

Abstract
Receptors for the Fc portion of immunoglobulin (Ig)G (FcγR) mediate phagocytosis of IgG-opsonized particles by a process that can be divided into four major steps: receptor–ligand binding, pseudopod extension, internalization, and lysosomal fusion. We have expressed single classes of FcγR in COS fibroblasts to examine the structural determinants necessary to complete the four steps of phagocytosis. Using phase contrast, fluorescence, confocal, and electron microscopy we have demonstrated that FcγR-expressing COS cells can phagocytose in a manner similar to that of professional phagocytes. We have further analyzed the capacity of the three classes of FcγR to phagocytose, placing special emphasis on the FcγRIA–γ chain complex, which allowed us to examine independently the roles of the ligand-binding unit (FcγRIA) and the signaling unit (γ chain). We found that receptor complexes containing a conserved tyrosine activation motif (ITAM), as found in the cytoplasmic domain of FcγRIIA and in the γ chain associated with FcγRIA and FcγRIIIA, readily internalized target particles. In contrast, FcγRIA alone, having no ITAM, was unable to internalize target particles efficiently, but did mediate pseudopod extension. Cotransfection of γ chain with FcγRIA restored the ability of the receptor to internalize target particles. A mutant FcγRIA in which the cytoplasmic domain had been deleted was also capable of mediating pseudopod extension, showing that neither the γ chain nor the cytoplasmic domain of FcγRIA were required for this step. Cytochalasin D, an inhibitor of actin polymerization, blocked particle internalization by all FcγR, but did not block pseudopod extension. Staining the FcγRIA COS cells for F-actin and for tyrosine phosphoproteins, we found that actin did not polymerize during FcγRIA-mediated pseudopod extension, nor were tyrosine kinases activated. Our data suggest that pseudopod extension and internalization are functionally distinct steps mediated through different pathways.
Fc receptor for IgG (FcγR)-mediated phagocytosis is the process whereby IgG-coated particulate pathogens are internalized and moved to lysosomes for degradation. The mechanism requires fine coordination of the action of a variety of molecules including cell membrane receptors, signaling molecules, and cytoskeleton components. The receptors initiate the process by a zipper mechanism whereby sequential interaction of particle-bound IgG with phagocyte FcγR stimulates the spreading of pseudopods around the particle resulting in its eventual engulfment (1). Complete circumferential coating of the target particle by ligand is required, and FcγR additional to those involved in the initial binding of the particle are recruited during the process (2).
Several characteristics of the intracellular mechanism of FcγR-mediated phagocytosis have been described. Engulfment of attached particles requires actin polymerization as demonstrated by the blockade of phagocytosis by inhibitors of actin polymerization such as cytochalasin D. Along with F-actin, tyrosine phosphoproteins accumulate beneath attached IgG-coated particles before FcγR-mediated internalization. Both F-actin accumulation and particle internalization can be blocked by tyrosine kinase inhibitors, indicating that the signal pathway requires tyrosine kinases (3). Indeed, several nonreceptor tyrosine kinases of the src and syk family have been coisolated with FcγR, and their activities have been shown to increase after FcγR clustering (4–6). Other signaling molecules such as protein kinase C and phosphoinositide-3 kinase have also been implicated in FcγR-mediated phagocytosis (7, 8).
FcγR comprise a family of integral membrane glycoproteins with three standard components, i.e., an extracellular ligand-binding portion consisting of two or three Ig-like domains, a short hydrophobic transmembrane region, and a cytoplasmic tail. In humans, three classes (I, II, and III) of these receptors have been characterized, differing in fine structure and affinity for IgG, and within these classes a total of eight subclasses (A, B, C) exist, each encoded by a separate gene (9). Of the eight FcγR in humans, three have convincingly been shown to mediate phagocytosis (FcγRIA, FcγRIIA, and FcγRIIIA; reference 10). Two of these three associate noncovalently with a common subunit, γ chain. A common amino acid sequence is present in the γ chain associating with FcγRIA and FcγRIIIA and in the cytoplasmic tail of FcγRIIA. This conserved motif, designated immunoreceptor tyrosine activation motif (ITAM)1, links receptor clustering to the activation of tyrosine kinases (11– 13). Upon FcγR clustering, the tyrosines of the ITAM are phosphorylated (6, 14, 15) and, in turn, serve as docking sites for signaling proteins containing src homology 2 domains (16, 17).
Analyzing the phagocytic mechanism of a single FcγR class has been difficult because monocytes and macrophages generally express multiple classes of FcγR. Therefore, structure–function studies of individual receptor classes have required expression of single FcγR classes into FcγR negative cells such as COS cell fibroblasts. This approach has allowed analysis of the phagocytic capacities of each receptor, the identification of receptor-associated molecules required for function (12, 18–20), and of receptor structural sequences necessary for phagocytosis (21–24).
However, we have questioned two aspects of this model; first, whether phagocytosis by COS cells mimics the process as it occurs in a professional phagocyte, and second whether phase contrast and fluorescence microscopy alone are adequate indicators of bona fide phagocytosis. Therefore, before embarking intently upon the study of the COS cell model of phagocytosis, we critically scrutinized in these cells the four major steps of FcγR-mediated phagocytosis: receptor–ligand binding, pseudopod extension, internalization, and fusion of the phagosome with lysosomes, with four microscopic methods: phase contrast, fluorescence, confocal, and electron microscopy. Here we show that COS cells indeed complete these four steps in a manner morphologically similar to professional phagocytes, and we validate that phase contrast and fluorescence microscopy are reasonably accurate methods for estimating phagocytosis.
We then focused on the signaling complex of FcγRIA with the γ chain to examine independently the effects of the ligand binding unit, FcγRIA, and the signaling unit, γ chain, in driving pseudopod extension and internalization. We found that although efficient particle internalization by the receptor complex requires the γ chain, pseudopod extension is mediated in the absence of γ chain and by a tailless FcγRIA. Moreover, pseudopod extension mediated by FcγRIA is not blocked by cytochalasin D, and F-actin does not polymerize nor do phosphotyrosine residues accumulate adjacent to pseudopod extensions. These data suggest that these two phases of phagocytosis, pseudopod extension and internalization, are functionally distinct steps mediated by different mechanisms.
Materials and Methods
Cells and Cultures.
COS-7 cells (American Type Culture Collection, Rockville, MD) were maintained in DMEM (GIBCO BRL, Gaithersburg, MD) supplemented with 10% fetal calf serum (Hyclone, Logan, UT), 2 mM l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The THP-1 monocytic cell line (gift from Dr. Paul Guyre, Dartmouth University, Lebanon, NH) was grown in RPMI 1640 supplemented as above with the addition of 2-mercaptoethanol at 5.5 × 10−2 mM. 48 h before experiments with THP-1 cells, 100 U/ml recombinant human interferon γ (Genentech, Inc., San Francisco, CA) was added to the culture medium to enhance FcγRI expression.
cDNAs and Transfection of COS-7 Cells.
The FcγRIA cDNA (25) was cloned into the pCDM expression vector for transfection into COS-7 cells. The human FcεRI–γ chain cDNA cloned into the expression vector pSVL was a gift from Dr. J.-P. Kinet (Harvard University, Cambridge, MA). The FcγRIIA cDNA cloned in the expression vector pCEXV3 and the FcγRIIIA cDNA were provided by Dr. J. Ravetch (Rockefeller University, New York). FcγRIIIA cDNA was further cloned into the expression vector pCDNA-1 (Invitrogen, San Diego, CA). The FcγRIA tailless mutant was constructed by deletion of the 3′ end of the cDNA corresponding to the 61 amino acids from the COOH terminus of the protein (26). COS-7 cells were transfected with 3 μg of plasmid DNA/10-cm dish by the diethyaminoethyl-dextran method as described previously (12) and analyzed for receptor expression and phagocytosis after 2 d.
Preparation of Phagocytic Targets.
Sheep RBCs, (Colorado Serum, Denver, CO) were washed three times in PBS (145 mM NaCl, 20 mM phosphate buffer, pH 7.4) to remove serum, and labeled by incubation with 0.1 mg/ml FITC in PBS overnight at 4°C. After four washes with PBS, FITC-labeled RBCs were incubated with a subagglutinating dose of rabbit anti–sheep RBC IgG (Diamedix, Miami, FL) at 37°C for 1 h. The RBCs were washed four times to remove excess IgG and were then used in phagocytosis assays.
Fluorescent latex beads, 2 μm in diameter, with a carboxylate coupled surface (Molecular Probes, Eugene, OR), were covalently coated with streptavidin (Pierce Chemical Co., Rockford, IL) using a carbodiimide procedure as specified by Molecular Probes. Purified human IgG (Cappel, Cochranville, PA) and BSA (GIBCO BRL) were biotinylated using NHS-LC-biotin (Pierce Chemical Co.) as specified by the manufacturer. The streptavidin-coated latex beads were mixed with either biotinylated IgG or BSA in PBS and incubated for 4 h. The beads were then washed four times in PBS to remove unbound protein, resuspended at 1% solids in PBS with 1 mg/ml BSA, and stored at 4°C until use. Nonfluorescent beads, 2 μm in diameter, (Polysciences, Warrington, PA) with a carboxylate-coupled surface were coated with human IgG or BSA using a carbodiimide procedure as specified by the manufacturer.
Phagocytosis Assay: Phase Contrast and Fluorescence Microscopy.
1 d after transfection, COS cells were seeded and grown overnight on glass coverslips in 24-well plates for analysis of phagocytosis. In other experiments, cells were grown on tissue culture plates for 2 d and then removed by trypsinization for analysis in suspension. FITC-labeled RBCs opsonized with IgG were gently pelleted onto the COS cells in culture medium by low speed centrifugation for 3 min. To study binding of RBCs to COS transfectants, cells were incubated together at 4°C for 1 h and were then washed in PBS and fixed with 1% paraformaldehyde in PBS. 300 cells were counted per condition by phase microscopy and the percentage of cells binding three or more RBCs was defined as rosetting activity. To study phagocytosis, cells were incubated with RBCs at 37°C for 1 h. The cells were then washed in PBS and subjected to a 45-s hypotonic shock to lyse externally bound RBCs; this treatment does not lyse COS cells or internalized RBCs. The cells were fixed in 1% paraformaldehyde and stained for fluorescence microscopy with 20 μg/ml ethidium bromide in PBS for 20 s. The cells were then mounted in glycerin-PBS media on slides and examined by phase contrast or fluorescence microscopy with a Zeiss microscope. 300 cells were examined per condition and scored for the number of phagocytosed RBCs. The data were expressed as a phagocytic index defined as the number of RBCs internalized per 100 FcγR-expressing COS cells. Expression of receptor was estimated by rosetting activity from parallel samples incubated at 4°C. In experiments where cytochalasin D (Sigma Chemical Co., St. Louis, MO) was used, the inhibitor was added at 1 μg/ml just before the RBC target. Cell viability was scored by trypan blue exclusion before and after incubation with cytochalasin D.
Phagocytosis Assay: Confocal Microscopy.
Phagocytosis assays were carried out in suspension as described above. The cells were then fixed in 1% paraformaldehyde and labeled with rhodamine- conjugated wheat germ agglutinin (Pierce Chemical Co.) to mark both the COS plasma membrane and any externally bound RBCs. The cells were immobilized on coverslips using poly- l-lysine. Cells were examined by confocal microscopy (MRC 600, BioRad Labs., Hercules, CA) and optical sections were taken in series every 0.5 μm in two fluorescent channels through single cells. 100 cells were examined per condition and the number of RBCs internalized was scored. Three-dimensional reconstructions using two-color fluorescence were accomplished using a Silicon Graphics Iris workstation with Voxelview software (Vital Images, Fairfield, IA). Measurements between internalized RBCs and the COS membrane were done on three-dimensional reconstructions in three axes of the cell (x, y, and z) from each individual RBC internalized for 10 representative COS cells.
F-actin Staining: Confocal Microscopy.
Transfected COS cells were grown overnight on glass coverslips in 24-well plates before the addition of nonfluorescent IgG-coated beads. Cells were incubated at 37°C with the bead targets for varying periods of time ranging from 10 to 40 min, and were then washed two times in DMEM and fixed in 3% paraformaldehyde in PBS for 15 min at 4°C. The cells were then permeabilized with 0.001% Triton X-100 in PBS for 7 min at room temperature, washed three times in PBS, and stained with FITC-phalloidin (Molecular Probes) diluted 1:20 in PBS for 45 min at room temperature. The cells were washed three times in PBS and were mounted in Mowiol medium (Polysciences) and viewed by confocal microscopy. To determine whether F-actin polymerization was triggered by particle binding, the density of stain was quantified by measuring the pixel intensity in the area 4 μm subjacent to the perimeter of the bound bead. This value was compared to background cortical F-actin staining taken at several areas of the cell devoid of particle binding to establish a baseline intensity value. Comparison of the values obtained from bead binding to baseline levels yields a numerical fold increase value which reflects the net increase in F-actin triggered by bead binding for each cell examined. Morphological criteria were also used to judge whether bead binding triggered assembly of F-actin extensions around target beads.
Lipid Labeling.
To examine pseudopod extension in parallel with F-actin measurements, transfected COS cells were labeled with a sulfonated DiI derivative (Molecular Probes) that is soluble in aqueous solutions. Cells were incubated with IgG-coated beads for various time periods and were washed in DMEM and fixed with 3% paraformaldehyde in DMEM for 15 min at 4°C. The lipid portion of the cell membranes was then labeled by incubation with 10 μM DiI in DMEM at 37°C for 20 min. After labeling, the cells were washed three times in DMEM and were postfixed in 1% paraformaldehyde before mounting in Mowiol and analysis by confocal microscopy.
Antiphosphotyrosine Staining.
Transfected COS cells adherent to glass coverslips were challenged with IgG-opsonized RBCs or IgG-coated nonfluorescent beads for 7 to 15 min at 37°C. The cells were then washed two times in DMEM and were fixed in cold acetone for 6 min. After fixation, the cells were washed with DMEM and incubated with 0.1% BSA in DMEM for 15 min at 4°C. Antiphosphotyrosine antibody 4G10 (UBI, Lake Placid, NY) or PY-20 (Santa Cruz Biotechnology, Santa Cruz, CA) were incubated with the cells at 2 μg/ml for 1 h at 4°C. The cells were then washed four times and incubated with a secondary FITC-conjugated F(ab)′2 donkey anti–mouse IgG (Jackson Immunoresearch, West Grove, PA) diluted 1:100 adsorbed against human, rabbit, and sheep serum proteins to prevent cross-reactivity. After 1 h at 4°C, the cells were washed three times and were mounted in Mowiol for viewing by confocal microscopy.
Phagocytosis Assay: Electron Microscopy.
Transfected COS cells detached from plates by trypsinization, or THP-1 cells, were mixed with either IgG- or BSA-coated fluorescent latex beads at 4°C for 30 min to allow binding and to synchronize phagocytosis. In other experiments, IgG-opsonized RBCs were used as targets and cell sorting was omitted. To test the specificity of binding, IgG- or BSA-coated beads were incubated with THP-1 cells and analyzed by flow cytometry. Results indicated that 92% of the THP-1 cells bound IgG beads, whereas only 4% bound BSA beads, confirming specificity. Mock-transfected COS cells bound IgG- or BSA-coated beads at background levels ranging from 1 to 4%, and did not internalize any beads. In experiments where cytochalasin D was used, the inhibitor was added before addition of target beads. After 30 min, the cells were then centrifuged to a pellet and the medium containing excess beads was removed. Cells were resuspended in complete DMEM and incubated at 37°C for 1 h to allow phagocytosis to occur, and were then washed in PBS three times to remove serum and unbound beads. Cells were fixed with 1% glutaraldehyde (EM grade; Polysciences) in 0.1 M sodium cacodylate buffer, pH 7.3, at 4°C for 1 h. Fixed cells were washed twice in sodium cacodylate and in PBS, and stored at 4°C before sorting. The cells were then sorted based on their ability to bind or internalize the fluorescent latex beads with an Elite EPICS fluorescence activated cell sorter (Coulter, Hialeah, FL) and collected for further processing. Sorted cells were labeled with a wheat germ agglutinin–horseradish peroxidase (HRP) conjugate (Pierce Chemical Co.) to mark the external membrane surface and then washed and attached with 2.5% glutaraldehyde to 3-cm culture dishes that had been coated with 0.25% poly-l-lysine for 1 h. To form an electron-dense label at the external membrane surface, the wheat germ agglutinin-HRP–labeled cells were reacted with a 0.5 mg/ml diaminobenzidine (DAB) solution containing 0.01% hydrogen peroxide in Tris-HCl, pH 7.6, buffer for 20 min and processed as described previously (27). The cells were then fixed in 1% osmium tetroxide (Polysciences) in 0.1 M cacodylate buffer for 1 h, and then washed in cacodylate buffer before dehydration in a graded ethanol series and embedding in EPON epoxy resin. Thin sections were cut on a Reichart ultramicrotome, counterstained with uranyl acetate and lead citrate (Polysciences), and then viewed on an electron microscope (Philips EM 300; Philips, Eindhoven, The Netherlands) operated at 60 kV.
Acid Phosphatase Localization.
Transfected COS cells were incubated as described above with IgG-coated fluorescent latex beads for 1.5 h at 37°C. The cells were washed twice in PBS followed by cacodylate buffer and subsequently fixed in 1% glutaraldehyde with 5% sucrose in 0.1 M cacodylate buffer for 30 min at 4°C. After fixation, the cells were washed in cacodylate buffer and then sorted for their ability to bind or internalize the fluorescent latex beads on a fluorescence-activated cell sorter (Coulter). Sorted cells were washed twice in 0.1 M sodium acetate, pH 5.0, buffer with 5% sucrose, and then incubated in a cerium-based reaction media to detect acid phosphatase and processed as previously described (28). In brief, the reaction medium consisted of 2 mM cerium chloride (Alfa Products, Danvers, MA), 2 mM of β-glycerophosphate as substrate, and 0.005% Triton X-100 to permeabilize the cells, in 0.1 M sodium acetate buffer, pH 5.0, with 5% sucrose. Samples were incubated without substrate to assess specificity of the reaction. The cells were then fixed with 2% osmium tetroxide for 45 min at room temperature and processed for electron microscopy as described above. IgG-coated bead targets were scored to determine whether they colocalized with the electron-dense reaction product produced during the enzyme cytochemical reaction. Control samples in which substrate was not present showed no reaction product, confirming specificity.
Results
FcγR-transfected COS Cells Complete All Steps of Phagocytosis in a Manner Similar to Professional Phagocytes
Using four different microscopic approaches, we found that FcγR-transfected COS cells indeed phagocytose in a manner similar to professional phagocytes. FcγR-transfected COS cells were incubated with IgG-opsonized FITC-labeled sheep RBCs under phagocytosing conditions, were subjected to a brief hypotonic lysis to remove external RBCs, and were then fixed, stained, and viewed by phase contrast and fluorescence microscopy to assess phagocytosis. By phase contrast microscopy, the RBC targets appeared internalized, compressed, and compacted, suggesting that they were contained within vesicles exerting a restrictive force on the RBC shape (Fig. 1 A). By fluorescence microscopy, the RBC targets, resistant to hypotonic lysis, appeared inside vesicular structures within the COS FcγR transfectants (Fig. 1 B).
Figure 1.

Analysis of FcγR-mediated phagocytosis by phase contrast and confocal microscopy. FcγR-transfected COS cells were incubated with FITC-labeled sheep RBCs opsonized with IgG at 37°C for 1 h to allow internalization to occur. The cells were then subjected to a brief hypotonic lysis to remove externally bound RBCs and were fixed and stained to mark the COS plasma membrane. A and B illustrate a single FcγRIIA-expressing cell stained orange with ethidium bromide examined by both phase contrast and fluorescence microscopy that has internalized several RBC targets (arrows) into large vesicles. In C and D, confocal sections in two axes of the same cells show green RBC targets that appear within the COS membrane labeled with a WGA–rhodamine conjugate appearing red. C shows several sections in the z-axis, whereas D shows the y-axis of the same cell. The third axis, x, is similar to D and is not shown. Bar, 10 μm.
To precisely distinguish internalized from surface-bound RBCs that may have survived the hypotonic lysis, a two-color fluorescence confocal strategy was used, labeling the RBCs with FITC and the COS cells with wheat germ agglutinin–rhodamine to mark the plasma membrane. The resulting sectional images in three axes show internalized RBCs clearly within the COS cell borders (two axes are shown in Fig. 1, C and D). Three-dimensional reconstruction, allowing measurement of the distance between the edge of internalized RBCs and the COS plasma membrane, further confirmed the intracellular location of the phagocytosed RBCs. 40% of the RBC targets were ⩾1 μm inside the COS plasma membrane in all three axes, clearly indicating that they were internalized. For the remaining 60%, the absence of overlap between the two fluorescence signals strongly suggested that the targets were internalized, but the distance between the RBCs and COS membrane was within 0.5 μm in at least one axis. Because the 0.5 μm measurement approaches the limit of resolution of our technique, internalization was not unequivocally demonstrable for 60% of the targets.
To determine definitively whether internalized targets were sealed within phagosomes, we evaluated phagocytosed IgG-coated latex bead targets by electron microscopy after labeling the exposed COS cell membrane surface with a wheat germ agglutinin–HRP conjugate and DAB peroxide. Such surface labeling of external membranes differentiates between truly internalized beads and beads that are surrounded by COS membrane but are not completely sealed inside a phagosome. Although, as illustrated in Fig. 2 A, some beads are found inside the cell border by position, they are not sealed inside the cell as indicated by the presence of the DAB label around the beads. However, other beads are completely internalized by the cell as indicated by the lack of the DAB staining, illustrated in Fig. 2 B. Thus, COS FcγR transfectants are capable of internalizing IgG-opsonized targets into sealed phagosomes, much like professional phagocytes, but not all targets that appear internalized by phase, fluorescent, confocal, and electron microscopy are necessarily sealed within phagosomes.
Figure 2.
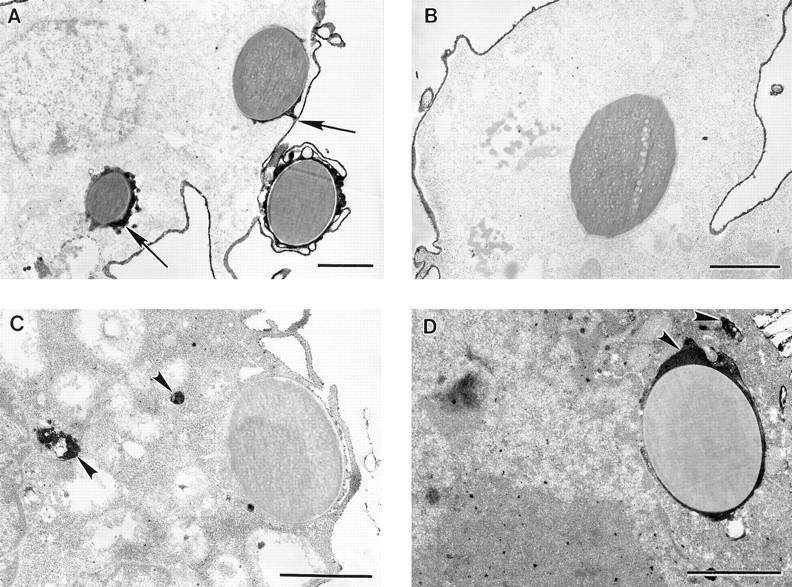
FcγR-transfected COS cells internalize targets into sealed phagosomes that subsequently fuse with lysosomes. Transfected COS cells were incubated with IgG-coated latex beads at 37°C to allow phagocytosis to occur. The cells were then fixed and processed for electron microscopy. In A and B, the plasma membrane was labeled with a WGA–HRP conjugate and reacted with a DAB peroxide solution resulting in an electron-dense band at the membrane surface. A illustrates target beads that have been bound but not sealed within the cell as indicated by the presence of the external membrane label around the beads (arrows). B shows a target bead that has been internalized within a sealed phagosome showing no membrane label, whereas the plasma membrane is densely stained. In C and D, the cells were processed to detect acid phosphatase activity, a lysosomal marker. C shows a bead target that has not been sealed within the cell and does not localize with acid phosphatase activity. D illustrates a bead target that has been internalized and does colocalize with the electron-dense reaction product of acid phosphatase (arrowheads), indicating lysosomal fusion. Bar, 1 μm.
To determine conclusively whether transfected COS cells could complete the final step of phagocytosis, fusion of phagosome with lysosome, we colocalized a lysosomal marker with target beads by electron microscopy. Transfected COS cells, after phagocytosing IgG-coated latex beads, were processed to detect acid phosphatase activity using an enzyme substrate and cerium metal to form an electron-dense precipitate at sites of enzyme activity. A majority of internalized bead targets, but not all, were found colocalizing with acid phosphatase (Fig. 2 D), demonstrating that FcγR COS transfectants sort targets to lysosomes to complete the process of phagocytosis.
Quantification of the Differential Ability of the Three FcγR to Mediate Phagocytosis in COS Cells
To examine whether each of the three classes of FcγR mediates phagocytosis, we transfected COS cells with the cDNA for each class of FcγR and analyzed the process of phagocytosis by phase contrast, fluorescence, confocal, and electron microscopy, quantifying the process at each stage.
FcγRIIA.
FcγRIIA COS transfectants were capable of phagocytosing IgG-opsonized targets, as shown by conventional fluorescence and confocal microscopy (Table 1), phagocytic indices (PIs) being similar by both methods, 277 ± 152 and 244 ± 81, respectively. Mock-transfected cells did not bind or phagocytose opsonized RBCs. Analysis of transfected cells by electron microscopy indicated that 22% (16 of 72 cells examined) of FcγRIIA expressing COS cells were capable of complete phagocytosis, i.e., capable of moving beads into sealed phagosomes showing no DAB stain.
Table 1.
Phagocytic Activity of the Three FcγR Classes Expressed in COS Cells
| COS transfectant | Phase and fluorescence | Confocal | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RA* | PI‡ | PI‡ | ||||||||
| Cytochalasin D | Cytochalasin D | |||||||||
| − | + | − | + | − | ||||||
| Mock | 0 | 0 | 0 | 0 | 0 | |||||
| FcγRIA | 20 ± 3 | 16 ± 1 | 61 ± 39 | 1 ± 1 | 28 ± 6 | |||||
| FcγRIA tailless | 18 ± 3 | 19 ± 2 | 40 ± 25 | 3 ± 1 | ND | |||||
| FcγRIA + γ chain | 10 ± 2 | 11 ± 4 | 270 ± 35 | 2 ± 1 | 135 ± 56 | |||||
| FcγRIA tailless + γ chain | 16 ± 4 | 15 ± 4 | 400 ± 133 | 4 ± 1 | ND | |||||
| FcγRIIA | 16 ± 2 | 16 ± 3 | 277 ± 152 | 3 ± 2 | 244 ± 81 | |||||
| FcγRIIIA + γ chain | 11 ± 2 | 9 ± 1 | 235 ± 68 | 0 | 178 ± 95 | |||||
COS cells were incubated in the presence or absence of cytochalasin D with FITC-labeled RBCs opsonized with IgG for 1 h and were subjected to a brief hypotonic lysis to remove external RBCs, and were then fixed. 300 cells were examined per condition by phase and fluorescence microscopy and were scored for the number of phagocytosed RBCs. Data represent the mean and SD of three experiments.
RA (rosetting activity) was determined from cells not subjected to hypotonic lysis and is defined as the percentage of cells that bound three or more RBCs.
PI is defined as the number of RBCs ingested by 100 FcγR-expressing COS cells.
Confocal PI is the PI derived from three experiments analyzed by confocal microscopy.
FcγRIIIA.
FcγRIIIA cotransfected with its associated subunit, γ chain, supported phagocytosis as determined by fluorescence and confocal microscopy, showing phagocytic indices of 235 ± 68 and 178 ± 95, respectively (Table 1). These values are similar to the results obtained with FcγRIIA (Table 1), indicating that both receptors mediate phagocytosis to a similar extent in the COS cell system.
FcγRIA.
On the other hand, while FcγRIA COS transfectants bound IgG-coated targets avidly both at 4°C and 37°C (not shown), they phagocytosed poorly, showing PIs of 28 ± 6 by confocal and 61 ± 39 by fluorescence microscopy (Table 1), clearly distinguishable from mock transfectants, which did not phagocytose at all. However, the efficiency of FcγRI-mediated phagocytosis was significantly increased when FcγRI was cotransfected with γ chain (PI of 270 ± 35), approaching levels achieved with FcγRIIA. Analysis by electron microscopy confirmed that only 3.6% (4 of 110 cells assessed) of FcγRIA-expressing cells were capable of phagocytosis, whereas COS cells cotransfected with both FcγRI and γ chain showed 27% (18 of 66 cells examined) of the transfectants to be phagocytosing beads. Using acid phosphatase activity as a lysosomal marker, we found that FcγRIA–γ chain cotransfectants localized threefold more target beads to lysosomes than FcγRIA alone (16 versus 5%).
To determine whether the cytoplasmic domain of FcγRIA collaborated with the γ chain to mediate phagocytosis, we used a tailless FcγRIA mutant cDNA. Expressing the tailless mutant alone in COS cells, we found avid binding of IgG opsonized targets, but minimal phagocytosis; the PI (40 ± 25) was similar to that observed with the intact FcγRIA (Table 1). However, cotransfection of the tailless receptor with γ chain greatly increased phagocytosis (PI of 400 ± 133, Table 1). Thus it would appear that the γ chain greatly enhances the FcγRI phagocytic signal and that the cytoplasmic domain of FcγRI is not required for this process.
Pseudopod Extension Is Independent of the Cytoplasmic Domain of FcγRIA
We analyzed by electron microscopy how FcγRIA and FcγRIIA direct the first three steps of phagocytosis, namely, (a) binding of ligand-coated beads to receptors on the cell surface, (b) pseudopod extension with formation of a phagocytic cup, and (c) internalization resulting in a sealed phagosome. The three phases, surface attachment, cup formation, and phagosome sealing, were defined (Table 2) by the position of the target beads relative to the COS cell plasma membrane identified by wheat germ agglutinin-peroxidase, and by the morphology of the surrounding structure. Using these criteria, we found that FcγRIA and FcγRIIA COS transfectants displayed a different distribution of cell-associated targets in these three phases of phagocytosis. FcγRIA COS cell transfectants showed minimal internalization of bound targets after 1 h (4%), but were capable of extending pseudopods around 52% of the targets (Table 2). Cotransfection of γ chain with FcγRIA increased internalization to 26%, yet the percentage of targets in the cupped phase remained high (63%) at the expense of surface-bound beads. In contrast, FcγRIIA COS cell transfectants internalized 20% of bound targets after 1 h, with only 5% of the targets remaining in the cupped phase, whereas the majority of targets (75%) were bound at the surface of the cells provoking no apparent pseudopod extension (Table 2). To determine if the cytoplasmic domain of FcγRIA was involved in directing pseudopod extension, we tested the mutant tailless receptor for the ability to induce cupping and found that it induced cupping of 48% of bound targets, similar to the 52% induced by the wild-type receptor (Table 2).
Table 2.
Location of Target Beads Relative to the COS Plasma Membrane after 1 h
| COS transfectant | Distribution of beads by location: % of total number | Total No. of beads associated with cells | ||||||
|---|---|---|---|---|---|---|---|---|
| Surface* | Cupped‡ | Sealed§ | ||||||
| FcγRIA | 44 | 52 | 4 | 125 | ||||
| FcγRIA tailless | 51 | 48 | 1 | 93 | ||||
| FcγRIA + γ chain | 11 | 63 | 26 | 96 | ||||
| FcγRIIA | 75 | 5 | 20 | 72 | ||||
COS cell transfectants were incubated with IgG-coated latex beads at 37°C for 1 h and were then fixed and processed for electron microscopy. The position of individual beads was determined relative to the labeled COS cell plasma membrane. The combined data from two experiments sort into three categories that define the position of the bead:
Surface: the bead is bound to the external membrane surface with little or no pseudopod advancement. The measured arc of contact between the COS cell membrane and bead was 90° ± 30° for 30 beads examined in this category.
Cupped: the bead is enveloped by pseudopods covering 50% or more of the bead circumference. The measured arc of contact between pseudopods and bead targets was 280° ± 60° for 30 beads examined in this category.
Sealed: the bead is sealed inside the cell as shown by a lack of external membrane staining around the bead.
To study the process of pseudopod extension in a professional phagocyte that naturally expresses FcγRIA, γ chain, and FcγRIIA, the monocytic cell line THP-1 was used. The monocytic cells were processed in the same manner as the COS cells using identical IgG-coated bead targets and were examined by electron microscopy. The THP-1 cells internalized 64% of targets bound after 1 h, and extended pseudopods around 34% of the targets (n = 70).
We also examined the process of pseudopod extension by electron microscopy using IgG-opsonized RBC targets in both THP-1 cells and COS transfectants to rule out any effects due to the nature of the IgG-coated latex beads. Both cell types were processed identically after 45-min and 1-h incubations to allow a direct comparison of morphological differences between monocytic cells and COS fibroblasts transfected with FcγRIA. In both cell types, morphologically similar pseudopod extensions were observed extending around the RBC target particles (Fig. 3). These extensions did not follow the contours of the RBC targets as tightly as those observed with the latex bead targets in both cell types. This slight difference may reflect the rigid nature of the beads and the possibility that the RBC targets may shrink during the electron microscopy fixation procedures, whereas the solid beads do not. We did not quantify the samples in which RBCs were used as targets since COS transfection efficiencies ranged from 20 to 30%, making analysis by electron microscopy very difficult. In experiments using fluorescent latex beads as targets, this problem was circumvented by sorting by flow cytometry only those cells that had bound beads. Regardless of whether IgG-opsonized RBCs or latex beads were used as target particles, pseudopod extensions were morphologically similar in both cell types.
Figure 3.
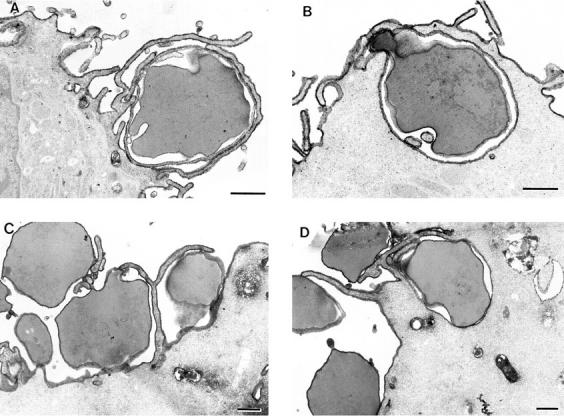
THP-1 monocytes and COS transfectants extend morphologically similar pseudopods. THP-1 monocytes (A and B) and FcγRIA COS transfectants (C and D) were incubated with IgG-opsonized sheep RBCs at 37°C for 45 min (A and C) or 1 h (B and D). The cells were then fixed, labeled with a WGA–HRP conjugate, and processed for electron microscopy as described in the legend to Fig. 2, A and B. In A, a THP-1 monocyte has extended fine pseudopods around an RBC target in an early phase of the phagocytic process. B shows a later phase of pseudopod extension in which the THP-1 cell has drawn the RBC target almost completely into the cell. In C, an FcγRIA COS transfectant has extended fine pseudopods around the RBC targets in an early phase of the process, whereas D shows a later terminal phase in which the RBC is surrounded by the cell, but not sealed within a phagosome. Bar, 1 μm.
The Role of Actin Polymerization in Phagocytosis
We examined the role of actin polymerization in the first three phases of phagocytosis by incubating COS cell transfectants with cytochalasin D. Cytochalasin D blocked internalization to near zero by all three classes of FcγR in COS cells, as measured by fluorescence microscopy (Table 1). Rosetting activity, however, which measures binding of IgG-opsonized targets, was unaffected (Table 1), and cells remained viable by the criterion of trypan blue exclusion (97% before, 96% after treatment). Remarkably, cytochalasin D had little or no effect on the ability of pseudopods to move around target particles, as evaluated by electron microscopy (Fig. 4). Although variations in the appearance of pseudopod extensions were common in both untreated (Fig. 4, A B) and cytochalasin D–treated samples (Fig. 4, C D), movement of the extensions around target particles was unaffected. We directly measured the quantitative extent of pseudopod formation in both untreated and cytochalasin D–treated cells expressing FcγRIA and FcγRIA γ chain, as illustrated in Fig. 4 D. The extent of pseudopod formation was measured in degrees as the arc of the bead target covered by pseudopods. Measurements of 50 beads showing pseudopod formation in both untreated and cytochalasin D–treated samples gave a mean and standard deviation of 270° ± 60° and 240° ± 70°, respectively, indicating no significant difference in the degree of pseudopod extension.
Figure 4.
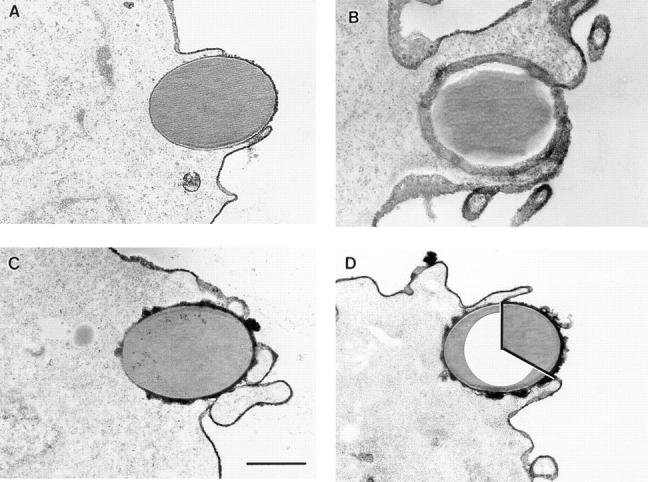
Electron micrographs of FcγRIA (A–C) and FcγRIA–γ chain (D) COS transfectants engaged in the cupping phase of phagocytosis. Cells were incubated with or without cytochalasin D for 1 h at 37°C and were then processed for electron microscopy. The cupping phase is observed in the absence (A and B) and presence (C and D) of cytochalasin D. Variations in morphology of the pseudopods were common, as illustrated in A and B in which both examples were not treated with cytochalasin D. D illustrates the method used to measure the extent of pseudopod formation. The circumference of the bead covered by pseudopods is measured in degrees, indicated by the inset white area. Bar, 1 μm.
In analyzing the qualitative effect of cytochalasin D on the phase of pseudopod extension, we determined the percentage of targets found in the cupped phase with and without treatment. COS cells transfected with FcγRIA alone displayed pseudopod extension in 63% of bound targets without cytochalasin D and 54% in the presence of the inhibitor (Table 3). Likewise, FcγRIA–γ chain cotransfectants exhibited 59% of bound targets cupped without inhibitor and 62% with cytochalasin D treatment. Consistent with results assessed by fluorescence microscopy, electron microscopy showed that cytochalasin D treatment blocked internalization from 30% down to 3% of cell-associated beads in the FcγRIA–γ chain cotransfectants (Table 3). FcγRIIA transfectants also showed a blockade of internalization from 20% down to 0% with cytochalasin D treatment, with an accompanying shift of targets to the cupped phase from 4% to 17% with treatment. Thus, in COS cells transfected with either the FcγRIA complex or FcγRIIA, cytochalasin D blocked internalization but did not block pseudopod extension.
Table 3.
Cupping Is Not Inhibited by Cytochalasin D in COS Cells
| COS transfectant | Cytochalasin D +/− | Distribution of beads by location: % of total | Total No. of beads associated with cells | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Surface | Cupped | Sealed | ||||||||
| FcγRIA | − | 36 | 63 | 1.5 | 80 | |||||
| FcγRIA | + | 46 | 54 | 0 | 95 | |||||
| FcγRIA + γ chain | − | 11 | 59 | 30 | 148 | |||||
| FcγRIA + γ chain | + | 35 | 62 | 3 | 107 | |||||
| FcγRIIA | − | 76 | 4 | 20 | 25 | |||||
| FcγRIIA | + | 83 | 17 | 0 | 53 | |||||
COS transfectants were incubated with IgG-coated latex beads with or without 1 μg/ml Cytochalasin D at 37°C for 1 h. Cells were processed as described in Materials and Methods, and were viewed by electron microscopy. The data sort into three categories, described in the legend to Table 2, that define the position of the bead relative to the COS cell membrane.
To determine whether pseudopod extension is necessarily associated with F-actin polymerization, we stained COS transfectants with FITC-phalloidin and analyzed by confocal microscopy both pseudopod extension and internalization during phagocytosis of IgG-coated beads. The density of actin filaments extending around the beads was quantified by measuring the pixel intensity of staining in a 4-μm area photometrically (Table 4). In COS cells expressing FcγRIA, only 1% of cell-associated beads showed subjacent accumulation of F-actin at 15 min of incubation. This same pattern was also observed at 40 min; only 1 of 241 beads was rimmed with polymerized F-actin. In contrast, 45% of beads associated with FcγRIIA cells were rimmed with F-actin at 15 min, with F-actin stain density being 9 ± 4-fold above background cortical F-actin density (Table 4). These differences are illustrated in Fig. 5 where F-actin bundles can be seen rimming beads in FcγRIIA-expressing cells (Fig. 5 A), whereas no such F-actin extensions are seen in FcγRIA-expressing cells (Fig. 5 C). To monitor pseudopod extension in this experiment, parallel samples were processed and fixed identically and then stained with a DiI derivative that labels lipid in membranes (Fig. 6). We were unable technically to combine lipid and F-actin staining in the same sample. Analysis by confocal microscopy revealed that FcγRIA-expressing COS cells showed vigorous pseudopod extension around attached beads (Fig. 6 C) similar to results observed by electron microscopy (Fig. 4). These data suggest that pseudopod extension mediated by FcγRIA does not require detectable F-actin polymerization.
Table 4.
Analysis of F-actin Polymerization following IgG-coated Bead Binding
| COS transfectant | Beads triggering F-actin | Total No. of bound beads | Fold increase in F-actin staining intensity* | |||||
|---|---|---|---|---|---|---|---|---|
| No. | % | |||||||
| FcγRIA | 6 | 1 | 432 | 3 ± 1 | ||||
| FcγRIIA | 104 | 45 | 230 | 9 ± 4 | ||||
COS cells were incubated with IgG-coated beads for 15 min at 37°C and were fixed and then stained with FITC-phalloidin to detect actin filaments. The cells were analyzed by confocal microscopy and F-actin staining subjacent to bound beads was analyzed for the presence of F-actin extensions around target beads. A 4-μm area beneath bound beads was also measured for F-actin staining intensity and was compared against background cortical F-actin of the same cell to determine a fold increase value. The data are derived from 35 cells for each condition from two experiments. The increase in F-actin staining intensity observed for FcγRIIA compared to FcγRIA is significant (P = 0.02).
Fold increase value derived only from beads which triggered F-actin accumulation.
Figure 5.
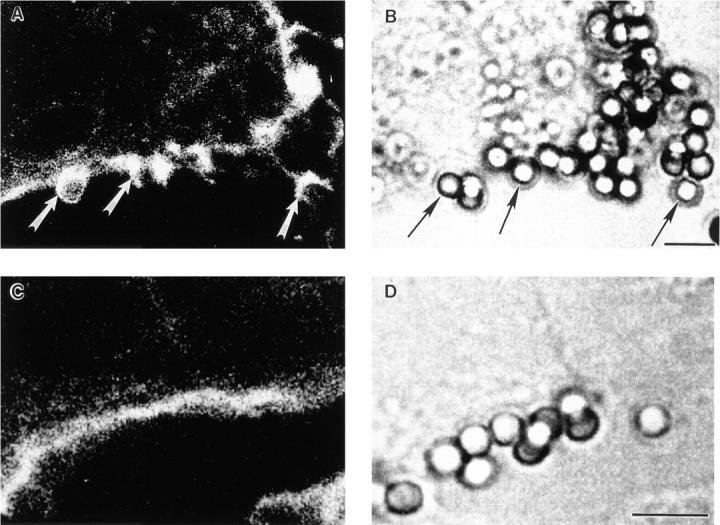
Confocal analysis of F-actin after IgG-coated particle binding to FcγR COS transfectants. Transfected COS cells were incubated with IgG-coated beads for 15 min at 37°C and were then fixed, permeabilized, and stained for F-actin using FITC-phalloidin. A and C show 0.5-μm–thick single confocal sections of F-actin staining, whereas B and D are companion nonconfocal transmitted light phase contrast sections. In A, an FcγRIIA transfectant shows F-actin extensions surrounding bound beads, whereas the parallel B provides the position of the beads. In C, an FcγRIA transfectant does not show an F-actin extensions or increases in F-actin due to target binding above background levels, whereas the parallel D shows the location of the beads. Cells were examined in all planes of focus to detect F-actin extensions as quantified in Table 4. Arrows, F-actin extensions in A, and associated beads in B. Bar, 5 μm.
Figure 6.
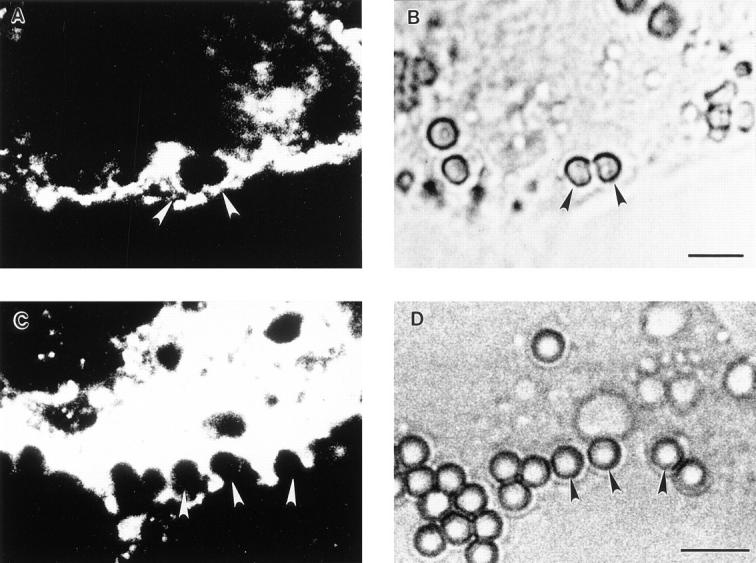
Analysis of pseudopod extension by confocal microscopy. Parallel samples of transfected COS cells from the experiment described in Fig. 5 were incubated with IgG-coated beads for 15 min at 37°C and were then fixed and stained with a DiI derivative to label the membranes of the cells. A and C show single confocal sections of DiI lipid staining, whereas B and D show companion nonconfocal phase contrast sections. In A and B, an FcγRIIA-transfected cell shows two beads with associated pseudopod extensions in the process of closure, whereas in C and D an FcγRIA-transfected cell shows several terminal pseudopod extensions around bound beads that do not fuse. Arrowheads, beads with associated pseudopod extensions. Bar, 5 μm.
FcγRIA-mediated Pseudopod Extension Is Not Associated with Tyrosine Phosphorylation
We next asked whether pseudopod extension is necessarily associated with the accumulation of tyrosine phosphoproteins beneath bound particles, assessing transfectants presented with either IgG-coated RBCs or beads. First, transfected COS cells were incubated with IgG-coated RBCs, fixed, stained with antiphosphotyrosine antibody, and then evaluated by confocal microscopy. We found no accumulation of tyrosine phosphoproteins around RBCs bound to 100 COS cells expressing FcγRIA (0/100) or the FcγRIA-tailless mutant (0/100). In contrast, of 100 FcγRIIA transfectants rosetting opsonized RBCs, 39 showed the accumulation of tyrosine phosphoproteins beneath bound targets (Fig. 7 A). Similar results were obtained using IgG-coated beads as targets. There was no accumulation of tyrosine phosphoprotein around beads bound to 25 FcγRIA-transfected cells (0%), despite our observation with the lipid stain that these beads are enveloped by pseudopods, whereas 9 out of 29 (31%) FcγRIIA-expressing cells showed beads with tyrosine phosphoprotein accumulation. These data would indicate that pseudopod extension need not be associated with the accumulation of tyrosine phosphoproteins.
Figure 7.

Antiphosphotyrosine staining of FcγR-transfected COS cells after IgG-opsonized particle binding. Transfected COS cells were incubated with IgG-opsonized RBCs for 10 min at 37°C and were then fixed and stained with antiphosphotyrosine mAb 4G10 followed by a secondary FITC-conjugated donkey anti–mouse IgG for detection. A and C show anti-phosphotyrosine staining, whereas B and D show companion nonconfocal phase contrast sections. In A, an FcγRIIA-expressing cell displays tyrosine phosphoprotein accumulation at sites of IgG-opsonized particle binding, indicating triggered tyrosine kinase activity. In B, several cells not expressing FcγR (n, nuclei) can be seen that show only background staining for phosphotyrosine in A, confirming specificity. C and D show an FcγRIA-expressing cell that has bound many IgG-opsonized RBCs, but does not show any phosphotyrosine staining above background levels. Bar, 10 μm.
Discussion
We derive three main points from the data presented in this study. First, we establish that FcγR-transfected COS cells phagocytose in a manner that appears similar both morphologically and biochemically to professional phagocytes. We have scrutinized the entire four-step process of phagocytosis using phase contrast, fluorescence, and confocal microscopy, and using electron microscopy we have applied two well-accepted criteria for phagocytosis, viz, external membrane staining to demonstrate internalization, and acid phosphatase colocalization to identify lysosome–phagosome fusion. All of these approaches converge to indicate that FcγR-transfected COS cells perform all four steps of phagocytosis, namely, receptor–ligand binding, pseudopod extension, particle internalization, and lysosome fusion in a manner that we cannot distinguish morphologically from the process observed in phagocytes.
Reconstitution of the FcγR-mediated phagocytic pathway in the COS cell, a simian kidney fibroblast, implies that the process in both phagocytes and fibroblasts may use common cellular components. Indeed, fibroblasts are considered nonprofessional phagocytes under certain conditions (29). For example, fibroblasts can phagocytose apoptotic neutrophils through a vitronectin receptor (30) and Con A–coated yeast by cross-linking of unidentified surface glycoproteins (31). Moreover, in murine macrophages, talin and paxillin, two fibroblast proteins associated with focal adhesion plaques, localize to phagocytic cups before internalization of IgG-opsonized targets (32, 33) suggesting the use of common pathways by the two cell types to mediate different functions. Another fibroblast cell line, the murine 3T3, has also been shown to mediate phagocytosis when transfected with FcγRIIA (34). However, not all fibroblast lines are capable of reconstituting FcγR-mediated phagocytosis; for instance, Chinese hamster ovary (CHO) cells transfected with FcγRIIA do not phagocytose (21), yet are capable of sorting the parasite Toxoplasma gondii to lysosomes after infection when expressing FcγRIIB (35). This suggests that although basic intracellular mechanisms can support phagocytosis, critical factors coupling FcγR with the phagocytic response may be missing in some cells. COS fibroblasts, however, appear to possess the necessary factors, and therefore may represent an appropriate model system for the analysis of the proximal steps of FcγR-mediated phagocytosis.
The second main point to be drawn from our data is that although previous studies of phagocytosis by FcγR-transfected COS cells (18–20, 36) have yielded valuable information, they have used methods that either assumed that particles appearing intracellular by phase contrast or fluorescence microscopy were indeed internalized, or failed to prove definitively that the targets were internalized. We have addressed and resolved these two issues. Quantitative measurements of COS cell phagocytosis by phase contrast, fluorescence, and confocal microscopy were similar when expressed as PIs. Confocal microscopy did not improve the results of phase and fluorescence in that 60% of the apparently internalized targets were so close to the COS plasma membrane as to be unresolvable. However, electron microscopy, with plasma membranes marked by wheat germ agglutinin-HRP, indicated that not all, but certainly the vast majority, of the apparently internalized beads were truly sealed from the exterior of the transfected COS cell. Likewise, colocalization of internalized beads and acid phosphatase by electron microscopy showed that most, but not all, bead-containing phagosomes had fused with lysosomes during the time course of the experiment. Thus, since our studies show that phase, fluorescence, or confocal microscopy come very close to approximating a true PI as determined by electron microscopy, we would conclude that the methodologic assumptions in these earlier studies are valid and that the results, therefore, are well grounded.
Our data support earlier studies showing that the ITAM is critical for phagocytosis (18, 22, 36). We show that efficient phagocytosis is mediated only by FcγR associated with the conserved tyrosine signaling motif, ITAM, implicated in signal transduction of several other immune system receptors (14, 37, 38). Others have shown that mutation of critical tyrosine residues in the ITAM of both γ chain and FcγRIIA block phagocytosis (22, 23, 39). Functionally, the ITAM provides a means of linking receptor clustering to tyrosine kinase activation required for FcγR-mediated phagocytosis (3). We have extended this observation by showing the accumulation of tyrosine phosphoproteins around opsonized particles being phagocytosed by FcγRIIA-transfected cells. Others have demonstrated that directly linking the tyrosine kinase syk to an FcγR extracellular domain results in a chimera capable of phagocytosis (40), reinforcing the central role of tyrosine kinase activity in directing phagocytosis.
The third and most novel aspect of our study is the use of the transfected complex of FcγRIA and γ chain to examine independently the effects of the ligand-binding unit, FcγRIA, and the signaling unit, γ chain, in driving pseudopod extension and internalization. We found that pseudopod extension and internalization could be uncoupled. COS cells expressing either FcγRIA alone or a tailless FcγRIA were capable of pseudopod extension, yet incapable of particle internalization. However, cotransfection of γ chain with FcγRIA to form an FcγRIA–γ chain complex increased dramatically the internalization of cell-associated particles while pseudopod extension remained essentially unchanged. Kinetic studies of phagocytosis >4 h (not shown) indicated that the percentage of phagocytic cells and the phagocytic index reached maximal levels at 30 min for FcγRIA and FcγRIIA COS transfectants, indicating that pseudopod extension was not an artifact of early sampling. Moreover, cupping by a monocytic cell line, THP-1, indicated that the phenomenon is not peculiar to transfected COS cells. Therefore, we would propose that the ITAM containing γ chain functions largely to mediate the internalization phase of phagocytosis, whereas pseudopod extension is primarily driven by the FcγRIA ligand-binding unit. In the case of FcγRIIA, where both the ITAM-signaling unit and the ligand-binding unit are contained in the same molecule, pseudopod extension and internalization are sequentially coupled events. At early time points, F-actin– rich pseudopods are observed by confocal microscopy (Fig. 5 A) extending around particles. As the process continues, these particles are internalized as observed by electron microscopy (Table 2) such that after 1 h, particles are either completely internalized or remain surface bound with no apparent pseudopod extension, indicating that the two steps are coupled for FcγRIIA.
The role of actin polymerization in pseudopod extension and particle internalization was examined using two approaches. First, we found that an inhibitor of actin polymerization, cytochalasin D, blocked internalization of IgG-coated targets by COS cells transfected with any of the three classes of FcγR, consistent with findings of others (18, 41). It was remarkable, however, that although cytochalasin D blocked internalization of particles, it did not block pseudopod extension mediated by either FcγRIA or FcγRIIA (Table 3). The effects of cytochalasin D on COS cell transfectants were seen in THP-1 cells as well, indicating that pseudopod extension in the presence of cytochalasin D is not cell-type specific. Second, we directly assayed F-actin polymerization in response to IgG–particle binding at several early time points by staining transfected cells with FITC-phalloidin and analyzing them by confocal microscopy. This approach revealed that FcγRIIA was capable of triggering F-actin polymerization, but FcγRIA was not (Table 4). Pseudopod extension around the particles was confirmed in parallel samples stained with the fluorescent DiI lipid label. Consideration of both the electron microscopy data and the F-actin confocal data for FcγRIA indicated that ∼50–60% of bound particles show pseudopod extension that is neither blocked by cytochalasin D nor associated with circumferential actin polymerization. The role of tyrosine kinases in the process of pseudopod extension and internalization was evaluated in similar fashion. We saw that tyrosine phosphoproteins were not found in the cups of FcγRIA-transfected cells, but were easily visible around particles being phagocytosed by FcγRIIA transfectants. These data suggest that pseudopods can extend in the absence of tyrosine kinase activity and subsequent F-actin polymerization.
Others have recently described the separation of pseudopod extension from internalization, underscoring the multistep nature of the phagocytic process. A recent study of macrophages has shown that the step from pseudopodia extension to fusion, resulting in particle internalization, requires phosphoinositide 3-kinase (PI3-K), but pseudopod extension itself is not blocked by PI3-K inhibitors (42). Similarly, a B lymphocyte line (DT40) transfected with either FcγRIIA or an FcγRIIIA–γ chain chimera manifests a block between pseudopod extension and internalization (43). These cells are able to form pseudopods and polymerize actin in response to an IgG-opsonized particle, yet are incapable of internalization, illustrating a further complexity in the stepwise mechanism of phagocytosis.
Work in another system has suggested that the two phases of phagocytosis, pseudopod extension and internalization, are functionally distinct. Studies in which the force of contraction in single granulocytes was measured during engulfment of large yeast pathogens found that the phagocytic response occurs as a sequential two-step process. During the first step, the phagocyte spreads rapidly over the yeast particle without contraction of the cell body. Then, when spreading has stopped, contraction force increases (44). The observed two-step process may reflect a functional division of the actions of a ligand-binding unit and a signaling unit of phagocytic receptors that is transmitted to the cell.
The simultaneous occurrence of F-actin accumulation and pseudopod extension in macrophages and B cells has been interpreted as indicating that F-actin polymerization drives pseudopod extension (3, 43). However, our findings would suggest that pseudopod extension is not necessarily dependent on the polymerization of F-actin. The formation of a pseudopod may result initially from the sequential interactions of receptor–ligand binding events that draw the cell membrane up around the particle. This process may only proceed to a certain degree without the support of the F-actin cytoskeleton, which is required to complete closure and internalize the particle. Whether this initial process reflects simply the receptor–ligand binding event or other possible signaling events that may be triggered through the receptor extracellular or transmembrane regions is not clear.
Methodological differences between our model and other models may explain why the separation of pseudopod extension from internalization has only recently been described. We and others (42) have used high-resolution techniques such as electron microscopy and DiI lipid labeling with confocal microscopy to observe fine membranous pseudopods, whereas earlier studies used phase contrast or fluorescence microscopy, which do not easily visualize these structures. The COS transfection system also allowed us to separate the effects of the ligand-binding unit from the signaling unit without the use of inhibitors that may have undesired effects on cellular functions.
This study provides further evidence for the functional separation of pseudopod extension from internalization during the process of phagocytosis. The case of the FcγRIA–γ chain complex highlights this point and raises several possibilities as to how pseudopod extension is driven. It will be of interest to examine how the ligand-binding unit of FcγRIA mediates pseudopod extension to gain insight into the various mechanisms at work during FcγR-mediated phagocytosis.
Acknowledgments
We thank Kathy Wolken and Ann Osterfeld for expert technical support with confocal and electron microscopy, and Wilson Burrows for assistance with computer imaging from the confocal microscope.
This work was supported by US Public Health Service award RO1-CA44983.
Footnotes
Abbreviations used in this paper: DAB, diaminobenzidine; FcγR, Fc receptor for IgG; HRP, horseradish peroxidase; ITAM, immunoreceptor tyrosine activation motif; PI, phagocytic index.
References
- 1.Griffin FM, Griffin JA, Leider JE, Silverstein SC. Studies on the mechanism of phagocytosis. I. Requirements for circumferential attachment of particle-bound ligands to specific receptors on the macrophage plasma membrane. J Exp Med. 1975;142:1263–1282. doi: 10.1084/jem.142.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffin FM, Jr, Griffin JA, Silverstein SC. Studies on the mechanism of phagocytosis. II. The interaction of macrophages with anti-immunoglobulin IgG-coated bone marrow–derived lymphocytes. J Exp Med. 1976;144:788–809. doi: 10.1084/jem.144.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg S, Chang P, Silverstein SC. Tyrosine phosphorylation is required for Fc receptor–mediated phagocytosis in mouse macrophages. J Exp Med. 1993;177:529–534. doi: 10.1084/jem.177.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salcedo TW, Kurosaki T, Kanakaraj P, Ravetch JP, Perussia B. Physical and functional association of p56lck with FcγRIIIA (CD 16) in natural killer cells. J Exp Med. 1993;177:1474–1480. doi: 10.1084/jem.177.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghazizadeh S, Bolen JB, Fleit HB. Physical and functional association of src-related protein tyrosine kinases with FcγRII in monocytic THP-1 cells. J Biol Chem. 1994;269:8878–8884. [PubMed] [Google Scholar]
- 6.Duchemin A-M, Ernst LK, Anderson CL. Clustering of the high affinity Fc receptor for immunoglobulin G (FcγRI) results in phosphorylation of its associated γ chain. J Biol Chem. 1994;269:12111–12117. [PubMed] [Google Scholar]
- 7.Zheleznyak A, Brown EJ. Immunoglobulin-mediated phagocytosis by human monocytes requires protein kinase C activation. J Biol Chem. 1992;267:12042–12048. [PubMed] [Google Scholar]
- 8.Ninomiya N, Hazeki K, Fukui Y, Seya T, Okada T, Hazeki O, Ui M. Involvement of phosphatidylinositol 3-kinase in Fcγ receptor signaling. J Biol Chem. 1994;269:22732–22737. [PubMed] [Google Scholar]
- 9.Ravetch, J.V., and C.L. Anderson. 1990. Fcγ receptor family: proteins, transcripts, and genes. In Fc Receptors and the Action of Antibodies. H. Metzger, editor. American Society for Microbiology, Washington DC. 211–235.
- 10.Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fcγ receptor classes on human leukocytes. J Exp Med. 1990;171:1333–1345. doi: 10.1084/jem.171.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reth MG. Antigen receptor tail clue. Nature. 1989;338:383. [PubMed] [Google Scholar]
- 12.Ernst LK, Duchemin A-M, Anderson CL. Association of the high affinity receptor for IgG (FcγRI) with the γ subunit of the IgE receptor. Proc Natl Acad Sci USA. 1993;90:6023–6027. doi: 10.1073/pnas.90.13.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ra C, Jouvin MHE, Blank U, Kinet JP. A macrophage Fcγ receptor and the mast cell receptor for IgE share an identical subunit. Nature. 1989;341:752–754. doi: 10.1038/341752a0. [DOI] [PubMed] [Google Scholar]
- 14.Paolini R, Jouvin MH, Kinet JP. Phosphorylation and dephosphorylation of the high-affinity receptor for immunoglobulin E immediately after receptor engagement and disengagement. Nature. 1991;353:855–858. doi: 10.1038/353855a0. [DOI] [PubMed] [Google Scholar]
- 15.Huang M-M, Indik Z, Brass LF, Hoxie JA, Schreiber AD, Brugge JS. Activation of Fc gamma RII induces tyrosine phosphorylation of multiple proteins including Fc gamma RII. J Biol Chem. 1992;267:5467–5473. [PubMed] [Google Scholar]
- 16.Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76:263–272. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 17.Chacko GW, Duchemin A-M, Coggeshall KM, Osborne JM, Brandt JT, Anderson CL. Clustering of the platelet Fcγ receptor induces noncovalent association with the tyrosine kinase p72 Syk. J Biol Chem. 1994;269:32435–32440. [PubMed] [Google Scholar]
- 18.Indik Z, Kelly C, Chien P, Levinson AI, Schreiber AD. Human FcγRII, in the absence of other Fcγ receptors, mediates a phagocytic signal. J Clin Invest. 1991;88:1766–1771. doi: 10.1172/JCI115496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J-G, Isaacs RE, Chien P, Schreiber AD. In the absence of other Fc receptors, FcγRIIIA transmits a phagocytic signal that requires the cytoplasmic domain of its γ subunit. J Clin Invest. 1993;92:1967–1973. doi: 10.1172/JCI116790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Indik Z, Hunter S, Huang MM, Pan XQ, Chien P, Kelly C, Levinson AI, Kimberly RP, Schreiber AD. The high affinity Fcγ receptor (CD64) induces phagocytosis in the absence of its cytoplasmic domain: the γ subunit of FcγRIIIA imparts phagocytic function to FcγRI. Exp Hematol (NY) 1994;22:599–606. [PubMed] [Google Scholar]
- 21.Odin JA, Edberg JC, Painter CJ, Kimberly RP, Unkeless JC. Regulation of phagocytosis and [Ca2+]iflux by distinct regions of an Fc receptor. Science. 1991;254:1785–1788. doi: 10.1126/science.1837175. [DOI] [PubMed] [Google Scholar]
- 22.Park J-G, Murray RK, Chien P, Darby C, Schreiber AD. Conserved cytoplasmic tyrosine residues of the γ subunit are required for a phagocytic signal mediated by FcγRIIIA. J Clin Invest. 1993;92:2073–2079. doi: 10.1172/JCI116804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell MA, Huang M-M, Chien P, Indik ZK, Pan XQ, Schreiber AD. Substitutions and deletions in the cytoplasmic domain of the phagocytic receptor FcγRIIA: effect on receptor tyrosine phosphorylation and phagocytosis. Blood. 1994;84:1753–1759. [PubMed] [Google Scholar]
- 24.Hutchinson MJ, Harrison PT, Floto RA, Allen JM. Fcγ receptor-mediated phagocytosis requires tyrosine kinase activity and is ligand independent. Eur J Immunol. 1995;25:481–487. doi: 10.1002/eji.1830250226. [DOI] [PubMed] [Google Scholar]
- 25.Ernst LK, van de Winkel JGJ, Chiu I-M, Anderson CL. Three genes for the human high affinity Fc receptor for IgG (FcγRI) encode four distinct transcription products. J Biol Chem. 1992;267:15692–15700. [PubMed] [Google Scholar]
- 26.Miller KL, Duchemin AM, Anderson CL. A novel role for the Fc Receptor γ subunit: enhancement of FcγR ligand affinity. J Exp Med. 1996;183:2227–2233. doi: 10.1084/jem.183.5.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simionescu N. Enzymatic tracers in the study of vascular permeability. J Histochem Cytochem. 1979;27:1120–1130. doi: 10.1177/27.8.90073. [DOI] [PubMed] [Google Scholar]
- 28.Robinson JM. Improved localization of intracellular sites of phosphatases using cerium and cell permeabilization. J Histochem Cytochem. 1985;33:749–754. doi: 10.1177/33.8.2991362. [DOI] [PubMed] [Google Scholar]
- 29.Rabinovitch M. Professional and non-professional phagocytes: an introduction. Trends Cell Biol. 1995;5:85–87. doi: 10.1016/s0962-8924(00)88955-2. [DOI] [PubMed] [Google Scholar]
- 30.Hall SE, Savill JS, Henson PM, Haslett C. Apoptotic neutrophils are phagocytosed by fibroblasts with participation of the fibroblast vitronectin receptor and involvement of a mannose/fucose-specific lectin. J Immunol. 1994;153:3218–3227. [PubMed] [Google Scholar]
- 31.Veras PS, de Chastellier C, Moreau MF, Villiers V, Thibon M, Mattei D, Rabinovitch M. Fusion between large phagocytic vesicles: targeting of yeast and other particulates to phagolysosomes that shelter the bacterium Coxiella burnetii or the protozoan Leishmania amazonensisin Chinese hamster ovary cells. J Cell Sci. 1994;107:3065–3076. doi: 10.1242/jcs.107.11.3065. [DOI] [PubMed] [Google Scholar]
- 32.Greenberg S, Burridge K, Silverstein SC. Colocalization of F-actin and talin during Fc receptor–mediated phagocytosis in mouse macrophages. J Exp Med. 1990;172:1853–1856. doi: 10.1084/jem.172.6.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greenberg S, Chang P, Silverstein SC. Tyrosine phosphorylation of the γ subunit of Fcγ receptors, p72syk, and paxillin during Fc receptor–mediated phagocytosis in macrophages. J Biol Chem. 1994;269:3897–3902. [PubMed] [Google Scholar]
- 34.Tuijnman WB, Cappel PJ, Van de Winkel JG. Human low affinity IgG receptor FcγRIIA (CD32) introduced into mouse fibroblasts mediates phagocytosis of sensitized erythrocytes. Blood. 1992;79:1651–1656. [PubMed] [Google Scholar]
- 35.Joiner KA, Fuhrman SA, Miettinen HM, Kasper LH, Mellman I. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor–transfected fibroblasts. Science. 1990;249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 36.Davis W, Harrison PT, Hutchinson MJ, Allen JM. Two distinct regions of FcγRI initiate separate signaling pathways involved in endocytosis and phagocytosis. EMBO (Eur Mol Biol Organ) J. 1995;14:432–441. doi: 10.1002/j.1460-2075.1995.tb07019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irving BA, Chan AC, Weiss A. Functional characterization of a signal transducing motif present in the T cell antigen receptor zeta chain. J Exp Med. 1993;177:1093–1103. doi: 10.1084/jem.177.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clark MR, Campbell KS, Kazlauskas A, Johnson SA, Hertz M, Potter TA, Pleiman C, Cambier JC. The B cell antigen receptor complex: association of Ig-α and Ig-β with distinct cytoplasmic effectors. Science. 1992;258:123–126. doi: 10.1126/science.1439759. [DOI] [PubMed] [Google Scholar]
- 39.Daeron M, Malbec O, Bonnerot C, Latour S, Segal DM, Fridman WH. Tyrosine containing activation motif–dependent phagocytosis in mast cells. J Immunol. 1994;152:783–792. [PubMed] [Google Scholar]
- 40.Greenberg S, Chang P, Wang DC, Xavier R, Seed B. Clustered syk tyrosine kinase domains trigger phagocytosis. Proc Natl Acad Sci USA. 1996;93:1103–1107. doi: 10.1073/pnas.93.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zigmond SH, Hirsch JG. Effects of cytochalasin B on polymorphonuclear leucocyte locomotion, phagocytosis and glycolysis. Exp Cell Res. 1972;73:383–393. doi: 10.1016/0014-4827(72)90062-6. [DOI] [PubMed] [Google Scholar]
- 42.Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3–kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox D, Chang P, Kurosaki T, Greenberg S. Syk tyrosine kinase is required for immunoreceptor tyrosine activation motif–dependent actin assembly. J Biol Chem. 1996;271:16597–16602. doi: 10.1074/jbc.271.28.16597. [DOI] [PubMed] [Google Scholar]
- 44.Evans E, Leung A, Zhelev D. Synchrony of cell spreading and contraction force as phagocytes engulf large pathogens. J Cell Biol. 1993;122:1295–1300. doi: 10.1083/jcb.122.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]