by way of active uracil excision. However, the double mutants, D145N/N204V and H268L/D145N, are likely to have a further 100–1,000-fold reduced activity (24) and probably do not reach the threshold activity that is required to restore CSR, as observed (6). The finding that the mouse counterpart of hUNG2-F251S is able to restore CSR in Ung-deficient mouse cells (6) apparently stands in contrast with our proposal that the F251S mutation is the underlying cause of HIGM in P2. However, mistargeting of UNG2-F251S seems to be linked intimately to the level of UNG1 expression in the cells. Thus, overexpression in HeLa of UNG2-F251S alone leads to partial nuclear translocation, whereas concomitant overexpression of UNG1 or UNG1-F251S leads to nearly complete mitochondrial translocation of UNG2. Overexpression of this mutant from a retroviral vector (6) in the absence of UNG1 likely results in nuclear accumulation of active enzyme, and thus, restores CSR.
It is not clear whether recognition and excision of uracil by UNG2 takes place while uracil is in the single-stranded conformation or to what extent uracil is generated in, and excised from, DNA:RNA hybrids. However, the present work demonstrates that deoxyuridine in ssDNA is an excellent substrate for B cell UNG2. This also is likely to be the case for UNG2 from other human cell types, and conforms to the results using recombinant UNG2 (Fig. 1 C). Furthermore, we previously showed that UNG from HeLa cells efficiently removes uracil from poly(dU):poly(rA) (25). Thus, uracil might well be excised from the template and nontemplate strand while still in the ssDNA conformation. Although in the present study we examined the substrate preference of UNG2 in the presence of EDTA to avoid nuclease activity in the extracts, the single-strand/double-strand DNA substrate specificity ratio of UNG2 increases ∼40-fold in the presence of physiological concentrations of Mg2+ (12). This clearly indicates that Uss is the preferred substrate in vivo. Removal of uracil from single-stranded Ig loci is substantiated further by our previous finding that UNG2 contains motifs within its NH2-terminal region that interact with the single-strand binding, replication protein A (RPA) (18, 19). Thus, RPA may enhance the recruitment of UNG2 to Uss in switch regions. Recently, it was demonstrated that AID also is recruited to transcribed Ig loci by the single-strand DNA binding protein, RPA (26); this raises the possibility that the individual steps in CSR are orchestrated within a multiprotein complex. The availability of UNG2 to such a complex is probably more important than its high catalytic turnover (which likely is adapted to cope with the speed of the replication machinery; reference 18). Thus, UNG2 may have two distinct functions in antibody affinity maturation: assembly of functional protein complexes and excision of uracil. This also would explain the apparent paradox that some UNG mutants with low catalytic activity sustain the function in CSR and SHM.
An integrated model for initiation of CSR
CSR has been proposed to occur in mice via an UNG-dependent pathway (major) and an UNG-independent pathway (backup); the latter involves mismatch recognition by MSH2/MSH6 (MutSα; reference 9). This model fails to explain how the initial strand breaks are formed in a UNG-independent pathway that ultimately leads to DSBs. In light of the present findings and recent reports in the literature, we propose that UNG2 also may be a major player in the pathway involving MutSα. However, the function of UNG2 in the MutSα-pathway likely is redundant, and may be compensated for by other factors (Fig. 5). Both pathways are initiated by AID that deaminates deoxycytidine in the template (27) and nontemplate strands (7) of transcribed switch regions. According to the proposed structure model (28) and in vitro activity analyses (16), AID dimers may accommodate only a single DNA strand within its active site. Thus, we speculate that deamination in the template strand takes place before formation of the RNA:DNA hybrid (e.g., in negative DNA supercoils upstream of the elongating RNA; reference 29). Recruitment of AID to the growing end of the transcription bubble may be facilitated by interactions with RPA (26), RNA POLII (30), or both. The nuclear isoform of UNG2 effectively excises uracil from the nontemplate strand (this paper), potentially aided by RPA (19). UNG2 also may act upon the DNA:RNA hybrid downstream of the polymerase, because we found previously that UNG effectively excises uracil from a poly(dU):poly(rA) substrate (25). When UNG2 is abundant (UNG +/+ cells), uracils are excised from both strands and ultimately lead to opposing AP sites at CSR (e.g., AGCT) hotspots (Fig. 5, left route). AP sites in the template strand also may destabilize the DNA:RNA hybrid, and thus, promote reannealing of the DNA strands to generate substrates for subsequent AP-endonuclease cleavage by APE1. Opposing AP sites resemble one type of clustered damage that often is observed after ionizing radiation, and that leads to a large number of DSBs (for review see reference 31). Furthermore, it was demonstrated recently that 40–60% of opposing AP sites located at the +1 position of each other (e.g., as would be expected in AGCT hotspots), were converted to DSBs within 60 min by mammalian nuclear extracts (32).
Figure 5.

Integrated model for initiation of CSR. Deamination of deoxycytidine in ssDNA close to the transcription complex initiates MutSα-independent (left) and MutSα-dependent (middle and right) pathways. The MutSα-independent pathway requires UNG2 that creates a large number of AP sites in both strands. After reannealing, opposing or closely spaced AP sites directly generate DSBs by AP-endonuclease (APE1) cleavage. When the cellular level (or function) of UNG2 is partially impaired, fewer AP sites are generated, and may not sustain DSB formation by direct cleavage. However, even a small number of AP sites generated by UNG2 would be sufficient to create nicks recognized by DNA mismatch repair factors and to initiate 5′-3′ processing toward MutSα bound to unprocessed U:G mismatches. If UNG2 is impaired fully, the MutSα-dependent pathway still may operate—although with strongly reduced efficiency—by using alternative nicking factors, such as ERCC1/XPF or TDG/NEIL1/SMUG1, followed by APE1. In such a scenario, the different pathways are likely to function in parallel, and the speed at which individual pathways operate may depend on the availability of the various factors at any given time point.
When the cellular UNG2 level is limited, the density of AP sites formed by uracil excision may decrease to less than the threshold sufficient to form DSBs by the direct (MutSα-independent) route. Instead, unprocessed U:G mismatches may be recognized by the MutSα heterodimer (Fig. 5, middle and right routes). Processing of such mismatches via the DNA mismatch repair system has been studied extensively using nicked substrates, and it was shown recently that human exonucleus (EXO)1—in conjunction with MutSα and RPA—supports 5′-3′ hydrolysis directed by a 5′ strand break (33). We hypothesize that these substrate 5′ strand breaks may be formed by the sequential action of UNG2 and the AP endonuclease, APE1. Thus, the two pathways may be collaborating by using common factors to generate the DSBs that mediate CSR. If UNG2 is totally impaired, the MutSα-dependent pathway may still operate (although considerably less effectively in humans than in mice) by recruitment of alternative proteins, such as ERCC1/XPF or NEIL1/SMUG1/TDG. The ERCC1/XPF endonuclease nicks double-stranded DNA immediately adjacent to 3′–single-stranded tails (34) and physically interacts with MSH2 (35); reduced CSR was demonstrated recently in murine Ercc1 −/− B cells (36). Such a model is consistent with the finding that murine Ung −/− B cells have a more pronounced loss of switch recombination than Msh2 −/− cells, and that it is essentially lost in the double knockouts. Notably, in the MutSα-dependent pathway, very few uracils need to be removed to generate the nicks that are recognized by EXO1-MutSα; these may be located at a considerable distance from the recognized mismatch itself, because mismatch-dependent hydrolysis observed in a purified MutSα-EXO1 system extended several thousand nucleotides from the strand break (33). Notably, a 3′-5′ exonucleolytic activity (Fig. 5, right route) was demonstrated recently for EXO1 in a reconstituted system made up of RFC, PCNA, MutSα, MutLα, EXO1, and RPA (33).
How the DSBs are processed downstream of these initiating steps has not been established. Several lines of evidence suggest that the nonhomologous end-joining apparatus plays a central role because mouse B cells that are deficient in ATM (37), H2AX (38), 53BP1 (39), Ku (40), or DNA-PKcs (41), and patients who have NBS1 or MRE11 deficiency (42) or ataxia telangiectasia (37) display varying degrees of perturbed class switching. How these events are orchestrated at the molecular level, and whether the initiating steps and downstream processing of the DSBs are organized within multiprotein complexes remain to be elucidated.
MATERIALS AND METHODS
Cell culture and preparation of total and nuclear cell extracts.
LCLs were prepared as described (10) and maintained in RPMI 1640 Glutamax I (GIBCO BRL) supplemented with 2.5 μg/ml fungizone, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated FBS (Euroclone) at 37°C and 5% CO2. Nuclear extracts were prepared from ∼107 cells (harvested during exponential growth) as described previously (12), aliquoted, snap frozen in liquid N2, and stored frozen at −80°C until use. Total cell extracts were prepared from 100 ml culture. Cells were harvested and resuspended in 500 μl buffer I (10 mM Tris pH 7.8, 200 mM KCl); thereafter, 500 μl buffer II (buffer I containing 2 mM EDTA, 40% glycerol, 0,5% NP-40, 2 mM DTT, 1× complete protease inhibitors; Roche) was added. The cell suspension was sonicated for 2 min (duty cycle 25, output 2.5). The extract was aliquoted, snap frozen in liquid N2, and stored at −80°C until use. Protein concentrations were measured using the Bio-Rad protein assay using BSA as standard.
UDG activity assays.
High-turnover activity assays using [3H]-labeled nick translated calf thymus DNA substrates (43) were performed essentially as described previously (12). In brief, activity was measured in 20 μl assay mixture containing (final) 20 mM Tris-HCl, pH 7.5, 60 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5 mg/ml BSA, 36 pmol [3H]dUMP-labeled calf thymus DNA substrate and 2 μg extract (measured as total protein) at 30°C for 10 min (or 4 μg protein, 37°C for 30 min in high-sensitivity assays). The amount of released uracil was measured as described previously (25). Single-stranded [3H]dUMP-labeled calf thymus DNA substrates were prepared by heating double-stranded substrate at 100°C for 10 min followed by rapid chilling on ice. Purified, recombinant UNG2-WT and the UNG2-F252S mutant was analyzed in the presence of 7.5 mM MgCl2, as described previously (12). Low turnover assay was performed using a 19-mer oligodeoxynucleotide U141 (5′-CATAAAGTGUAAAGCCTGG-3′; MWG Biotech). U141 was [33P]5′-end labeled, and used as single-stranded substrate (Uss). Double-stranded substrates were prepared by annealing the labeled strand to 19-mer complementary strands containing either A or G opposite uracil (U:A and U:G, respectively). UDG activity was measured in 10 μl assay mixture containing the same buffer as above. 0.2 pmol [33P]-labeled oligo-substrate and 2 μg nuclear extract (measured as total protein) were incubated at 37°C for 1 h. Purified recombinant UNG2 (0.2 fmol to 2 pmol) and SMUG1 (0.2 fmol to 2 pmol) were incubated with 0.2 pmol [33P]-labeled oligo-substrate, 0.2 pmol APE1, and 7.5 mM MgCl2 in the buffer described above at 37°C for 15 min. Reactions were stopped and abasic sites cleaved by addition of 50 μl 10% piperidine and incubation at 90°C for 20 min. Uracil excision was analyzed as described previously (12).
Immunoprecipitation and Western blot analysis.
Immunoprecipitation of UNG and SMUG1 was performed by mixing 2 mg (measured as protein) of total or nuclear extract with 20 μl of paramagnetic protein A Dynabeads (Dynal) covalently coupled with polyclonal antibodies against the catalytic domain of UNG (PU101; reference 12) or against SMUG1 (PSM1; reference 12), respectively. Samples were separated on the NuPage electrophoresis system (Invitrogen) and electro-blotted onto Immobilon PVDF membranes (Millipore). UNG and SMUG1 were detected using PU101 or PSM1 primary antibodies, respectively, HRP swine anti–rabbit (1:5,000; DakoCytomation) secondary antibodies, Super Signal West Femto substrate (Pierce Chemical Co.), and analyzed on the Kodak Image Station 2000R.
Heterologous expression and purification of UNG2-WT, UNG2-F251S, and SMUG1.
The F251S mutation was prepared by mutating pET28a-UNG2 (44) (expressing UNG2 with an NH2-terminal 6xHis-tag) using the Quick-change site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. hSMUG1 cloned into pET11a (Invitrogen) has been published previously (12). The open reading frame of the hSMUG1 cDNA was excised from pET11a-hSMUG1 as NdeI–BamHI fragment and subcloned into pET28a (Novagen). pET28a-hSMUG1 encodes hSMUG1 with a NH2-terminal 6xHistidine-tag. The APE1 expression vector (45) was a gift from I. Hickson (Cancer Research UK Laboratories, Oxford, England). The His-tagged proteins were expressed in E. coli BL21 codon plus-RIL. Cells were lysed by sonication at 4°C in the presence of 1 mg/ml lysozyme and Complete (Roche) protease inhibitors. UNG2-WT and UNG2-F251S were purified using Dynabeads Talon according to the manufacturer's protocol. His-tagged SMUG1 and His-tagged APE1 were purified by Ni-NTA superflow chromatography (QIAGEN). Both proteins were purified further by MonoS (HR5/5) chromatography (Amersham Biosciences).
Mammalian expression constructs and confocal microscopy.
pUNG1-EYFP, pUNG1-ECFP, pUNG2-ECFP, and pUNG2-EYFP were prepared by replacing the EGFP-tag (AgeI–NotI fragment) in pUNG1-EGFP and pUNG2-EGFP with the corresponding fragment from pECFP-N1 and pEYFP-N1 (CLONTECH Laboratories, Inc.; reference 20). In this vector system, transcription is regulated by the human CMV immediate early promoter, and thus, allows overexpression of the fusion proteins. The site-specific mutation, F251S, was made using the Quick-change site-directed mutagenesis kit. All constructs were verified by DNA sequencing. Cells were transfected using the calcium phosphate method (Profection, Promega) according to the manufacturer's protocol. Fluorescent images of transfected, freely cycling HeLa cells (1 μm thickness) were produced using a Zeiss LSM Meta laser scanning microscope equipped with a plan-apochromate 63×/1.4 oil immersion objective. ECFP fusions were excited at λ = 458 nm and detected at λ = 470–500 nm, EYFP fusions were excited at λ = 514 nm, and detected at λ > 530 nm. Mitochondria were visualized with a monoclonal mouse anti–human mitochondria primary antibody (p110, Calbiochem) and a rhodamine (tetra-methyl) conjugated goat anti–mouse secondary antibody (Molecular Probes) on cells fixed with 2% paraformaldehyde (5 min) followed by cold methanol (−20°C) on ice for 10 min. Rhodamine fluorescence was excited at λ = 543, detected at λ > 560, and visualized using a 63×/1.4 oil immersion objective.
Comet assay.
Cultured B cells were pelleted at 400 g or 5 min and embedded in 1% low melting point agarose. After lysis in ice-cold alkaline lysis solution for 1 h (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, adjusted to pH 10, 1% Triton X-100). Single-cell gel electrophoresis (comet assay) was performed as described previously (46). Comets were quantified by visual scoring by the same observer in all experiments. 100 comets were selected randomly from each slide and given a value from 0 (undamaged) to 4 (maximum damage); overall scores ranged from 0 to 400 arbitrary units. Each cell line was analyzed in at least four separate experiments, each time in duplicate; each sample was evaluated two to five times.
Acknowledgments
We wish to thank O. Sundheim, IKM, for preparing the graphic representations of UNG and B. Monterotti and H. Sahlin Pettersen, IKM, for technical assistance.
This work was supported by The National Programme for Research in Functional Genomics in Norway (FUGE) in The Research Council of Norway, the Norwegian Cancer Association, The Cancer Fund at St. Olav's Hospital, Trondheim, the Svanhild and Arne Must Fund for Medical Research, l'Institut National de la Santé et de la Recherche Médicale (INSERM), l'Association de la Recherche Contre le Cancer (ARC), la Ligue Contre le Cancer, the European Community (EURO_POLICY PID 006411), the French Rare Disease Program (GIS), the Assistance-Publique, Hopitaux de Paris, and the Uehara Memorial Foundation.
The authors have no conflicting financial interests.
Abbreviations used: AID, activation-induced cytidine deaminase; APE, apurinic/apyrimidinic endonuclease; CSR, class switch recombination; DSB, DNA double strand break; EXO, exonucleus; HIGM, hyper IgM; LCL, lymphoid cell line; MBD4, methyl-CpG binding domain 4 glycosylase; MSH, MutS homologue; NEIL, Nei-like glycosylase; RPA, replication protein A; SHM, somatic hypermutation; SMUG1, single-strand–specific monofunctional uracil-DNA glycosylase; ssDNA, single-stranded DNA; TDG, thymine-DNA glycosylase; UDG, uracil-DNA glycosylase; Uss, uracil in single-stranded DNA.
References
- 1.Krokan, H.E., H. Nilsen, F. Skorpen, M. Otterlei, and G. Slupphaug. 2000. Base excision repair of DNA in mammalian cells. FEBS Lett. 476:73–77. [DOI] [PubMed] [Google Scholar]
- 2.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 3.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 4.Honjo, T., M. Muramatsu, and S. Fagarasan. 2004. AID: how does it aid antibody diversity? Immunity. 20:659–668. [DOI] [PubMed] [Google Scholar]
- 5.Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. [DOI] [PubMed] [Google Scholar]
- 6.Begum, N.A., K. Kinoshita, N. Kakazu, M. Muramatsu, H. Nagaoka, R. Shinkura, D. Biniszkiewicz, L.A. Boyer, R. Jaenisch, and T. Honjo. 2004. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science. 305:1160–1163. [DOI] [PubMed] [Google Scholar]
- 7.Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 9.Rada, C., J.M. Di Noia, and M.S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the a/t-focused phase of somatic mutation. Mol. Cell. 16:163–171. [DOI] [PubMed] [Google Scholar]
- 10.Imai, K., G. Slupphaug, W.I. Lee, P. Revy, S. Nonoyama, N. Catalan, L. Yel, M. Forveille, B. Kavli, H.E. Krokan, et al. 2003. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 4:1023–1028. [DOI] [PubMed] [Google Scholar]
- 11.Nilsen, H., K.A. Haushalter, P. Robins, D.E. Barnes, G.L. Verdine, and T. Lindahl. 2001. Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J. 20:4278–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kavli, B., O. Sundheim, M. Akbari, M. Otterlei, H. Nilsen, F. Skorpen, P.A. Aas, L. Hagen, H.E. Krokan, and G. Slupphaug. 2002. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 277:39926–39936. [DOI] [PubMed] [Google Scholar]
- 13.Haushalter, K.A., M.W. Todd Stukenberg, M.W. Kirschner, and G.L. Verdine. 1999. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 9:174–185. [DOI] [PubMed] [Google Scholar]
- 14.Masaoka, A., M. Matsubara, R. Hasegawa, T. Tanaka, S. Kurisu, H. Terato, Y. Ohyama, N. Karino, A. Matsuda, and H. Ide. 2003. Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry. 42:5003–5012. [DOI] [PubMed] [Google Scholar]
- 15.Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- 16.Bransteitter, R., P. Pham, M.D. Scharff, and M.F. Goodman. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA. 100:4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bharati, S., H.E. Krokan, L. Kristiansen, M. Otterlei, and G. Slupphaug. 1998. Human mitochondrial uracil-DNA glycosylase preform (UNG1) is processed to two forms one of which is resistant to inhibition by AP sites. Nucleic Acids Res. 26:4953–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otterlei, M., E. Warbrick, T.A. Nagelhus, T. Haug, G. Slupphaug, M. Akbari, P.A. Aas, K. Steinsbekk, O. Bakke, and H.E. Krokan. 1999. Post-replicative base excision repair in replication foci. EMBO J. 18:3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagelhus, T.A., T. Haug, K.K. Singh, K.F. Keshav, F. Skorpen, M. Otterlei, S. Bharati, T. Lindmo, S. Benichou, R. Benarous, and H.E. Krokan. 1997. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J. Biol. Chem. 272:6561–6566. [DOI] [PubMed] [Google Scholar]
- 20.Nilsen, H., M. Otterlei, T. Haug, K. Solum, T.A. Nagelhus, F. Skorpen, and H.E. Krokan. 1997. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 25:750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otterlei, M., T. Haug, T.A. Nagelhus, G. Slupphaug, T. Lindmo, and H.E. Krokan. 1998. Nuclear and mitochondrial splice forms of human uracil-DNA glycosylase contain a complex nuclear localisation signal and a strong classical mitochondrial localisation signal, respectively. Nucleic Acids Res. 26:4611–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beger, R.D., S. Balasubramanian, S.E. Bennett, D.W. Mosbaugh, and P.H. Bolton. 1995. Tertiary structure of uracil-DNA glycosylase inhibitor protein. J. Biol. Chem. 270:16840–16847. [DOI] [PubMed] [Google Scholar]
- 23.Kavli, B., G. Slupphaug, C.D. Mol, A.S. Arvai, S.B. Peterson, J.A. Tainer, and H.E. Krokan. 1996. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- 24.Mol, C.D., A.S. Arvai, G. Slupphaug, B. Kavli, I. Alseth, H.E. Krokan, and J.A. Tainer. 1995. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell. 80:869–878. [DOI] [PubMed] [Google Scholar]
- 25.Krokan, H., and C.U. Wittwer. 1981. Uracil DNa-glycosylase from HeLa cells: general properties, substrate specificity and effect of uracil analogs. Nucleic Acids Res. 9:2599–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaudhuri, J., C. Khuong, and F.W. Alt. 2004. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 430:992–998. [DOI] [PubMed] [Google Scholar]
- 27.Arudchandran, A., R.M. Bernstein, and E.E. Max. 2004. Single-stranded DNA breaks adjacent to cytosines occur during Ig gene class switch recombination. J. Immunol. 173:3223–3229. [DOI] [PubMed] [Google Scholar]
- 28.Xie, K., M.P. Sowden, G.S. Dance, A.T. Torelli, H.C. Smith, and J.E. Wedekind. 2004. The structure of a yeast RNA-editing deaminase provides insight into the fold and function of activation-induced deaminase and APOBEC-1. Proc. Natl. Acad. Sci. USA. 101:8114–8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen, H.M., and U. Storb. 2004. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc. Natl. Acad. Sci. USA. 101:12997–13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nambu, Y., M. Sugai, H. Gonda, C.G. Lee, T. Katakai, Y. Agata, Y. Yokota, and A. Shimizu. 2003. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 302:2137–2140. [DOI] [PubMed] [Google Scholar]
- 31.Pierce, A.J., J.M. Stark, F.D. Araujo, M.E. Moynahan, M. Berwick, and M. Jasin. 2001. Double-strand breaks and tumorigenesis. Trends Cell Biol. 11:S52–59. [DOI] [PubMed] [Google Scholar]
- 32.Lomax, M.E., S. Cunniffe, and P. O'Neill. 2004. Efficiency of repair of an abasic site within DNA clustered damage sites by mammalian cell nuclear extracts. Biochemistry. 43:11017–11026. [DOI] [PubMed] [Google Scholar]
- 33.Dzantiev, L., N. Constantin, J. Genschel, R.R. Iyer, P.M. Burgers, and P. Modrich. 2004. A defined human system that supports bidirectional mismatch-provoked excision. Mol. Cell. 15:31–41. [DOI] [PubMed] [Google Scholar]
- 34.de Laat, W.L., E. Appeldoorn, N.G. Jaspers, and J.H. Hoeijmakers. 1998. DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 273:7835–7842. [DOI] [PubMed] [Google Scholar]
- 35.Lan, L., T. Hayashi, R.M. Rabeya, S. Nakajima, S. Kanno, M. Takao, T. Matsunaga, M. Yoshino, M. Ichikawa, H. Riele, et al. 2004. Functional and physical interactions between ERCC1 and MSH2 complexes for resistance to cis-diamminedichloroplatinum(II) in mammalian cells. DNA Repair (Amst.). 3:135–143. [DOI] [PubMed] [Google Scholar]
- 36.Schrader, C.E., J. Vardo, E. Linehan, M.Z. Twarog, L.J. Niedernhofer, J.H. Hoeijmakers, and J. Stavnezer. 2004. Deletion of the nucleotide excision repair gene Ercc1 reduces immunoglobulin class switching and alters mutations near switch recombination junctions. J. Exp. Med. 200:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan-Hammarstrom, Q., S. Dai, Y. Zhao, I.F. van Dijk-Hard, R.A. Gatti, A.L. Borresen-Dale, and L. Hammarstrom. 2003. ATM is not required in somatic hypermutation of VH, but is involved in the introduction of mutations in the switch mu region. J. Immunol. 170:3707–3716. [DOI] [PubMed] [Google Scholar]
- 38.Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward, I.M., B. Reina-San-Martin, A. Olaru, K. Minn, K. Tamada, J.S. Lau, M. Cascalho, L. Chen, A. Nussenzweig, F. Livak, et al. 2004. 53BP1 is required for class switch recombination. J. Cell Biol. 165:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casellas, R., A. Nussenzweig, R. Wuerffel, R. Pelanda, A. Reichlin, H. Suh, X.F. Qin, E. Besmer, A. Kenter, K. Rajewsky, and M.C. Nussenzweig. 1998. Ku80 is required for immunoglobulin isotype switching. EMBO J. 17:2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manis, J.P., D. Dudley, L. Kaylor, and F.W. Alt. 2002. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 16:607–617. [DOI] [PubMed] [Google Scholar]
- 42.Lahdesmaki, A., A.M. Taylor, K.H. Chrzanowska, and Q. Pan-Hammarstrom. 2004. Delineation of the role of the Mre11 complex in class switch recombination. J. Biol. Chem. 279:16479–16487. [DOI] [PubMed] [Google Scholar]
- 43.Wittwer, C.U., G. Bauw, and H.E. Krokan. 1989. Purification and determination of the NH2-terminal amino acid sequence of uracil-DNA glycosylase from human placenta. Biochemistry. 28:780–784. [DOI] [PubMed] [Google Scholar]
- 44.Scaramozzino, N., G. Sanz, J.M. Crance, M. Saparbaev, R. Drillien, J. Laval, B. Kavli, and D. Garin. 2003. Characterisation of the substrate specificity of homogeneous vaccinia virus uracil-DNA glycosylase. Nucleic Acids Res. 31:4950–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothwell, D.G., B. Hang, M.A. Gorman, P.S. Freemont, B. Singer, and I.D. Hickson. 2000. Substitution of Asp-210 in HAP1 (APE/Ref-1) eliminates endonuclease activity but stabilises substrate binding. Nucleic Acids Res. 28:2207–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nilsen, H., I. Rosewell, P. Robins, C.F. Skjelbred, S. Andersen, G. Slupphaug, G. Daly, H.E. Krokan, T. Lindahl, and D.E. Barnes. 2000. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell. 5:1059–1065. [DOI] [PubMed] [Google Scholar]