

Abstract
p52 is a subunit of nuclear factor (NF)-κB transcription factors, most closely related to p50. Previously, we have shown that p52, but not p50 homodimers can form transactivating complexes when associated with Bcl-3, an unusual member of the IκB family. To determine nonredundant physiologic roles of p52, we generated mice deficient in p52. Null mutant mice were impaired in their ability to generate antibodies to T-dependent antigens, consistent with an absence of B cell follicles and follicular dendritic cell networks in secondary lymphoid organs, and an inability to form germinal centers. Furthermore, the splenic marginal zone was disrupted. These phenotypes are largely overlapping with those observed in Bcl-3 knockout animals, but distinct from those of p50 knockouts, supporting the notion of a physiologically relevant complex of p52 homodimers and Bcl-3. Adoptive transfer experiments further suggest that such a complex may be critical in accessory cell functions during antigen-specific immune reactions. Possible roles of p52 and Bcl-3 are discussed that may underlie the oncogenic potential of these proteins, as evidenced by recurrent chromosomal translocations of their genes in lymphoid tumors.
The nuclear factor (NF)-κB transcription factors have been implicated in the induction of numerous genes in response to pathogen-derived or stress signals. In general, NF-κB factors lie dormant in the cytoplasm of cells until appropriate cellular stimulation results in their activation. The prototypical mechanisms of activation entails rapid proteolytic degradation of IκB proteins (including -α, -β, -ε), a family of closely related inhibitors of NF-κB, which avidly bind to the transcription factors to retain them in the cytoplasm (for general reviews see references 1–4). Their signal-induced degradation is mediated by first phosphorylation and then ubiquitination (5, 6). Upon proteolysis-aided release from the IκB inhibitors, NF-κB factors are subject to further phosphorylation-mediated control to enhance their activation potential in the nucleus (7, 8).
NF-κB factors are homo- or heterodimeric complexes composed of members of the NF-κB/Rel family of polypeptides, which in mammalian cells includes Rel (c-Rel), p65 (RelA), RelB, p50 (NF-κB1), and p52 (NF-κB2) (for general reviews see references 1–4). p50 and p52 are the two most closely related members. Although p50 is highly ubiquitous and usually quite abundant, p52 is expressed primarily in hematopoietic cells. Both proteins are generated from precursors by proteolytic processing; p52 derives from p100, and p50 from p105. As precursors, both proteins behave essentially like IκB inhibitors owing to the presence of ankyrin repeats characteristic of IκB family members. During processing, these inhibitory domains are degraded and the resulting p50 and p52 proteins become bona fide subunits of transactivating NF-κB heterodimers with Rel, p65, or RelB. Unlike these latter proteins, however, p50 and p52 do not carry identifiable transactivation domains and as homodimers do not transactivate by themselves; instead they usually function as inhibitors in various transfection assays, competing with transactivating complexes for κB sites. Nevertheless, the physiologic roles of p50 or p52 homodimers in the context of other transcription factors within endogenous promoters/enhancers can not be readily assessed.
The physiologic role of these homodimers is particularly intriguing, not least because the homodimers appear to be subject to regulation as well. Instead of by conventional IκB proteins (-α, -β, -ε), these homodimers are regulated by Bcl-3, an unusual member of the IκB family that is not degraded, but is induced in response to signals (9–16). In addition, unlike the typical cytoplasmic retention of complexes seen with conventional IκB proteins, Bcl-3, p50, p52 homodimers are readily observed in nuclei (2, 10, 11, 14, 16, 17). As shown previously by us, a ternary complex of DNA, p52 homodimers, and Bcl-3 can be demonstrated, which has transactivation potential due to transactivating domains on Bcl-3 (14). To what extent a similar transactivating complex involving p50 homodimers forms may depend on cell type and signals. In some assays, Bcl-3 rapidly disassociates p50 homodimers from DNA upon binding, but this is not true in all cases (10–16, 18, 19). To shed light on what may be the physiologic roles and mechanisms of Bcl-3 and p52, we generated knockout mice to identify potentially unique and critical functions of these proteins. Identification of critical functions may also help to elucidate the oncogenic potential of both proteins, as chromosomal translocations involving their respective genetic loci have been found in some lymphoid tumors (9, 20–23). Previously, p50 (24), IκB-α (25, 26), Rel (27), RelB (28, 29), and p65 (30, 31) have been knocked out, and various immunologic functions were shown to be impaired in the mutant mice. Recently we reported that Bcl-3 knockout mice are impaired with respect to several immunologic parameters (32). They are defective in generation of a switched isotype antibody response and in germinal center formation, which parallels loss of B cell follicular structures and follicular dendritic cell networks in secondary lymphoid organs. In addition they have a disrupted splenic marginal zone and they fail to mount a protective immune response to Toxoplasma gondii. Loss of germinal centers and impaired antibody responses have also been reported by others who further noted some impairment in handling of certain bacterial infection (33). We now show that p52 knockouts share many (but not all) phenotypes with Bcl-3 knockouts, supporting the idea that p52 and Bcl-3 form a functional, physiologically relevant complex. The data suggest a role for such a complex in accessory cell function during immune activation.
Materials and Methods
Targeted Disruption of the NF-κB2 Locus.
Two overlapping phage clones (LC60, ∼17 kb; and LC94, ∼17.5 kb) spanning a 31-kb genomic region containing the NF-κB2 locus were isolated from a 129SV mouse genomic library (Stratagene Corp., La Jolla, CA) and cloned into the NotI restriction site of Bluescript SK− (Stratagene Corp.). Inserts were then mapped for restriction sites and partially sequenced. For construction of the targeting vector, the 4.1-kb BamHI–SpeI fragment of BS-LC94 (containing exon 20 [8th ankyrin] and sequences further 3′) was inserted into the BamHI site of the pPNT vector (34), immediately 3′ to the neomycin (neo) gene and 5′ to the hsv-tk gene. Subsequently, the 1.8-kb BamHI fragment derived from BS-LC60 was inserted into the XhoI site (just 5′ of the neo gene; after filling in by Klenow reaction) to generate pPNT-P52KO. In this way exons 11 to 19, encoding for the dimerization domain and the first seven ankyrin repeats of p100, were replaced with the neo cassette. 50 μg of the targeting construct (pPNT-P52KO) were linearized with NotI and electroporated into 107 J1, embryonic stem (ES)1 cells (passage 12) as detailed elsewhere (35). Positive/negative selection was started 24 h after transfection using G418 (GIBCO BRL, Gaithersburg, MD; 350 μl/ml) and ganciclovir (2 μM). Screening of 76 G418/ganciclovir-resistant clones revealed two correctly recombined clones, and each was used to generate independent knockout lines (see Results).
Western Blot Analyses and Antibodies.
Whole-cell extracts were prepared as previously described (36) and proteins were separated by SDS-PAGE. Proteins were then blotted onto nitrocellulose membranes by using a semidry blotter (Bio Rad Labs., Hercules, CA), and analyzed with the enhanced chemiluminescence (ECL) Western blotting detection system (Amersham Corp., Arlington Heights, IL), as instructed by the manufacturer. The antibodies used were a rabbit antipeptide antibody directed against the first 14 amino acids after the initiating methionine of the murine p50 (37), a rabbit polyclonal antibody directed against amino acids 1–398 of the human p52 (38), a rabbit antipeptide antibody to the COOH terminus of the human p65 (39), a commercial rabbit antibody recognizing amino acids 152–176 of the murine c-Rel (Santa Cruz Biotechnology, Santa Cruz, CA) and a commercial rabbit anti–Rel-B COOH terminal peptide antibody (amino acids 540–558, Santa Cruz Biotechnology).
Flow Cytometric Analysis.
Three-color flow cytometric (FCM) analysis of single-cell suspensions of spleen and lymph nodes was performed with anti-B220 PE–, anti-IgD-FITC–, anti–CD3-FITC–, and anti-IgM-biotin–conjugated antibodies (PharMingen, San Diego, CA) followed by streptavidin–Red 670 (GIBCO BRL) as previously described (40).
Immunoglobulin Isotype-specific Inhibition ELISA.
NUNC Maxisorb Immuno-plates (Thomas Scientific, Swedesboro, NY) were coated with myeloma proteins representing various isotypes (Litton Bionetics, Inc., Charlestown, SC, or Organon Teknika, Durham, NC) in PBS, 100 μl/well, 0.5 μg/ml, overnight, and then blocked with 1% FCS-PBS-azide. In separate microtiter plates, serum samples to be tested were titrated in diluent (PBS, 0.1% Tween, 0.05% BSA, 0.02% sodium azide). Goat anti– mouse isotypes conjugated to alkaline phosphatase (Southern Biotechnology Assoc., Birmingham, AL) were added at dilutions predetermined by titration to be on the steep slope of the dilution curves. These mixtures were incubated for 1.5 h and then transferred to immunoglobulin-coated ELISA plates washed with PBS-Tween. Plates were incubated for 1 h, washed, and then incubated with substrate (1 mg/ml) p-nitrophenyl phosphate in 50 mM Na-bicarbonate buffer, 1 mM MgCl2, pH 9.8; Sigma Chemical Co., St. Louis, MO). Optical density was measured with Titertek MultiScan ELISA plate reader at 414 nm.
Preparation of TNP (2,4,6 trinitro-phenyl)-Conjugates and Injection Protocols.
TNP-KLH or TNP-BSA (bovine serum albumin) were prepared as previously described (41). In brief, 20 mg of lyophilized KLH (Pierce Chemical Co., Rockford, IL) or 4.8 mg of BSA (Pierce Chemical Co.) were dissolved in 4 ml of potassium borate buffer (0.25 M, pH 9.2), and 3 or 6 mg of TNBS (2,4,6 trinitro-benzyl sulfonic acid; Sigma Chemical Co.), respectively, were added along with 16 μl of sodium carbonate (1 M). The reaction was allowed to take place overnight, after which the protein derivatives were dialyzed against PBS, pH 7.4. TNP-protein derivatives were frozen at −20°C until use. TNP-LPS was purchased from Sigma Chemical Co., and TNP-Ficoll was a gift of Dr. John Inman (Laboratory of Immunology, National Institutes of Health). For the isotype-specific determination of anti-TNP antibodies, 5–30-wk-old mice of both sexes were injected intraperitoneally with 100 μg of TNP-KLH, 50 μg of TNP-LPS, or 25 μg of TNP-Ficoll. For cryosections, mice were injected intraperitoreally with TNP-KHL (100 μg) absorbed to alum, and spleens were collected 8 or 17 d after injection.
Assay for Anti–TNP-specific Antibodies.
Anti-TNP–specific antibodies were tested in sera of animals 24 h before and 14 d after injection of the TNP conjugates by isotype-specific ELISA. 96-well plates (Immunlon-4; Dynatech Laboratories, Inc., Vienna, VA) were coated overnight at 4°C with 50 μg/ml of TNP-BSA in PBS (pH 7.4) and then washed three times with PBS-Tween 20 (0.5%). Subsequently, serial dilutions of murine sera in PBS-Tween were added to the plates and incubated overnight at 4°C. After three washes in PBS-Tween, goat anti–murine IgM, IgG1, IgG2a, IgG2b, and IgG3 conjugated to horseradish peroxidase (HRP; 1:1,000; Southern Biotechnology Assoc.) were added, incubated for 1 h at room temperature, and washed three times with PBS-Tween. The HRP substrate ABTS (2,2′ azino-di [3-ethyl-benzothiazoline sulfonate], Kirkeguard & Perry Labs., Inc., Gaithersburg, MD) was finally added and left at room temperature in the dark. The reaction was stopped by addition of 1% SDS and OD measured at 405 nm.
Immunoperoxidase Staining of Paraffin-embedded Tissues.
Spleens were fixed in Bouin's fixative for 24 h, fixed, and transferred into 70% ethanol. Tissues were then processed through alcohols and xylene, embedded in paraffin, sectioned at 5 μm, and used for immunohistochemistry. Sections were stained with an anti–human CD3 rabbit polyclonal antibody (Dako Corp., Carpinteria, CA) or an anti–mouse CD45R/B220, biotin-conjugated rat monoclonal antibody (Boehringer Mannheim, Indianapolis, IN), as described previously (32). An anti–rabbit, biotin-conjugated secondary antibody was subsequently used for the CD3 stain (Vector Laboratories, Burlingame, CA). Slides were then incubated with streptavidin-conjugated HRP and the avidin–biotin complexes were revealed with the DAB (3,3′-diaminobenzidine) tetrahydrochloride chromogen (Sigma Chemical Co.).
Immunoperoxidase Staining of Frozen Sections.
Spleens were extracted, placed in OCT freezing medium (Miles Laboratories Inc., Elkhart, IN), and flash frozen. 10 μm acetone-fixed sections were stained as described previously (32, 42). In brief, tissue sections were rehydrated in PBS containing 0.1% BSA (fraction V, PBS/BSA; Sigma Chemical Co.), and then blocked for 30 min with 20% normal mouse serum and 20% goat or rabbit serum (same species as secondary antibody) in PBS (PBS/serum). After blocking, sections were incubated for 60 min with the primary antibody prepared in PBS/serum, washed in PBS/BSA, and then (except for those stained with biotinylated peanut agglutinin [PNA] and biotinylated anti-CD35 antibody) incubated for an additional 30 min with the biotinylated secondary antibody in PBS/serum. After quenching endogenous peroxidase activity, tissue sections were incubated for 30 min with streptavidin conjugated-HRP (Vector Laboratories), washed in PBS/BSA, and the avidin– biotin complexes were revealed with the DAB chromogen (Vector Laboratories), according to the manufacturer's instructions. Finally, slides were rinsed, counterstained with methyl green (Vector Laboratories) and permanently mounted with Permount (Fisher Scientific, Pittsburgh, PA). Biotinylated PNA (Pierce Chemical Co.) and the following primary antibodies were used: biotinylated-8C12 (anti-CD35; PharMingen), FDC-M1 (anti-FDCs; a gift of Dr. Kosco-Vilbois, Glaxo-Wellcome Research and Development S.A., Switzerland, and Dr. Burton, Virginia Commonwealth University, Richmond, VA), BM8 (anti–red pulp macrophages; BACHEM Bioscience Inc., Philadelphia, PA), MOMA-1 (anti-metallophilic macrophages [MMs]; BACHEM Bioscience Inc.), ERTR-9 (anti– marginal zone macrophages [MZMs]; BACHEM Bioscience Inc.).
Double Staining of Frozen Sections.
Double immunohistochemical staining was performed on acetone-fixed cryosections. In brief, tissue sections were rehydrated in PBS/BSA, blocked for 20 min with Dako® Protein Block (Dako Corp.), and subsequently incubated for 1 h with biotinylated anti-CD45RB220 antibodies (PharMingen) followed by a 30-min incubation with alkaline phosphatase (AP) streptavidin (Vector Laboratories). Sections were then incubated for 1 h with HRP-conjugated PNA (Dako Corp.) and endogenous peroxidase activity quenched with 1% H202 in PBS for 15 min. Washings were as described above. Finally, AP and HRP enzymatic activities were revealed with the Fast Red (Dako Corp.) and DAB (Vector Laboratories) chromogens, respectively, and specimens mounted in aqueous mounting medium (Dako Corp.).
Bone Marrow Adoptive Transfer.
107 bone marrow cells, isolated from femurs of 11-wk-old p52 (−/−), (+/−), and (+/+) donor mice, were injected intravenously into recombination activation gene 1 (RAG-1)–deficient mice (Jackson Laboratory, Bar Harbor, ME), which had been lethally irradiated at 900 Rads 24 h earlier. 15 wk later, adoptively transferred RAG-1–deficient mice were injected intraperitoneally with TNP-KLH (100 μg) to assay for serum levels of anti-TNP antibodies and bled before and 14 d after injection. For cryosections, mice were injected intraperitoneally with TNP-KLH (100 μg) adsorbed to alum and spleens were collected 9 d after injection.
Results
Generation of p52/p100–deficient Mice.
Mice deficient in expression of functional p52 or p100 protein were generated by targeted gene disruption in ES cells. Homologous recombination of the targeting vector resulted in loss of exons encoding a major part of the dimerization domain, the nuclear translocation sequence, and the first seven ankyrin repeats of the p52/p100 protein; these regions were replaced with a neo cassette (Fig. 1 A). Elimination of these regions assures that no functional protein can be generated, since an intact dimerization domain is essential for homo- or heterodimerization and DNA binding, and an intact ankyrin domain is essential for IκB-related inhibitory activities of p100 (for review see references 1–4). Two independently derived mutant ES clones with a disrupted p52/p100 gene (Fig. 1 B, left) were microinjected into C57Bl/6 blastocysts, and resulting male chimeras were bred to C57Bl/6 females to generate mice heterozygous for the mutated allele. Matings of these animals generated p52/p100 +/−, +/+, and −/− mice (Fig. 1 B, right). Both mutant ES lines transmitted their germlines, and all results reported here could be observed in either line.
Figure 1.

Targeted disruption of the p52/p100 gene. (A) The targeting vector, a partial restriction map of the p52/p100 gene locus, and the mutated allele after homologous recombination of the targeting vector are shown. Probes used and fragments detected in Southern blots are indicated. kb, kilobase pairs; tk, thymidine kinase; B, BamHI; Sm, SmaI; Sp, SpeI; X, XbaI. (B) Southern analyses of ESs (left) and of the offsprings from the resulting mice heterozygous for the mutated allele (PROGENY; right). Correct homologous recombination generated a new Xba I restriction fragment B (7.4 kb) in addition to the wild-type allele-derived fragment A (9.4 kb), and both were detected with probe α (3.4-kb BamHI– XbaI fragment of BS-LC94). Correct insertion of the targeting plasmid was confirmed with SpeI digestion of genomic ES cell DNA and probe β (1.1-kb SmaI fragment of BS-LC60) that detected fragments C and D (wild-type allele [10.0 kb] and mutated allele [7.1 kb], respectively). A neo probe was also used to exclude additional nonhomologous integration of the targeting vector (data not shown). Matings of heterozygous animals then generated p52/p100 (+/−), (+/+), and (−/−) mice that were discerned with probe γ (1.8-kb BamHI fragment of LC60). This probe hybridizes to the BamHI fragments E (wild-type, 1.8 kb) and F (mutant, 3.7 kb) (PROGENY). (C) Western blot analyses of spleens of p52/p100 (+/ +) and (−/−) mice are shown. Antibodies to various NF-κB family members were used as indicated.
Western analyses for expression of p52/p100 demonstrated the absence of p52 (and p100) in the −/− mutant mice, whereas expression of other members of the NF-κB family (p50/p105, p65 [RelA], c-Rel, RelB) appeared unperturbed (Fig. 1 C). As expected from this, all NF-κB complexes that did not contain p52 were readily detected in electrophoretic mobility shift assays of unstimulated and/or stimulated splenic cell extracts (data not shown). These data suggest that the absence of p52 and p100 did not obviously alter other NF-κB complexes. Mutant mice deficient in other components of this transcription factor family also do not show significant perturbations in the expression of the remaining family members (24–27, 29, 30, 33).
Reduction in the B to T Cell Ratio.
p52/p100–deficient animals (hereafter referred to as p52 deficient) were born with the expected frequency and did not exhibit obvious macroscopic defects. However, FCM analyses indicated a significant reduction of B220+ B cells in spleen and lymph nodes (analysis of spleen only shown in Fig. 2 A), accompanied by an increase in levels of CD3+ T cells (Fig. 2 A). Therefore, the ratio of B/T was dramatically reduced in the mutant mice. Of note, a further reduction of B cells was observed with more advanced age (data not shown). Among the B cells the reduction in numbers was equally apparent regardless of whether they were positive for IgM and/or IgD (Fig. 2 A).
Figure 2.


Reduction in the B/T cell ratio and reduced levels of total serum antibodies in p52/ p100 (−/−) mice. (A) Representative FCM analysis of splenocytes from 16-wk-old p52/ p100 (−/−) and (+/−) mice. IgM–Red 670 versus IgD-FITC two-color profiles are displayed in the top panels. Single-color profiles depict B220-PE and CD3-FITC staining on total splenocytes (middle and bottom, respectively). Numbers reflect the percentage of positively stained spleen cells. (B) Reduced levels of IgG1, IgG2b, and IgA in sera of p52/p100 (−/−) mice. Titers of total immunoglobulin isotypes from 7–10-wk-old mice of each genotype are shown as indicated. IgG1, IgG2a, and IgA titers differed significantly (P <0.002, P <0.05, and P <0.001) among the three groups (+/+, +/−, and −/−) by one-way analysis of variance. (−/− differed significantly from both other groups.) The reduction in levels of IgG3 observed in p52-deficient mice was not statistically significant (P = 0.079).
Impaired Antibody Responses.
The loss of B cell follicles prompted us to investigate for potential defects in antibody production. Although total serum levels of IgM and IgG2a appeared essentially normal in unchallenged mutant mice, levels of IgG1, IgG2b, and IgA were partially reduced (Fig. 2 B). (Apparent reduction in IgG3 was not statistically significant.) We then challenged animals with the T-dependent (TD) antigen TNP-KLH. Administration of this antigen to mutant mice (in the absence of adjuvants) failed to elicit significant levels of antigen-specific switched immunoglobulin isotypes, whereas littermate controls were able to raise such antibodies (Fig. 3 A). The mutant mice nevertheless recognized and responded to antigen, as evidenced by the even higher than normal antigen-specific IgM levels. It is noteworthy that heterozygous animals exhibited a partial reduction in antigen-specific switched Ig isotypes. Together, these data suggest that p52-deficient animals are impaired in generating full antibody responses to TD antigen (in the absence of adjuvants) and that loss of one p52 allele already has a measurable effect in this assay. Consistent with these results, mutant mice were also impaired in their antibody response to a physiologically relevant challenge. Infection with influenza virus, which presents as a TD antigen, resulted in impaired flu-specific IgG2a production (with IgG2a being the predominant flu-specific switched isotype produced in wild-type mice [43]; data not shown). Therefore, p52-dependent functions are important in this infectious model.
Figure 3.
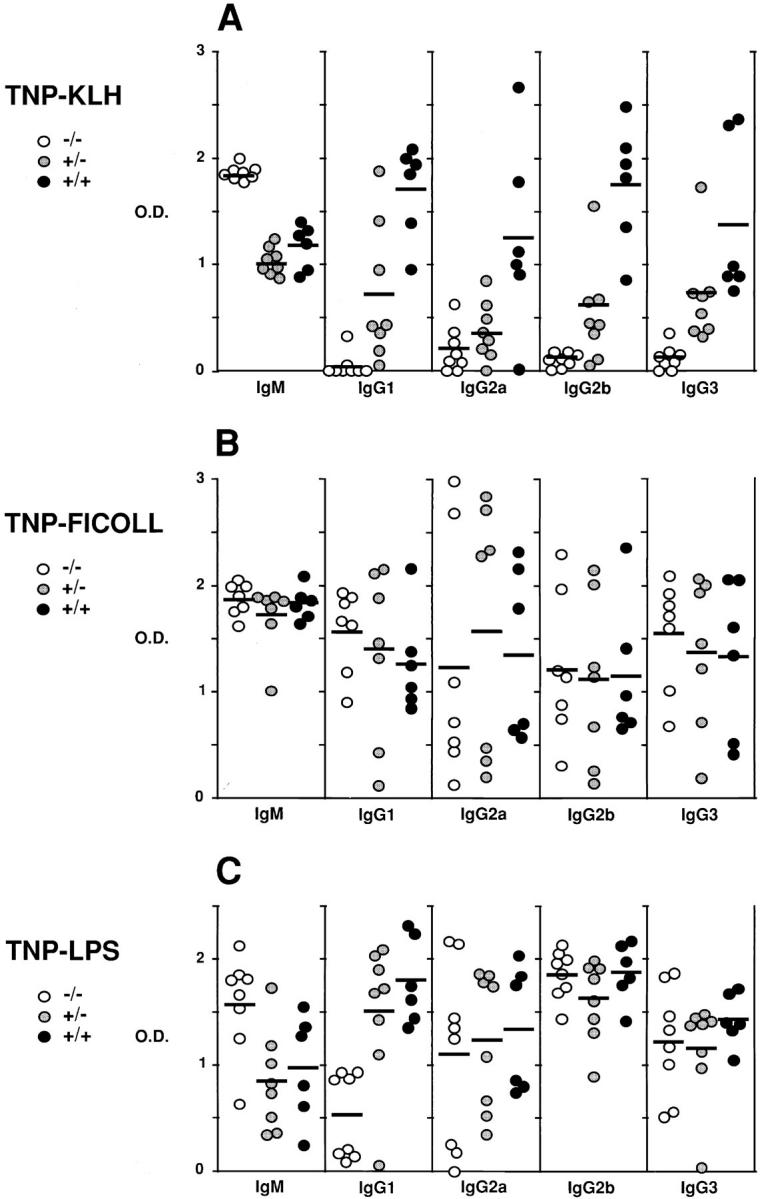
Defective antibody response to T-dependent antigens in p52-deficient mice. 5–30-wk-old mice of both sexes were injected intraperitoneally with (A) 100 μg of TNP (2,4,6 trinitro-phenyl)–KLH (TD), (B) 25 μg of TNP-Ficoll (TI-2) or (C) 50 μg of TNP-LPS (TI-1). Serum levels of anti-TNP specific antibodies were determined by isotype-specific ELISA 14 d after injection of the TNP conjugates. Antibody levels, expressed as OD, are shown for each genotype, as indicated. Anti-TNP antibodies were undetectable in sera of animals before challenge (data not shown). OD values differed significantly among the following groups of animals by one-way analysis of variance. (A) (−/−) versus (+/+) all isotypes tested; (−/−) versus (+/−), IgM, IgG1, IgG2b and IgG3; (+/−) versus (+/+), IgG1, IgG2a, IgG2b. (C) (−/−) versus (+/−) and (+/+), IgM, and IgG1. All other groups were not significantly different.
The mutant mice do not have absolute defects in the ability of B cells to undergo switch recombination or to properly respond to a TD antigen, since administration of TNP-KLH adsorbed to alum resulted in normal or near normal antibody responses (data not shown; the antibody levels of wild-type controls were at least one order of magnitude higher in the presence of adjuvant as compared to without adjuvant). The presence of an adjuvant appeared to have bypassed a critical defect in null mutant mice, a phenomenon that has been observed in other mutant mice (44).
We also investigated T-independent (TI) antigens. The response to TNP-Ficoll (TI-2) was normal in the mutant mice, confirming that there are no absolute defects with respect to the ability of B cells to undergo switching (Fig. 3 B). The response to TNP-LPS (TI-1) appeared normal as well, except with respect to the IgG1 isotype, which was partly reduced (Fig. 3 C).
In summary, these results are consistent with an impaired presentation/handling of antigen by APCs and/or impaired cellular signaling in response to TD antigen in p52-deficient animals, a defect which is overcome with stronger adjuvant-aided stimulation.
Loss of B Cell Follicles and Germinal Centers.
To characterize the reduction of B cells in more detail and to search for defects that may be consistent with problems in antigen presentation, handling, and/or associated cell signaling, we performed immunohistochemical analyses on paraffin-embedded splenic tissue sections of null mutant mice and their littermate controls (heterozygous littermates were generally indistinguishable from wild-type littermates). B220+ B cell follicles were severely reduced in p52-deficient animals as compared to littermate controls (Fig. 4; the B220 marker and the CD3 marker define B and complementary T cell zones, respectively, within the splenic white pulp islands). Although B cell follicular areas were depleted or absent, B cells were still present in the marginal zone, as evidenced by the “ring” of B220+ cells surrounding the white pulp within the spleen. Consistent with a lack of clearly defined B cell follicular areas, the mutant animals also did not contain germinal centers that normally form within B cell follicles in response to TD antigens. Such germinal centers could be found in littermate controls, probably in response to environmental antigens (Fig. 4, upper right). In contrast to p52-deficient animals, p50-deficient animals exhibited normal B cell follicles and germinal centers (Fig. 4, bottom), suggesting clearly distinct roles for the two proteins (see Discussion).
Figure 4.
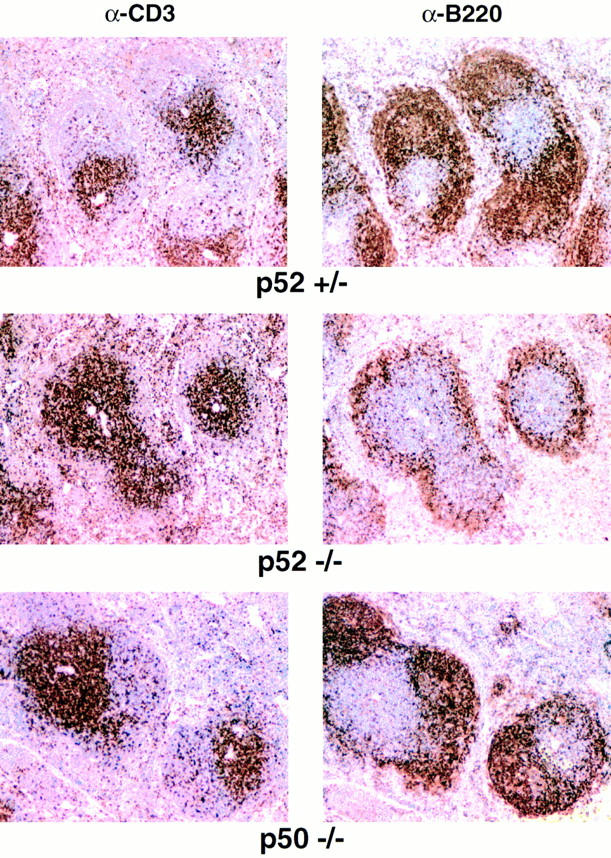
Germinal centers and B cell follicles are absent in spleens of p52 (−/−) mice, but are present in p50 (−/−) mice. Analyses of spleens taken from an unchallenged, 21-wk-old p52/p10 (−/−) mouse, a (+/−) littermate control (top and middle, respectively), and an unchallenged 15-wk-old p50/ p105 (−/−) mouse (bottom). Bouin-fixed, paraffin-embedded sections were processed with anti-CD3 or anti-B220 antibodies, as indicated.
The results obtained with p52-deficient mice are similar to those obtained with Bcl-3 null mice (32). In the latter animals, B cell follicular areas were generally diffuse and could not be clearly defined, the switched antibody response to influenza virus was defective, and germinal centers were largely missing. To better define the defects in p52-deficient mice and to allow a comparison with Bcl-3– deficient mice, we investigated the apparent loss of germinal centers and the overall splenic microarchitecture in more detail. Mice were challenged with TNP-KLH adsorbed to alum to stimulate a vigorous response. Splenic cryosections were stained with PNA, a marker for germinal center B cells. Although littermate controls contained large, intensely staining PNA positive regions within B cell follicles, the mutant mice did not (Fig. 5 A, top). Furthermore, anti-CD35 and FDCM1, two markers for follicular dendritic cells (FDCs), failed to stain sections of mutant mice, but clearly stained the FDC-containing B cell follicular region in littermate controls (Fig. 5 A, middle and bottom). Since FDCs are a hallmark of the light zone of germinal centers, these data demonstrate that p52-deficient mice not only lack defined B cell follicular areas, but that they are also totally unable to form germinal centers. Although small, weakly staining PNA positive foci were sometimes observed, they were located within the T cell area, as shown by double staining with PNA and anti-B220 antibodies (Fig. 5 B). It is possible that these cells represent pre–germinal center B cells that are no longer able to or are otherwise prevented from forming germinal centers. It remains to be explored if such cells could be responsible for the adjuvant-aided antibody response of mutant mice to TD antigens.
Figure 5.
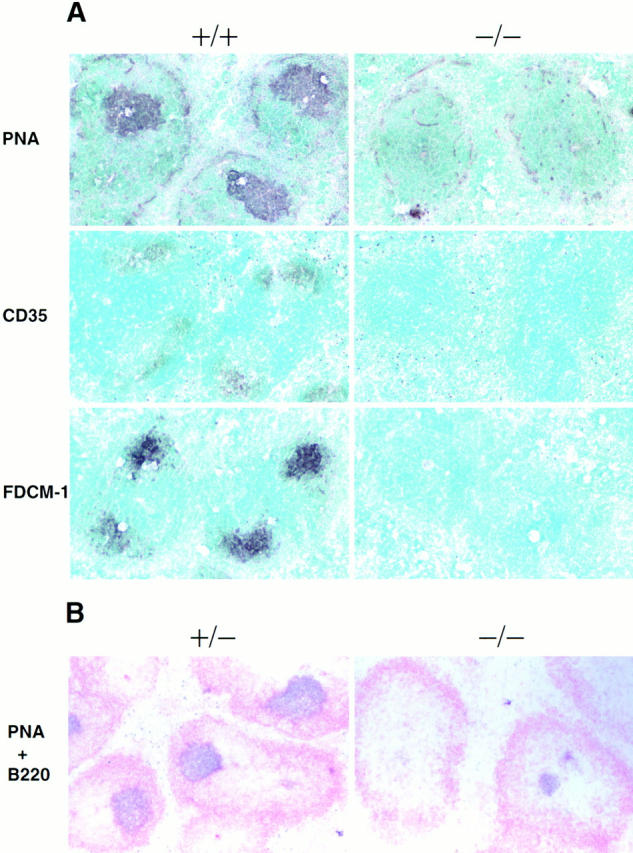
Absence of germinal centers and dendritic cell networks in spleens of challenged p52/p100 (−/−) mice. 13–15-wk-old p52/p100 (−/−), (+/−), and (+/−) control mice were challenged by intraperitoneal injection of 100 μg of alum-adsorbed TNP-KLH. Frozen sections of spleens were obtained 8 or 17 d after injection and stained (A) with PNA, anti-CD35(CR1), or FDC-M1 antibodies and (B) with PNA ( purple) and anti-B220 antibodies ( pink), as indicated. Stained cryosections from representative pairs of littermates are shown.
Adoptive Transfer.
The above data indicate that p52-deficient animals are defective in some aspects of antigen-specific activation of B cells, either in the presentation/handling of antigen by accessory cells, or in the signaling between accessory cells and B and T cells, or both. To better define those cells of mutant mice that contain biologically relevant defects, we adoptively transferred bone marrow of p52 null mutant mice into irradiated RAG-1–deficient animals, which lack both B and T cells (45). Thereafter, we challenged these mice with TNP-KLH adsorbed to alum and we investigated their ability to form germinal centers. As seen in Fig. 6, simultaneous staining with anti-B220 antibodies and PNA now revealed the presence of germinal center PNA+ B cells within B220+ follicular areas. Furthermore, challenge of adoptively transferred animals with TNP-KLH in the absence of alum now elicited a switched antibody response, albeit a weaker one than that seen with transfer of wild-type bone marrow (data not shown). Therefore, the inability to form germinal centers and the impaired switched antibody response to a TD antigen in p52-deficient mice cannot be explained by absolute defects within the p52-deficient B or T cells; in RAG-1 mice these p52-lacking lymphocytes were capable of participating in germinal center reactions and generation of switched antibodies in the absence of adjuvants. Adoptively transferred Bcl-3–deficient lymphocytes could also participate in germinal center reactions (Franzoso, G., L. Poljak, and U. Siebenlist, unpublished observations).
Figure 6.

Adoptively transferred p52-deficient lymphocytes support formation of germinal centers in RAG–1-deficient mice. Lethally irradiated RAG-1–deficient mice were injected with bone marrow cells isolated from 11-wk-old p52 (−/−), (+/−), and (+/+) donor mice, and 15 wk later challenged intraperitoneally with TNP-KLH (100 μg) adsorbed to alum. Splenic cryosections were obtained 9 d after challenge and stained with PNA ( purple) and anti-B220 antibodies (pink). Stained cryosections from a representative p52/p100 (−/−) marrow transferred animal is shown.
Impaired Marginal Zone Architecture.
To further investigate for possible similarities with Bcl-3–deficient mice, we looked at the splenic marginal zone surrounding the white pulp by staining splenic cryosections of p52–deficient mice with markers for local macrophage populations. MMs constitute a subset of macrophages that reside in the inner ring of the marginal zone (MZ) and they can be detected with the MOMA-1 antibody (46). When sections of mutant mice were stained with MOMA-1, no MMs were detected, whereas the MZ of littermate controls was clearly stained (Fig. 7, bottom). This is reminiscent of the data obtained with Bcl-3–deficient animals, where MMs are severely reduced in numbers (32). In contrast, MZMs, which are located in the outer ring of MZs and which are detected with the ERTR-9 marker (46), were readily observed in p52– deficient animals (Fig. 7, middle), but are lacking in Bcl-3– deficient animals (32). Nevertheless, some p52 null mutant mice exhibited reduced numbers of these cells as well (not shown).
Figure 7.

Altered MZ microarchitecture in spleens of p52/p100 (−/−) mice. Splenic cryosections were derived from the same group of p52/p100 (−/ −), (+/−), and (+/+) mice that were used for Fig. 5. Sections were stained with monoclonal antibodies specific to red pulp macrophages (RPM; BM8), MMs (MOMA-1) or MZMs; (ERTR-9), as indicated. Similar results were obtained with spleens from unchallenged animals (data not shown).
The MZ is considered critical to the control of cell trafficking into the white pulp (47). Consistent with an apparently impaired MZ architecture and thus impaired traffic control in mutant mice, the white pulp of these animals appeared to have been infiltrated with macrophage subpopulations that are normally completely excluded (Fig. 7, top, BM8). The same phenomenon has been observed in Bcl-3–deficient mice (32). Together, the data further document the close similarities, but also some differences between p52– and Bcl-3–deficient mice, implying that these proteins share many, but not all functions.
Infection with T. gondii.
p52-deficient animals were infected with T. gondii, an infectious model for T cell dependent immunity. We previously demonstrated that Bcl-3–deficient animals succumb to infection with an avirulent T. gondii strain within 3–5 wk of parasite exposure, and that this defect is related to an inability to mount an adequate host response; in particular, T helper 1 cells appeared not to be primed in vivo, resulting in absence of IFN-γ production from these cells (32). By contrast, p52-deficient animals displayed a protracted pattern of mortality with some animals succumbing as early as 1 wk after infection and others surviving beyond the 16-wk period of observation (Fig. 8). The basis for this partial impairment is unknown. The data suggest that Bcl-3 and p52 serve somewhat distinct roles in determining resistance to T. gondii.
Figure 8.

p52/p100 (−/−) mice are more susceptible to T. gondii infection. p52/p100 (−/−) mice and (+/−) and (+/+) littermates were injected intraperitoneally with ∼20 cysts of the avirulent T. gondii strain ME49 (initially provided by Dr. J. Remington, Palo Alto Research Foundation, Palo Alto, CA), as detailed previously (62). 10–23-wk-old mice of both sexes were used as follows: 17 (−/−), 15 (+/−), and 4 (+/+) animals. Data are shown as percent survival calculated from two independent experiments.
Discussion
We have demonstrated that p52–deficient animals (a) have reduced numbers of B cells, consistent with a loss of B cell follicles within their spleens and lymph nodes, (b) are unable to form germinal centers and are impaired in antibody responses to TD antigens, (c) lack follicular dendritic cell networks, (d) lack MMs in the splenic MZ and are impaired in exclusion of cells from the white pulp. We have also demonstrated that the inability to generate germinal centers does not track with lymphocytes, as adoptively transferred p52-deficient lymphocytes can form germinal centers in RAG-1–deficient mice. Together, these data reveal a defect in p52 null mutant mice in select aspects of antigen-dependent activation of lymphocytes, and they implicate accessory cells as primary effectors of unique and critical functions for p52 in wild-type animals (especially in follicular dendritic cells; see below).
Many defects observed in p52 null mice are very similar to those observed in Bcl-3–deficient animals (32). This supports the notion that a ternary complex of DNA-binding p52 homodimers and Bcl-3 (14) may regulate expression of some critical genes. Nevertheless, these two proteins are likely to have separate functions as well. Mice deficient in p52 not only lack p52 homodimers, they also lack the p100 precursor and any heterodimeric complexes containing p52. If these latter complexes have critical, nonredundant functions, we would expect to see defects independent of Bcl-3. Similarly, Bcl-3 may have critical, nonredundant functions, independent of p52 (see below). As shown here, Bcl-3 and p52 knockouts differ somewhat with respect to resistance to infection with T. gondii and loss of MZMs and MMs (32).
In contrast to mice deficient in Bcl-3 or p52, mice lacking p50 form apparently normal B cell follicles, normal germinal centers, normal follicular dendritic networks, and exhibit no loss of macrophages of the marginal zone (Fig. 4, and Franzoso, G., L. Poljak, and U. Siebenlist, unpublished observations; reference 33). Therefore, the data have revealed several specific biologic functions that depend on both Bcl-3 and p52, but not on p50, despite the fact that p50 is quite homologous to p52, and despite the fact that Bcl-3 is known to interact with p50 homodimers (although this interaction may differ from that with p52 homodimers; see Introduction) (10–13, 15, 16). Clear biologic contexts in which a complex between p50 homodimers and Bcl-3 is uniquely critical have yet to be described, although mice deficient in p50 or in Bcl-3 have been reported as impaired in their overall ability to contain certain bacterial infections (24, 32, 33). It remains possible, of course, that p50 homodimers and Bcl-3 have antagonistic activities (see Introduction).
Given the present data, it appears that a complex of p52 and Bcl-3 may be particularly critical for some accessory cell functions during antigen-dependent stimulation of lymphocytes. Adoptively transferred lymphocytes lacking p52 (or lacking Bcl-3) can form germinal centers in RAG-1– deficient mice and generate at least some T-dependent antibody responses, even in the absence of adjuvants. This implies that the primary defects with respect to these biologic processes do not reside within the mature, p52–deficient B or T cell lineages, which implicates accessory cells. In this regard, the apparent loss of follicular dendritic cell networks in both p52– and Bcl-3–deficient mice is particularly noteworthy and could account for the impairment in B cell follicular structure and the lack of germinal center formation (Figs. 4 and 5; reference 32). On the other hand, FDCs may not always be absolutely necessary for germinal center formation (48). It remains to be shown if defects lie within FDCs themselves or if they lie elsewhere, for example, in some other long-lived non–T, non–B cells or in nonhematopoietic cells. Previous data suggest high expression of p52 in FDCs (49), supporting the notion that this protein has important functions within FDCs. It is possible that p52 and Bcl-3 are needed for the development/differentiation of these cells, acting directly or indirectly, just like RelB is required for formation of mature interdigitating dendritic cells (IDCs; references 28, 29); (IDCs are present in both p52 and Bcl-3 knockout mice [Franzoso, G. and U. Siebenlist, unpublished observations].) It is worth noting also that the absence of p52 or Bcl-3 may well have direct effects in more than one cell type. In any case, a correlation between the failure to form FDC networks and impaired antibody response has also been noted in lymphotoxin (LT)α–deficient mice (50).
Adjuvant-aided presentation of antigen in p52 knockout mice appears to be able to compensate for the dramatic failure to respond to TD antigens without adjuvants. Although even then, proper germinal centers were still not observed. (Antibody production in the absence of germinal centers has been previously observed in LTα knockout mice [51].) This indicates a largely normal behavior of mutant lymphocytes once they are stimulated, implying that defects in the mutant mice must lie in other cell types. Strong adjuvant-aided antigenic stimulation may be associated with inflammation and thus production of cytokines in the mutant mice, which may overcome what is otherwise a defective priming of lymphocytes by accessory cells.
Which genes could be the unique targets of p52 and Bcl-3? Such genes must be regulated, at least in part, by nonredundant functions of these proteins. One clue may come from mutant mice that present with partially related phenotypes. For example, mice deficient in TNF-α, TNF receptor I, LTα, or LTβ display disrupted lymphoid microarchitecture including lack of germinal centers and FDC networks (52– 56). An absence of germinal centers has also been noted in mutant mice deficient in various proteins known to be involved in costimulatory signaling during antigen presentation, such as CD40 and CD40 ligand, but these mice have apparently normal lymphoid architecture (47, 57–59). All of these null mutant mice do have additional phenotypes that are not shared with p52– and Bcl-3–deficient mice. Still, it is quite likely that specific aspects of the signaling cascades that occur during in vivo antigenic stimulations are impaired in p52– or Bcl-3–deficient mice and/or that the specific in vivo microenvironments are disrupted that are needed to set up proper antigen-stimulated signaling cascades between cells. It remains to be shown if p52 and Bcl-3 lie on some of the same signaling pathways disrupted in the knockouts discussed above. In this regard, it is important to note that NF-κB complexes control, as well as are controlled by various members of the TNF ligand and TNF receptor families (2, 4, 60, 61), allowing for multiple ways in which these particular molecules could be functionally connected. Identification of molecular targets involved will be essential to understanding how these proteins regulate gene expression and how antigen priming is accomplished in vivo.
Since both p52 and Bcl-3 have been found to be associated with recurrent translocations (9, 20–23), it is noteworthy that an absence of either protein leads to losses in B cell numbers, in particular with age and with immune challenges. This suggests that p52 and Bcl-3 normally help to maintain B cells. When overexpressed and/or inappropriately expressed, these properties of p52 and Bcl-3 may contribute to tumor formation.
Acknowledgments
We thank A. Sher and K. Kelly for review of the manuscript and A.S. Fauci for continued support. We are grateful to W. Sha and D. Baltimore for the p50 (−/−) mice, A. Sher and B. Kelsall for helpful advice, M.H. Kosco Vilbos and G. Burton for the FDC-M1 antibody, J. Inmann for the TNP-Ficoll, and M. Rust for help with the preparation of this manuscript. We are deeply indebted to H. Westphal for use of his facilities in generating the knockout mice.
Footnotes
Abbreviations used in this paper: DAB, 3,3′-diammobenzidine; ES, embryonic stem; FCM, flow cytometric; FDC, follicular dendritic cell; HRP, horseradish peroxidase; LT, lymphotoxin, MM, metallophilic macrophage; MZ, marginal zone; MZM, MZ macrophage; neo, neomycin; NF, nuclear factor; PNA, peanut agglutinin; RAG-1, recombination activation gene 1; TD, T-dependent; TI, T-independent.
References
- 1.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 2.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 3.Verma IM, Stevenson JK, Schwarz EM, Antwerp DV, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 5.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of IκB-α proteolysis by site-specific signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 6.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IκB-α by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 7.Naumann MF, Scheidereit C. Activation of NF-κB in vivo is regulated by multiple phosphorylations. EMBO (Eur Mol Biol Organ) J. 1994;13:4597–4607. doi: 10.1002/j.1460-2075.1994.tb06781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 9.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicate in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 10.Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB–mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- 11.Franzoso G, Bours V, Azarenko V, Park S, Tomita-Yamaguchi M, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 can facilitate NF-κB–mediated transactivation by removing inhibiting p50 homodimers from select κB sites. EMBO (Eur Mol Biol Organ) J. 1993;12:3893–3901. doi: 10.1002/j.1460-2075.1993.tb06067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatada EN, Nieters N, Wulczyn FG, Naumann M, Meyer R, Nucifora G, McKeithan TW, Scheidereit C. The ankyrin repeat domains of the NF-κB precursor p105 and the proto-oncogene bcl-3 act as specific inhibitors of NF-κB DNA binding. Proc Natl Acad Sci USA. 1992;89:2489–2493. doi: 10.1073/pnas.89.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wulczyn FG, Naumann M, Scheidereit C. Candidate proto-oncogene bcl-3 encodes a subunit-specific inhibitor of transcription factor NF-κB. Nature. 1992;358:597–599. doi: 10.1038/358597a0. [DOI] [PubMed] [Google Scholar]
- 14.Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 15.Naumann M, Wulczyn FG, Scheidereit C. The NF-κB precursor p105 and the proto-oncogene product Bcl-3 are IκB molecules and control nuclear translocation of NF-κB. EMBO (Eur Mol Biol Organ) J. 1993;12:213–222. doi: 10.1002/j.1460-2075.1993.tb05647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nolan GP, Fujita T, Bhatia K, Huppi K, Liou H-C, Scott ML, Baltimore D. The bcl-3 proto-oncogene encodes a nuclear IκB-like molecule that preferentially interacts with NF-κB p50 in a phosphorylation-dependent manner. Mol Cell Biol. 1993;13:3557–3566. doi: 10.1128/mcb.13.6.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Didonato JA, Karin M, McKeithan TW. BCL3 encodes a nuclear protein which can alter the subcellular localization of NF-κB proteins. Mol Cell Biol. 1994;14:3915–3926. doi: 10.1128/mcb.14.6.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita T, Nolan GP, Liou H-C, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers. Genes Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- 19.Caamano JH, Perez P, Lira SA, Bravo R. Constitutive expression of Bcl-3 in thymocytes increases the DNA binding of NF-κB1 (p50) homodimers in vivo. Mol Cell Biol. 1996;16:1342–1348. doi: 10.1128/mcb.16.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neri A, Chang CC, Lombardi L, Salina M, Corradini P, Maiolo AT, Chaganti RS, Dalla-Favera R. B cell lymphoma-associated chromsomal translocation involves candidate oncogene lyt-10 homologous to NF-κB p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- 21.Neri A, Fracchiolla NS, Migliazza A, Trecca D, Lombardi L. The involvement of the candidate proto-oncogene NF-κB2/lyt-10 in lymphoid malignancies. Leuk Lymphoma. 1996;23:43–48. doi: 10.3109/10428199609054800. [DOI] [PubMed] [Google Scholar]
- 22.Fracchiolla NS, Lombardi L, Salina M, Migliazza A, Baldini L, Berti E, Cro L, Polli E, Maiolo AT, Neri A. Structural alterations of the NF-κB transcription factor lyt-10 in lymphoid malignancies. Oncogene. 1993;8:2839–2845. [PubMed] [Google Scholar]
- 23.Michaux L, Dierlamm J, Wlodarska I, Bours V, Berghe HVD, Hagemeijer A. t(14;10)/BCL3 rearrangements in lymphoproliferative disorders: a review of 23 cases. Cancer Genet Cytogenet. 1997;94:36–43. doi: 10.1016/s0165-4608(96)00247-6. [DOI] [PubMed] [Google Scholar]
- 24.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 25.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 26.Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt PH, Chen CH, Rosen CA, Stewart CL. IκBα deficiency results in sustained NF-κB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 28.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 29.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 30.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 31.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-κB RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–961. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franzoso G, Carlson L, Sharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, et al. Critical roles for the Bcl-3 oncoprotein in T cell–mediated immunity, splenic microarchitecture and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 33.Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- 34.Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphoenia in mice with homozygous disruption of the c-abl proto-oncogene. Cell. 1991;2:223–238. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 35.Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, Drago J, Noguchi M, Grinberg A, Bloom ET, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity. 1995;2:223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 36.Dignam JD, Lebovitz RM, Roeder RC. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-κB: homology to rel and dorsal. Cell. 1990;62:1019–1029. doi: 10.1016/0092-8674(90)90276-k. [DOI] [PubMed] [Google Scholar]
- 38.Bours V, Burd PR, Brown K, Villalobos J, Park S, Ryseck R, Bravo R, Kelly K, Siebenlist U. A novel mitogen-inducible gene product related to p50-p105-NF-κB participates in transactivation through a κB site. Mol Cell Biol. 1992;12:685–695. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franzoso G, Biswas P, Poli G, Carlson LM, Brown KD, Tomita-Yamaguchi M, Fauci AS, Siebenlist U. A family of serine proteases expressed exclusively in myelomonocytic cells specifically processes the nuclear factor-κB subunit p65 in vitro and may impair human immunodeficiency virus replication in these cells. J Exp Med. 1994;180:1445–1456. doi: 10.1084/jem.180.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shores EW, Huang K, Tran T, Lee E, Grinberg A, Love PE. Role of TCR ζ chain in T cell development and selection. Science. 1994;266:1047–1050. doi: 10.1126/science.7526464. [DOI] [PubMed] [Google Scholar]
- 41.Stüber E, Strober W. Distinct populations of dendritic cells are present in the subepithelial dome and T cell regions of the murine Peyer's patch. J Exp Med. 1996;183:979–989. doi: 10.1084/jem.183.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelsall BL, Strober W. Distinct populations of dendritic cells are present in the subepithelial dome and T cell regions of the murine Peyer's patch. J Exp Med. 1996;165:64–69. doi: 10.1084/jem.183.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coutelier JP, van der Logt JT, Heessen FW, Warnier G, Van Snick J. IgG2a restriction of murine antibodies elicited by viral infections. J Exp Med. 1987;165:64–69. doi: 10.1084/jem.165.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- 45.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1–deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 46.Leenen PJ, DeBruijin MF, Voerman JS, Campbell PA, van Ewijk W. Markers of mouse macrophage development detected by monoclonal antibodies. J Immunol Methods. 1994;174:5–19. doi: 10.1016/0022-1759(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y-J, Banchereau J. Mutant mice without B lymphocyte follicles. J Exp Med. 1996;184:1207–1211. doi: 10.1084/jem.184.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxins α and β revealed in lymphotoxin β–deficient mice. Immunity. 1997;6:491–500. doi: 10.1016/s1074-7613(00)80292-7. [DOI] [PubMed] [Google Scholar]
- 49.Feuillard JM, Koerner M, Israel A, Vassy J, Raphael M. Differential nuclear localization of p50, p42, and relB proteins in human accessory cells of the immune response in situ. Eur J Immunol. 1996;26:2547–2551. doi: 10.1002/eji.1830261102. [DOI] [PubMed] [Google Scholar]
- 50.Fu YX, Molina H, Matsumoto M, Huang G, Min J, Chaplin DD. Lymphotoxin-α (LTα) supports development of splenic follicular structure that is required for IgG responses. J Exp Med. 1997;185:2111–2120. doi: 10.1084/jem.185.12.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsumoto M, Lo SF, Carruthers CJ, Min J, Mariathasan S, Huang G, Plas DR, Martin SM, Geha RS, Nahm MH, Chaplin DD. Affinity maturation without germinal centres in lymphotoxin-α deficient mice. Nature. 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 52.Pasparakis ML, Alexopoulou V, Episkopou V, Killias G. Immune and inflammatory responses in TNF-α–deficient mice: a critical requirement for TNF-α in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeTogni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Schoenberger J. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 54.LeHir M, Bluethmann H, Kosco-Vilbois MH, Muller M, di Padova F, Moore M, Ryffel B, Eugster HP. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor–1 signaling. J Exp Med. 1996;183:2367–2372. doi: 10.1084/jem.183.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. Role of lymphotoxin and the type 1 TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 56.Ettinger R, Browning J, Michie SA, VanEwijk W, McDevitt HO. Disrupted splenic architecture, but normal lymph node development in mice expressing a soluble lymphotoxin-β receptor–IgG1 fusion protein. Proc Natl Acad Sci USA. 1996;93:13102–13107. doi: 10.1073/pnas.93.23.13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune response in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 58.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 59.Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 60.Tewari M, Dixit VM. Recent advances in tumor necrosis factor and CD40 signalling. Curr Opin Genet Dev. 1996;6:39–44. doi: 10.1016/s0959-437x(96)90008-8. [DOI] [PubMed] [Google Scholar]
- 61.Ware CF, VanArsdale S, VanArsdale TL. Apoptosis mediated by the TNF-related cytokine and receptor families. J Cell Biochem. 1996;60:47–55. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C47::AID-JCB8%3E3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 62.Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Showe L, Grunvald E, Hieny S, Gazzinelli RT, Sher A. In the absence of endogenous IFN-γ mice develop unimpaired IL-12 response to Toxoplasma gondiiwhile failing to control acute infection. J Immunol. 1996;157:4045–4054. [PubMed] [Google Scholar]