

Abstract
The nfkb2 gene is a member of the Rel/NF-κB family of transcription factors. COOH-terminal deletions and rearrangements of this gene have been associated with the development of human cutaneous T cell lymphomas, chronic lymphocytic leukemias, and multiple myelomas. To further investigate the function of NF-κB2, we have generated mutant mice carrying a germline mutation of the nfkb2 gene by homologous recombination. NF-κB2–deficient mice showed a marked reduction in the B cell compartment in spleen, bone marrow, and lymph nodes. Moreover, spleen and lymph nodes of mutant mice presented an altered architecture, characterized by diffuse, irregular B cell areas and the absence of discrete perifollicular marginal and mantle zones; the formation of secondary germinal centers in spleen was also impaired. Proliferation of NF-κB2–deficient B cells was moderately reduced in response to lipopolysaccharide, anti-IgD-dextran, and CD40, but maturation and immunoglobulin switching were normal. However, nfkb2 (−/−) animals presented a deficient immunological response to T cell–dependent and –independent antigens. These findings indicate an important role of NF-κB2 in the maintenance of the peripheral B cell population, humoral responses, and normal spleen architecture.
The nfkb2 gene was originally cloned as a mitogen-inducible cDNA encoding a protein with strong homology to NF-κB1 (p105/p50), a member of the Rel/NF-κB family of transcription factors (1–3). Neri et al. isolated nfkb2 as the protooncogene involved certain human B cell lymphomas (4). More recently, several groups have documented that truncations in the COOH-terminal region of this gene are associated with the development of cutaneous T cell lymphomas, chronic lymphocytic leukemias, and multiple myelomas (5–8).
The Rel/NF-κB family of transcription factors plays a major role as an early mediator of immune and inflammatory responses (for reviews see references 9–14). This group of proteins contains a highly conserved region named the Rel homology domain that is responsible for both DNA binding and protein dimerization. In mammalian cells, this protein family can be divided into two classes: one class includes the NF-κB1 (p105/p50) and NF-κB2 (p100/p52) proteins that are synthesized as precursor molecules (p105 and p100, respectively) that remain in the cytoplasm. Upon proteolytic processing they generate the DNA binding subunits p50 and p52, respectively (1, 15–17). The second class of proteins comprises RelA (p65), c-Rel, and RelB. These proteins do not undergo proteolytic processing, and they harbor transcriptional activation domains. The different biological functions of p50, RelA, RelB, and c-Rel have recently been addressed by the generation of null mice (18–22).
In unstimulated cells, the Rel/NF-κB dimers associate with members of the family of inhibitor proteins called IκBs and remain as an inactive pool in the cytoplasm. Upon stimulation by different agents, IκB molecules are rapidly phosphorylated and degraded, allowing the NF-κB dimers to translocate to the nucleus and regulate transcription through binding to κB sites (9, 12, 23–26). The in vivo function of two members of the IκB family, IκBα and Bcl-3, have been recently analyzed in knockout mice (27–30).
The biological role of NF-κB2 (p100/p52) remains unclear despite the evidence that abnormal NF-κB2 expression correlates with cell transformation (31). Terminally differentiated plasmacytoma and long term LPS-treated pre-B and B cell lines express p52-containing complexes, implying that p52–c-Rel and p52/–RelB dimers play a role in the final stages of B cell differentiation (32–33). Bcl-3, an oncoprotein overexpressed in human chronic lymphocytic leukemias, associates with p50 and p52 homodimers (34– 37). Transfection experiments show that these interactions lead to transactivation of κB-dependent reporter constructs (8, 35–36).
To further investigate the function of NF-κB2, we have generated mice carrying a germline mutation of the nfkb2 gene. NF-κB2-null mice have a marked reduction of the B cell compartment in spleen, bone marrow, and lymph nodes and are defective in the formation of secondary germinal centers. B cells from mutant mice have mild proliferative defects in response to LPS, mIg, and CD40, but have normal maturation and immunoglobulin switching. However, nfkb2 (−/−) mice have impaired humoral responses to T cell–independent and T cell–dependent antigens when compared to controls. These findings indicate an important role for NF-κB2 in the maintenance of the peripheral B cell population, germinal center formation, and B cell immune function.
Materials and Methods
Targeting Vector.
To generate the pNF-κB2 targeting vector, we isolated genomic fragments corresponding to the murine nfkb2 locus from a D3 embryonic stein (ES)1 cell genomic DNA library in λ Dash II (Stratagene, La Jolla, CA). The probe used for the screening contained nucleotides +1 to +250 of the murine nfkb2 cDNA. 4 overlapping phages containing the promoter region and the first 20 exons of the nfkb2 gene were obtained. As a targeting strategy we introduced a mouse phosphoglycerate kinase– promoter neo resistance cassette (38) into an EcoRI site present in exon 4 of the nfkb2 gene. For that purpose, a 10.5-kb EcoRI fragment containing the 5′ region, exons 1–3 and part of exon 4 was subcloned in the XhoI–NotI sites of the pPNT vector (39). Subsequently, a 3.9-kb EcoRI fragment containing part of exon 4 to exon 15 was introduced in the EcoRI site of the pPNT.
ES Cell Culture and Generation of Mutant Mice.
CJ7 ES cells were cultured as previously described (40). The electroporation was performed using 25 μg of NotI-linearized pNF-κB2 per 107 ES cells with a gene pulser (Bio-Rad Laboratories, Hercules, CA). Cells were grown under double selection with G418 and fialuridine. Colonies were picked 10 d after selection and expanded for freezing and DNA extraction. Homologous recombinants (2 out of 250 neo-resistant clones) were confirmed by Southern blot analysis with a 3′ external probe. Targeted ES cells were identified by the appearance of a 5.3-kb recombinant band in addition to the 8.0-kb wild-type band in SpeI-digested DNA (Fig. 1 A and B). nfkb2 (+/−) ES cells were injected into C57BL/6 blastocysts which were transferred into pseudopregnant ICR females. Resulting chimeric mice were mated with C57BL/6 mice and heterozygous offspring were interbred to obtain nfkb2-null animals. The studies presented here were performed with line 3C and subsequently confirmed using mice from line 5A. These studies were performed according to guidelines established by the Bristol-Myers Squibb Animal Care and Use Control Committee.
Figure 1.

Generation of nfkb2-null mice. (A) The targeting vector pNF-κB2 is shown at the top. The thin line depicts plasmid sequences, the thick line represents intron sequences, and the closed boxes show exons of the nfkb2 gene. The open boxes correspond to the PGK-neo and PGK-tk cassettes. The long arm contains a fragment of the 5′ region, exons 1 to 3, and 33 nucleotides of exon 4 (EcoRI site). The short arm contains the rest of exon 4 to exon 15. A homologous recombination event between the targeting vector and wild-type genomic DNA introduces the PGK-neo cassette into exon 4, disrupting the open reading frame of the gene. (B) Southern Blot analysis of genomic DNA isolated from mouse tails. 10 μg of DNA from mice of different genotypes were digested with SpeI, run in 0.7% agarose gels, blotted according to standard procedures, and hybridized with the 3′ external probe A. The 8.0- and 5.3-kb fragments correspond to the wild-type and targeted alleles respectively. (C) Thymocytes from control (+/+) and nfkb2-null (−/−) mice were labeled with [35S]methionine for 3 h in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of PMA/PHA (see Materials and Methods). Cell extracts were immunoprecipitated with NF-κB2–specific antiserum and analyzed by SDS-PAGE. (D) EMSA profiles of the κB binding complexes in whole cell extracts of thymus (Th) and spleen (Sp) from control (+/+) and nfkb2-null mice (−/−). Extracts were incubated with preimmune serum (p.i.), or antisera against p50 (αp50), p52 (αp52), or both, before the addition of the κB palindromic oligo.
Histopathological Analysis and Immunohistochemistry.
Histopathological analysis was performed on a minimum of five animals per age group and genotype. Animals were killed and organs were fixed by immersion in 10% neutral buffered formalin. Tissues were processed by standard methods, embedded in paraffin blocks, sectioned at 4–6 μm, stained with hematoxylin and eosin, and finally examined by light microscopy. For immunohistochemical analysis, frozen tissue sections were stained with anti-IgD (Southern Biotechnology Associates, Birmingham, AL; 1:200 dilution), MOMA-1 (Serotec Ltd., Oxford, UK; 1:100 dilution), peanut agglutinin (PNA)-biotin (Vector Labs., Burlingame, CA; 80 μg/ml) and FDC-M1 (1:100).
Cell Labeling and Immunoprecipitation.
Single-cell suspensions from thymus were prepared as previously described (41) and cultured in RPMI 1640 with 10% heat-inactivated fetal calf serum. 2 × 107 cells were labeled for 3 h with 500 μCi/ml of [35S]methionine in methionine-free medium with or without the addition of PMA (10 ng/ml) and PHA (1 μg/ml). Cells were washed with PBS and lysed in 1 ml of radioimmunoprecipitation assay buffer without SDS. Extracts were precleared with preimmune serum (3 μl), immunoprecipitated for 4 h with specific antiserum (3 μl), and then incubated with Protein A–Sepharose CL-4B (Pharmacia Biotech, Piscataway, NJ) for 3 h on a roller at 4°C. The immunocomplexes were washed twice with buffer A (0.2 NP-40, 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1 mM EDTA); once with buffer B (0.2 NP-40, 10 mM Tris-HCl, pH 7.5, 500 mM NaCl, and 2 mM EDTA); and once with buffer C (10 mM Tris-HCl, pH 7.5). Samples were boiled in 30 μl of Laemmli buffer, and separated on a 12.5% polyacrylamide gel (SDS-PAGE) at 12 milliAmperes for 16 h. Gels were fixed and incubated with Entensify (DuPont-NEN, Boston, MA). After drying, gels were exposed to X-Omat AR film (Eastman Kodak Co., Rochester, NY) at −70°C overnight.
Preparation and Culture of B Cells.
The culture medium and reagents used for the B cell proliferation assays have been previously described (42). Single cell suspensions from spleen were treated with ammonium chloride buffer to lyse red blood cells. Cells were stained with FITC-labeled rat IgG2a anti–mouse B220 mAb (clone RA3-6B2; PharMingen, San Diego, CA) plus PE-labeled hamster anti–mouse CD3e mAb (clone 145-2C11; PharMingen). Small, resting B cells (B220+CD3−, low forward and side scatter profile) were obtained by electronic cell sorting on an EPICS Elite cytometer (Coulter Corp, Hialeah, FL). Reanalysis of sorted cells immediately after isolation showed B cell purity of >99%. Cells were cultured at 37°C in a humidified incubator containing 6% CO2 at a cell density of 2 × 105 cells/ml.
Electrophoretic Mobility Shift Assays.
Whole cell extracts were prepared as previously described (43). Protein concentrations were determined with the Bio-Rad protein assay kit according to the manufacturer's instructions and were further analyzed by Western blot analysis. Electrophoretic mobility shift assays (EMSAs) were performed using 3 μg of protein extract and 5 × 104 cpm of 32P-labeled probe (κB palindromic; reference 37).
Measurement of DNA Synthesis by [3H]thymidine Incorporation.
Purified B cells (105) were cultured for 72 h in a final volume of 0.2 ml in complete RPMI in flat-bottomed 96-well plates. [3H]thymidine was added to the cultures for an additional 18 h. Cultured cells were then harvested onto glass fiber filter paper with a Wallac 1295-001 cell harvester (LKB, Uppsala, Sweden). Specific incorporation of [3H]thymidine was analyzed by scintillation spectroscopy, and results are expressed as the arithmetic mean ± SEM of triplicate cultures.
Flow Cytometric Analysis.
Cells were resuspended in PBS containing 1% bovine serum albumin before staining with the following conjugated antibodies: CD3, CD4, CD8, CD25, α/β TCR, Thy 1.2, CD44, CD45R (B220), IgD, IgM, HSA, Ter-119, Mac-1, Gr-1, F4/80, 7/4, and ICAM (intracellular adhesion molecule)–1. Antibodies were obtained from GIBCO-BRL (Gaithersburg, MD) and PharMingen. Flow cytometry was performed on a FACScalibur flow cytometer (Becton-Dickinson Co., Mountain View, CA) and 104 events were collected in each case.
Quantitation of Secreted Ig Isotype Concentrations in Culture Supernatants.
Purified B cells were stimulated with LPS (20 μg/ml), IL-4 (4.2 ng/ml) + IL-5 (150 U/ml), sCD40L (10 μg/ml), anti– IgD-dextran (3 ng/ml), TGF-β (3 ng/ml); and IFN-γ (10 U/ml). Ig isotype concentrations were measured by an ELISA assay as previously described (44).
Immunization and ELISA Assays.
To induce a T cell–dependent response and analyze the formation of germinal centers in the spleen, five mice of each genotype were immunized by intraperitoneal injection of either sheep red blood cells (SRBCs) or (4-hydroxy-3-nitrophenyl) acetyl (NP-KLH). Each mouse was injected with 250 μl of phosphate-buffered saline containing 10% SRBCs (2.5 × 108 cells) or with 100 μg of alum-precipitated NP-KLH. Animals were killed 10 d after immunization and spleens were frozen in O.C.T. (Tissue-Tek, Miles Inc., Elkhart, IN) for immunohistochemistry. Additional mice were also boosted with a second injection of the same antigens 10 d after the first immunization, and the same assays were performed. The nfkb2-null mice that were used to evaluate the basal immunoglobulin levels and the antigen-specific immunoglobulin production have been previously backcrossed for eight generations to the C57 Bl/6 strain.
Basal levels of immunoglobulins were measured in sera collected from control and nfkb2 null mice before immunizations. For T cell–dependent responses, mice were immunized by intraperitoneal injection of 100 μg of alum-precipitated NP-KLH. In the case of the T cell–independent responses, mice were immunized with 10 μg of NP coupled to LPS (20, 44). Serum samples were collected before immunizations and at 7-d intervals during 3 wk. To determine the levels of NP-specific immunoglobulins, we used ELISA plates coated with NP17-BSA for capture, and goat anti–mouse isotype-specific antibodies conjugated to horseradish peroxidase (Southern Biotechnology Associates) as previously described (45). The level of each antigen-specific isotype was determined by comparison with a standard curve. NP-specific Ig levels in unchallenged mice were below detection. One symbol represents one mouse. Statistical analysis was performed using the unpaired Student's t test to calculate second two-tailed P values.
Results
Generation of the nfkb2-null Mice.
The strategy to generate the nfkb2-null allele in ES cells involved the introduction of a PGK-neo cassette in exon 4 of the mouse nfkb2 locus in an EcoRI site in nucleotide 54 of the nfkb2 open reading frame (Fig. 1 A). Heterozygous nfkb2 (+/−) animals, containing the 8.0-kb wild-type and 5.3-kb recombinant SpeI fragments, were crossed to obtain homozygous mice (−/−) (Fig. 1 B). No differences in size, behavior, or life span were observed between nfkb2-null mice and wild-type littermates. These animals were born at the expected Mendelian ratios, which demonstrates that NF-κB2 is not required for normal embryo development. The presence of the NF-κB2 protein was analyzed in thymocytes of control and mutant mice. While thymic cells from wild-type mice expressed p100/p52 upon stimulation, neither p100 nor p52 were present in cells from the mutant mice, demonstrating that the targeted disruption of the nfkb2 locus resulted in a complete loss of both proteins (Fig. 1 C). The nfkb2 (−/−) mice had no gross developmental deficiencies or health abnormalities while maintained in a pathogen free facility. No significant difference in protein levels of other Rel/NF-κB family members was detected in thymus and spleen whole cell extracts from mutant mice compared to control littermates by Western blot analysis (data not shown). EMSAs using whole cell extracts from thymus of control and nfkb2 (−/−) mice showed no remarkable differences in their DNA binding (Fig. 1 D). In contrast, whole cell extracts from splenocytes have a decrease DNA binding activity corresponding to p52-containing complexes compared to control cells (Fig. 1 D).
Histopathological Analysis of the nfkb2-null Mice.
Light microscopic evaluation of all major tissues from nfkb2 (−/−) mice of varying ages (3, 9, 12, 24, and 36 wk) showed that histopathological changes were largely restricted to lymphoid organs. The thymus of the null mice was normal in size, and corticomedullary ratios were similar to those observed in wild-type age-matched animals (data not shown). The spleen of the naive mutant mice was grossly similar to wild-type controls. However, microscopic evaluation of the spleen of nfkb2 (−/−) mice showed a disruption of the normal architecture characterized by diffuse, irregular follicular (B cell) areas with the absence of a discrete, perifollicular marginal zone and germinal centers (compare Fig. 2, A and C). These morphologic abnormalities were substantiated after immunohistochemical detection of B cells. In contrast to wild-type spleen where numerous B220 positive (B220+) cells were observed within discrete germinal centers, the spleen from nfkb2 (−/−) mice presented B220+ cells scattered among indiscrete germinal centers (Fig. 2, B and D). No differences were observed in the number of cells undergoing apoptosis in the red or white pulp of the spleen of unstimulated control and nfkb2 (−/−) mice as determined by Apoptag (Oncor, Inc., Gaithersburg, MD) histochemical staining (data not shown). However, the number of Apoptag-positive cells in the splenic white pulp of SRBC-stimulated nfkb2 (−/−) mice was decreased by ∼70% as compared to the white pulp of SRBC-stimulated control mice. The number of cells undergoing apoptosis in the splenic red pulp of SRBC-stimulated nfkb2 (−/−) and SRBC-stimulated control mice were similar.
Figure 2.
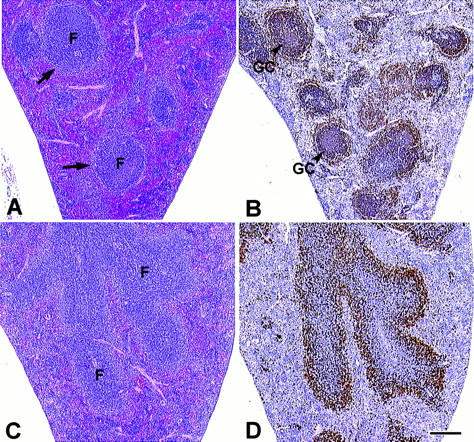
Histopathologic and immunohistochemical analysis of the spleen of naive control and nfkb2 (−/−) mice. (A and C) Spleen sections stained with hematoxylin and eosin from 8-wk-old control and nfkb2 (−/−) mice, respectively. F, follicles. Perifollicular marginal zones are indicated by arrows. (B and D) Immunohistochemistry of consecutive spleen sections from A and C, respectively, stained with the B cell–specific mAb B220. GC, germinal center. Bar equals 100 μm.
Morphologic changes similar to those observed in B cell areas in the spleen also occurred in lymph nodes of mutant mice (data not shown).
Reduction of the B Cell Population in the Spleen, Bone Marrow, and Lymph Nodes from Mutant Mice.
To further characterize the histopathological changes observed in the nfkb2 (−/−) mice, we performed an extensive survey of surface marker expression on hemopoietic cells from mutant mice. Immunofluorescence staining and flow cytometric analysis of thymocytes with anti-CD3, -CD4, -CD8, -CD25, and -Thy 1.2 showed no remarkable changes between control and mutant mice (data not shown). No difference in the total cell number was observed in the thymus of null mice with respect to the controls. Consistent with the initial histopathologic and immunohistochemical observations, the spleen of the naive nfkb2-null mice presented a marked decrease in the B cell population. A 50–80% reduction in the total number of B cells was detected in the spleen of mutant mice as early as 1 wk after birth (Fig. 3 A). A concomitant increase of the T cell compartment comprising both CD4 and CD8 single positive cells was detected in the spleen of mutant mice (Fig. 3 A). A similar decrease in the number of B cells and increase in the T cell population were observed in older mice (Fig. 3, B and C). We also examined the stages of B cell differentiation by surface B220, CD43, HSA, IgM, IgD, and CD19 expression. Both immature and mature B cells were present in the spleen of the nfkb2 (−/−) mice, indicating that a general reduction of all the B cell lineages rather than a maturation blockage occurred in the absence of NF-κB2. Similar results were observed in the lymph nodes from mutant mice.
Figure 3.

Reduction of the B cell population in the spleen of the nfkb2-deficient mice. (A) Scatter plots of splenocytes stained with B220 versus Thy 1.2; CD4 versus CD8; IgM versus B220; and IgD versus B220 antibodies. Single cell suspensions of splenocytes from 12-wk-old control (+/+) and mutant (−/−) mice were analyzed by flow cytometry as described in Materials and Methods. Representative diagrams from four independent experiments are shown with the percentage of positive cells in relevant quadrants. (B) Diagram showing the decrease in the total B cell population in spleen of the nfkb2 (−/−) mice respective to the control littermates at different time points. (C) Total T cells in the spleen of the nfkb2-null mice at different time points.
The bone marrow of the 6-wk-old nfkb2 (−/−) mice have a normal number of B cells (Fig. 4 A). The total number of bone marrow cells recovered from control and nfkb2 (−/−) mice showed no significant differences. However, mutant mice had a decline of 50% in the absolute number of B cells between 8 and 12 wk of age in the bone marrow (Fig. 4 B).
Figure 4.

Changes in the B cell population in the bone marrow of the nfkb2-deficient mice. Scatter plots of bone marrow cells stained with IgM versus B220 from 6-wk-old (A) or 10-wk-old (B) control (+/+) and mutant (−/−) mice were analyzed by flow cytometry as described in Materials and Methods.
NF-κB2-deficient B Cells have Modest Proliferative Defects in Response to LPS, mIg, or CD40 Signaling.
Normally, B cells stimulated with LPS, anti-CD40, or anti–IgD-dextran proliferate, and these responses can be further augmented by IL-4 and/or IL-5 addition. The signal transduction pathways mediating these effects are distinct for each molecule, and hence could be differentially affected by the absence of NF-κB2 (45). In this context, and in light of the distinctive roles for p50, c-Rel, and RelB in murine B cell proliferation (20, 21, 42, 45), we determined whether any of these mitogenic pathways were dependent upon the expression of NF-κB2. Highly purified resting B cells from naive nfkb2 (−/−) and control mice were obtained by electronic cell sorting and stimulated for 72 h with various concentrations of LPS, anti–IgD-dextran, or anti-CD40 mAb in the presence of IL-4 and IL-5. Similar results were obtained in three independent experiments, which demonstrated only moderate reduction in the proliferative response of NF-κB2–deficient cells compared to control B cells (Fig. 5, A–C). At concentrations between 2 and 10 ng/ml of anti–IgD-dextran, NF-κB2–deficient B cells showed a nearly twofold reduction in their proliferative response compared to controls. These results prompted us to analyze whether the NF-κB2–deficient B cells were more prone to undergo apoptosis upon stimulation. Splenocytes from control and nfkb2-deficient mice have a similar number of B cells undergoing apoptosis upon stimulation with anti–IgD-dextran, anti-CD-40 mAb, or TNF-α as detected by Facs® analysis using anti-B220 and anti–Annexin V (data not shown).
Figure 5.

In vitro proliferation of B cells lacking NF-κB2. Purified resting B cells from nfkb2-null (open circles) and control mice (closed circles) were cultured during 72 h at 105 cells/ml in the presence of different concentrations of (A) LPS (0.09–20 μg/ml); (B) anti–IgD-dextran (0.1–10 ng/ml); or (C) anti-CD40 mAb (0.08–5 μg/ml) + IL-4 (3,000 U/ml) + IL-5 (150 U/ml). [3H]thymidine was added for 12 h and cells were then harvested for determination of [3H]thymidine incorporation. Values shown represent the means of triplicate cultures ± SEM.
B cells from nfkb2-null Mice have Normal Maturation to Ig Secretion and Switching to Downstream Ig Isotypes In Vitro.
NF-κB2 has been shown to be expressed late during B cell maturation, suggesting that it may regulate B cell maturation to Ig secretion (32, 33). Furthermore, multiple Rel/ NF-κB binding sites have been identified in the Ig heavy chain locus, including promoters and enhancers involved in regulating Ig isotype switching (46, 47). Indeed, B cells lacking NF-κB1 (p105/p50) have distinctive and selective defects in Ig isotype switching (45). Thus, we tested the relative ability of B cells lacking NF-κB2 (p100/p52) to undergo B cell maturation to IgM secretion and switch to the expression of downstream Ig isotypes in response to various combinations of B cell activators and cytokines.
IL-4, IFN-γ, and TGF-β induce class switching to IgG1 and IgE, IgG3 and IgG2a, and IgG2b and IgA, respectively, in appropriately activated B cells (48). Combinations of stimuli optimal for induction of class switching to different sets of isotypes were used to assess switch recombination in NF-κB2–deficient B cells. In two similar sets of experiments, maturation to IgM secretion was essentially comparable between B cells lacking NF-κB2 and similarly activated control B cells, in response to a variety of distinct stimuli (Fig. 6). Likewise, induction of all six non-IgM, non-IgD isotypes in NF-κB2–deficient B cells was comparable to that observed for control B cells. The production of IgM in NF-κB2-deficient B cells was two times higher than in controls. Thus, in contrast to NF-κB1, NF-κB2 is not essential for Ig isotype switching.
Figure 6.

NF-κB2 (−/−) B cells have normal maturation to Ig secretion and class switching in vitro. Control (+/+) and NF-κB2 (−/−) B cells were stimulated as described in Materials and Methods. Concentrations of secreted immunoglobulins were measured in culture supernatants by ELISA 6 d after initiation of the cuture. The values represent the mean of triplicate cultures.
Impaired Antigen-specific Immunoglobulin Production in nfkb2-null Mice.
Mice rendered deficient for c-Rel, RelB, NF-κB1 (p105/p50), or Bcl-3 have defective basal immunoglobulin levels and antibody production upon antigenic challenge (20, 21, 29, 30, 49). To evaluate the humoral immunity of the nfkb2-null mice, we measured the basal production of immunoglobulins in naive mutant and control littermates and the statistical analysis was performed using the Student's t test (Fig. 7 A). While IgG1 and IgG2b showed similar levels in control and nfkb2 (−/−) mice, IgM (4.5-fold; P <0.0005) and IgG2a (2-fold; P <0.008) were increased in null mice. In contrast, a threefold reduction in the levels of IgG3 (P <0.01) and twofold lower levels of IgA (P <0.000004) were detected in the nfkb2-null mice compared to wild-type littermates.
Figure 7.

Decreased immunoglobulin production in nfkb2-null mice. (A) Resting immunoglobulin isotype levels in unimmunized mice. Serum immunoglobulin levels in naive 7-wk-old littermates were determined by isotype-specific ELISA. Closed circles correspond to control (+/+), open circles to heterozygous (+/−), and open squares to mutant (−/−) mice. (B) Immune response to the T cell–dependent antigen NP-KLH. Five littermates of control (+/+, closed circles) and mutant (−/−, open squares) mice were used for this assay. Serum samples were collected 7, 14, and 21 d after immunization as indicated. (C) Immune response to the T cell–independent antigen NP-LPS. Five control (+/+) and five mutant (−/−) mice were used in this assay. Serum samples were collected at 7, 14, and 21 d after immunization. Horizontal lines indicate the mean values.
We next examined the humoral responses of control and mutant mice to specific antigenic challenges. To assess the T cell–dependent immune responses, mice were immunized with keyhole limpet hemocyanin coupled to (NP-KLH) and bled 7, 14, and 21 d after immunization. The production of NP-specific isotype IgG1 by nfkb2 (−/−) mice was reduced fourfold at day 7 (P <0.008), and decrease to 10-fold (P <0.00001) at 21 d after treatment (Fig. 7 B).
The evaluation of the T cell–independent response was performed using NP-LPS to immunize control and nfkb2-null mice and the serum levels of NP-specific IgG3 were measured at 7, 14, and 21 d after immunization (Fig. 7 C). Under these conditions, the mutant mice had a 10-fold lower production of specific antibody at day 7 (P <0.01) and a 30-fold decrease 21 d after immunization (P <0.02). NP-specific Ig levels in unchallenged mice were below detection. These results demonstrate that NF-κB2 is required for normal levels of antigen-specific immunoglobulins, in particular, in the absence of T cell help.
Impairment of Germinal Center Formation in nfkb2 (−/−) Mice.
The abnormal architecture of the spleen in the nfkb2 (−/−) mice suggests that immune responses dependent on cellular interactions in the follicles may not be fully functional. To test this hypothesis we immunized mice with SRBCs and evaluated the formation of germinal centers (GCs) 10 d after immunization. The spleen of wild-type mice had numerous GCs characterized by B cell areas that bound PNA surrounded by IgD+ cells (Fig. 8, A and C). In contrast, the spleen of mutant mice had very few cells stained by PNA and a diffuse perifollicular staining of cells expressing surface IgD (Fig. 8, B and D). To further evaluate the alterations of spleen follicular structure in the nfkb2 (−/−) mice, tissue sections from the immunized mice were stained with MOMA-1, a specific antibody for metallophilic macrophages (Fig. 8, E and F). While the spleen of control mice had a ring-like zone of MOMA-1 + cells demarcating the marginal zone, the spleen of nfkb2 (−/−) mice contained reduced numbers of loosely organized MOMA-1–positive cells scattered throughout the white pulp. MOMA-1 staining in the outer PALS showed no differences between controls and nfkb2 (−/−) mice. The presence of antigen presenting follicular dendritic cells (FDCs), that promote B cell maturation, was analyzed by immunostaining with the FDC-M1 monoclonal antibody (50, 51). While clusters of FDCs could be clearly seen in the GCs of the control mice, very few FDCs could be detected in the mutant mice (Fig. 8, G and H).
Figure 8.

Impaired formation of GC in nfkb2 (−/−) mice. Control (+/+) and mutant (−/−) mice were injected with SRBCs. Animals were killed 10 d after immunization, and then spleens were collected and processed for frozen tissue sections. A and B correspond to PNA, C and D to anti-IgD, E and F to MOMA-1, and G and H to FDC-M1 staining of spleen sections. A–D have the same magnification (bar in D equals 75 μm). E and F have the same magnification (bar in F equals 100 μm). Magnification in G and H is identical (bar in H equals 50 μm).
Similar defects in the formation of GCs were observed when mice received a second immunization boost of SRBC 10 d after the first one. In addition, nfkb2 (−/−) mice immunized with NP-KLH also displayed a lack of well defined GC formation.
Discussion
Since the description of the NF-κB binding activity more than 10 y ago (52), an extensive body of information has been gathered describing the importance of this family of proteins in the regulation of genes involved in cell growth, stress, inflammation, and immune responses. An increasing number of NF-κB target genes have been identified to date, including cytokines, cytokine receptors, interferons, growth factors, MHC proteins, and other genes. However, whether or not particular members of the NF-κB family regulate specific genes in different cell types, and the distinct biological function of each protein, remain to be elucidated. In an attempt to answer some of these questions, several laboratories have generated mice with mutated genes of the NF-κB family. The data obtained indicate that the Rel/NF-κB proteins play an important role in the hematopoietic system. In brief, relA-deficient mouse embryos die at embryonic day 15 following massive apoptosis of hepatocytes in the liver (18). Recent studies indicate that RelA has a role in preventing TNF-α–induced apoptosis (53–55). Secretion of IgG1 and IgA is reduced in RelA-deficient B cells (56). Also, T and B cells lacking RelA show a marked reduction in their proliferative response. RelB-deficient mice exhibit a T cell–dependent multiorgan inflammation, myeloid hyperplasia, and splenomegaly associated with extramedullary hemopoiesis (19, 22, 57). RelB-deficient B cells have defective proliferative responses, although they undergo normal cell maturation, Ig secretion and isotype switching (42). On the other hand, c-Rel has been shown to be required for lymphocyte activation and cytokine expression, as demonstrated by the B and T proliferative defects and IL-2 production by T cell in c-Rel (−/−) mice (20). The immune responses to T cell–dependent antigens were defective in these mice.
Based on the studies mentioned above, the NF-κB proteins can be divided into two groups according to the role they play in the hematopoietic system. One of the groups includes p50 and c-Rel, which regulate genes involved in rapid immune responses. The other group comprises RelA and RelB, which control the expression of genes implicated in immune responses and also hemopoietic development and housekeeping functions.
Immune Defects of nfkb2-null Mice.
Here, we show that NF-κB2 is required for B cell development and function. First, nfkb2 (−/−) mice have a dramatic reduction in the absolute number of B cells in peripheral lymphoid organs. Second, NF-κB2 (−/−) B cells exhibited reduced in vitro proliferation. Third, the number of B cells in the bone marrow of the nfkb2 (−/−) mice undergoes a reduction of 50% between 8 and 12 wk of age.
In vitro, NF-κB2 (−/−) B cells display normal cell maturation to immunoglobulin secretion and class switching. The number of splenic T cells is increased although stimulation-induced proliferation is normal in the absence of NF-κB2. In addition, the nfkb2 (−/−) mice have an abnormal splenic microarchitecture, with diffuse B cell areas, lack of lymphoid follicles, and a discrete marginal zone, an area where nfkb2 transcripts have been previously shown (58).
Challenges with T cell–dependent or –independent antigens showed that the nfkb2 (−/−) mice are impaired in the production of antigen-specific antibodies. Consistent with these defective antibody responses, the spleen of nfkb2 (−/−) mice lacks well-defined GC structures. The absence of centrocytes and a decreased number of metallophilic macrophages and FDCs are some of the characteristics of the GCs in the mutant mice. Such centers are the primary sites where T–B cell communication takes place, together with B cell expansion, somatic hypermutation, isotype switching, and affinity maturation (59–62).
Our results suggest that either the production or the lifespan of B cells is defective in nfkb2 (−/−) mice. In addition, the migration of the B cells to the peripheral lymphoid organs may be deficient. Moreover, the low number of splenic B cells in these mutant mice coupled with the marked reduction of FDCs and the lack of GCs results in impaired immune responses. Thus, NF-κB2 regulates the expression of genes involved in B cell production, formation of GCs, and normal levels of antigen-specific antibodies. Although the genes regulated by NF-κB2 are still unknown, we are starting to understand the pathways in which this protein is involved. Further studies will be necessary to clarify whether nfkb2-null B cells are intrinsically defective or exhibit abnormalities as a consequence of impaired T lymphocytes, dendritic cells, or the microenvironment of the spleen.
Similar defects in the formation of GCs have been described in mice deficient in different ligands and receptors including CD40, CD40L, TNF-α, LT-α, TNF receptor I, MHC class II, CD28, the putative chemokine receptor BLR1, the complement receptor locus Cr II, and OBF-1, the B cell–specific coactivator of Oct-1 and Oct-2 (63–72). Interestingly, CD40, TNF-α, and LT-α signaling induce NF-κB binding activity, suggesting that p52 may have a role in the signal transduction pathways triggered by these molecules (73–75).
Several phenotypic defects are common between bcl-3 (−/−) and nfkb2 (−/−) mice (29, 30). Mice rendered deficient for Bcl-3 also have a decreased B cell population, impaired formation of GCs, and severe defects in the production of antigen specific antibodies. Such strong similarities argue for a synergistic effect of these two proteins in transcription (35).
Functional Redundancy between p105/p50 and p100/ p52.
The phenotype of the nfkb2-deficient mice is quite distinct from that of mice rendered deficient in other members of the NF-κB family of transcription factors. Although NF-κB2 (p100) is 52% homologous to NF-κB1 (p105; references 1, 3, 4), their pattern of expression is different. While nfkb1 is ubiquitously expressed in mouse tissues, nfkb2 expression is more restricted to lymphoid organs, and the highest mRNA levels are found in thymic medulla, marginal zone, and outer region of the periarterial sheath of the spleen (58). A comparison between the phenotypic changes displayed by mice lacking NF-κB1 and NF-κB2 reveals significant differences but also some similarities. The nfkb2-null mice have an abnormal splenic architecture and impaired formation of secondary GCs. In contrast, the nfkb1-null mice do not have any of these deficiencies (21, 30). B cells from nfkb1-null mice have a very poor proliferative response to LPS, but respond normally to anti–IgD-dextran and anti-CD40. In the case of nfkb2-null mice, B cells exhibit a moderate decrease in proliferation in the presence of similar stimuli. These data indicate that NF-κB1/p50 has a key role in the LPS stimulatory pathway, whereas NF-κB2/p52 has a modest effect on proliferation independent of the pathway. Another important difference in the roles of NF-κB1/p50 and NF-κB2/p52 is that nfkb1-null B cells have defects in their maturation and immunoglobulin secretion, germ line CH transcription, and isotype switching (21, 45), whereas nfkb2 (−/−) B cells do not exhibit any of these deficiencies. However, the production of antigen-specific antibodies is affected in mice mutant for either NF-κB1 or NF-κB2. T cell–dependent responses are impaired in nfkb1 mutant mice and reduced in nfkb2-null mice. T cell–independent immune responses are severely impaired in nfkb2-null mice.
These phenotypic differences between nfkb1 and nfkb2 mutant mice clearly indicate that p105/p50 and p100/p52 have distinct functions in the B cell lineage. However, we still cannot rule out a functional redundancy for these proteins. Further experiments generating double mutant mice should help to clarify this matter.
Acknowledgments
We are grateful to Mavis Swerdel, Alice Lee, and Sergio Lira in the Transgenic Unit and all the staff in Veterinary Sciences at Bristol-Myers Squibb. We would like to thank Kenneth Class for flow cytometry; Donna Dambach for comments and FDC immunostaining; James K. Loy for photoimaging; and Carol Ryan, Anne Lewin, and Michelle French for excellent technical assistance. We are indebted to Ricardo Attar, Daniel Carrasco, and Falk Weih for technical help and valuable comments of this manuscript. We also thank Albert Bianchi, Anne Gaëlle Borycki, Violetta Iotsova, Heather Macdonald-Bravo, and Rolf-Peter Ryseck for helpful suggestions and comments concerning this manuscript. We are grateful to Marie Kosco-Vilbois for kindly providing the FDC-M1 antibody.
Footnotes
Abbreviations used in this paper: EMSA, electrophoretic mobility shift assays; ES, embryonic stem; FDC, follicular dendritic cell; GC, germinal center; PNA, peanut agglutinin; SRBC, sheep red blood cell.
References
- 1.Schmid RM, Perkins ND, Duckett CS, Andrews PC, Nabel GJ. Cloning of an NF-κB subunit which stimulates HIV transcription in synergy with p65. Nature. 1991;352:733–736. doi: 10.1038/352733a0. [DOI] [PubMed] [Google Scholar]
- 2.Mercurio F, Didonato J, Rosette C, Karin M. Molecular cloning and characterization of a novel Rel/NF-κB family member displaying structural and functional homology to NF-κB p50/p105. DNA Cell Biol. 1992;11:523–537. doi: 10.1089/dna.1992.11.523. [DOI] [PubMed] [Google Scholar]
- 3.Bours V, Burd PR, Brown K, Villalobos J, Park S, Ryseck R-P, Bravo R, Kelly K, Siebenlist U. A novel mitogen-inducible gene product related to p50/p105–NF-κB participates in transactivation through a κB site. Mol Cell Biol. 1992;12:685–695. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neri A, Chang CC, Lombardi L, Salina M, Corradini P, Maiolo AT, Chaganti RS, Dalla-Favera R. B cell lymphoma–associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- 5.Migliazza A, Lombardi L, Rocchi M, Trecca D, Chang C-C, Antonacci R, Fracchiolla NS, Ciana P, Maiolo AT, Neri A. Heterogeneous chromosomal aberrations generate 3′ truncations of the NFKB2/lyt-10 gene in lymphoid malignancies. Blood. 1994;84:3850–3860. [PubMed] [Google Scholar]
- 6.Thakur S, Lin H-C, Tseng W-T, Kumar S, Bravo R, Foss F, Gélinas C, Rabson AB. Rearrangement and altered expression of the NFKB-2gene in human cutaneous T-lymphoma cells. Oncogene. 1994;9:2335–2344. [PubMed] [Google Scholar]
- 7.Fracchiolla NS, Lombardi L, Salina M, Migliazza A, Baldini L, Berti E, Cro L, Polli E, Maiolo AT, Neri A. Structural alterations of the NF-κB transcription factor lyt-10 in lymphoid malignancies. Oncogene. 1993;8:2839–2845. [PubMed] [Google Scholar]
- 8.Chang C-C, Zhang J, Lombardi L, Neri A, Dalla-Favera R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol. 1995;15:5180–5187. doi: 10.1128/mcb.15.9.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finco TS, Baldwin AS. Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 10.Kopp EB, Ghosh S. NF-κB and Rel proteins in innate immunity. Adv Immunol. 1995;58:1–27. doi: 10.1016/s0065-2776(08)60618-5. [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto S, Verma IM. Rel/NF-κB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 12.Thanos D, Maniatis T. NF-κB: A lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 13.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 14.Baldwin AS. The NFκB and Iκβ proteins, new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-κB: homology to rel and dorsal. . Cell. 1990;62:1019–1029. doi: 10.1016/0092-8674(90)90276-k. [DOI] [PubMed] [Google Scholar]
- 16.Kieran M, Blank V, Logeat F, Vandekerckhove J, Lottspeich F, LeBail O, Urban MB, Kourilsky P, Baeuerle PA, Israel A. The DNA binding subunit of NF-κB is identical to factor KBF1 and homologous to the reloncogene product. Cell. 1990;62:1007–1018. doi: 10.1016/0092-8674(90)90275-j. [DOI] [PubMed] [Google Scholar]
- 17.Mercurio F, DiDonato JA, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-κB–mediated signal transduction. Genes Dev. 1993;7:705–718. doi: 10.1101/gad.7.4.705. [DOI] [PubMed] [Google Scholar]
- 18.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–169. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 19.Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 20.Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-relproto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 21.Sha WC, Liou H-C, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 22.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 23.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of IκB-α proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 24.Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 25.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of the Iκ-Bα by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 26.DiDonato J, Mercurio F, Rosette C, Wu-Li J, Suyang H, Ghosh S, Karin M. Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol Cell Biol. 1996;16:1295–1304. doi: 10.1128/mcb.16.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 28.Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt H, Chen C-H, Rosen CA, Stewart CL. IκBα deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, et al. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 30.Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- 31.Gilmore TD, Koedood M, Piffat KA, White DW. Rel/NF-κB/IκB proteins and cancer. Oncogene. 1996;13:1367–1378. [PubMed] [Google Scholar]
- 32.Liou H-C, Sha WC, Scott ML, Baltimore D. Sequential induction of NF-κB/Rel family proteins during B-cell terminal differentiation. Mol Cell Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grumont RJ, Gerondakis S. The subunit composition of NF-κB complexes changes during B-cell development. Cell Growth Differ. 1994;5:1321–1331. [PubMed] [Google Scholar]
- 34.Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- 35.Bours V, Franzoso G, Azarenko V, Park S, Kannno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 36.Fujita T, Nolan GP, Liou H-C, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers. Genes Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- 37.Caamaño JH, Perez P, Lira SA, Bravo R. Constitutive expression of Bcl-3 in thymocytes increases the DNA binding of NF-κB (p50) homodimers in vivo. . Mol Cell Biol. 1996;16:1342–1348. doi: 10.1128/mcb.16.4.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McBurney MW, Sutherland LC, Adra CN, Leclair B, Rudnicki MA, Jardine K. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 1991;20:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 40.Swiatek PJ, Gridley T. Perinatal lethality and defects in hindbrain development in mice homozygous for a targeted mutation of the zinc finger gene Krox20. Genes Dev. 1993;7:2071–2084. doi: 10.1101/gad.7.11.2071. [DOI] [PubMed] [Google Scholar]
- 41.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober. 1992. Isolation and fractionation of mononuclear cell populations. In Current Protocols in Immunology. Greene Publishing Associates & Wiley-Interscience, John Wiley and Sons, New York. 3.0.1–3.1.5.
- 42.Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, Weih F. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med. 1996;184:1537–1541. doi: 10.1084/jem.184.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lernbecher T, Müller U, Wirth T. Distinct NF-κB/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365:767–770. doi: 10.1038/365767a0. [DOI] [PubMed] [Google Scholar]
- 44.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B cells from p50/ NF-κB knockout mice have selective defects in proliferation, differentiation, germ-line CHtranscription, and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 45.Lalor PA, Nossal GJV, Sanderson RD, McHeyzer-Williams MG. Functional and molecular characterization of single, (4-hydroxy-3-nitrophenyl) acetyl (NP)- specific, IgG1 +B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur J Immunol. 1992;22:3001–3011. doi: 10.1002/eji.1830221136. [DOI] [PubMed] [Google Scholar]
- 46.Snapper, C.M., and F.D. Finkelman. 1993. Immunoglobulin class switching. In Fundamental Immunology, 3 ed., W.E. Paul, editor. Raven Press, New York. pp. 837–863.
- 47.Snapper CM, Mond JJ. A model for induction of T cell–independent humoral immunity in response to polysaccharide antigens. J Immunol. 1996;157:2229–2233. [PubMed] [Google Scholar]
- 48.Stavnezer J. Antibody class switching. Adv Immunol. 1996;61:79–147. doi: 10.1016/s0065-2776(08)60866-4. [DOI] [PubMed] [Google Scholar]
- 49.Weih F, Warr G, Yang H, Bravo R. Multifocal defects in immune responses in RelB-deficient mice. J Immunol. 1997;158:5211–5218. [PubMed] [Google Scholar]
- 50.Kosco-Vilbois MH, Scheidegger D. Follicular dendritic cells: antigen retention, B cell activation, and cytokine production. Curr Top Microbiol Immunol. 1995;201:69–82. doi: 10.1007/978-3-642-79603-6_4. [DOI] [PubMed] [Google Scholar]
- 51.Clark EA. Regulation of lymphocytes by dendritic cells. J Exp Med. 1997;185:801–803. doi: 10.1084/jem.185.5.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 53.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α–induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 54.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α–induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 55.Wang C-Y, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 56.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-κB RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–961. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent. J Immunol. 1996;157:3974–3979. [PubMed] [Google Scholar]
- 58.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-κB activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 59.Kelsoe G. Life and death in germinal centers. Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- 60.Liu Y-J, Banchereau J. Mutant mice without B lymphocyte follicles. J Exp Med. 1996;184:1207–1211. doi: 10.1084/jem.184.4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y-J, Banchereau J. The paths and molecular controls of peripheral B-cell development. The Immunologist. 1996;4:55–66. [Google Scholar]
- 62.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381:751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- 63.Ahearn JM, Fischer MB, Croix D, Georg S, Ma M, Zia J, Zhou X, Howard RG, Rothstein TL, Carroll MC. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–262. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- 64.Förster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 65.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 66.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DC. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 67.Nielsen PJ, Georgiev O, Lorenz B, Schaffner W. B lymphocytes are impaired in mice lacking the transcriptional co-activator Bob1/OCA-B/OBF1. Eur J Immunol. 1996;26:3214–3218. doi: 10.1002/eji.1830261255. [DOI] [PubMed] [Google Scholar]
- 68.Renshaw BR, Fanslow WCI, Armitage RJ, Campbell KA, Liggitt D, Wright B, Davison BL, Maliszewski CR. Humoral immune responses in CD40 ligand-deficient mice. J Exp Med. 1994;180:1889–1900. doi: 10.1084/jem.180.5.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schubart DB, Rolink A, Kosco-Vilbois MH, Botteri F, Matthias P. B-cell–specific coactivator OBF-1/ OCS-B/Bob1 required for immune response and germinal centre formation. Nature. 1996;383:538–542. doi: 10.1038/383538a0. [DOI] [PubMed] [Google Scholar]
- 70.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand. Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 71.Kim U, Qin X-F, Gong S, Stevens S, Luo Y, Nussenzweig M, Roeder R. The B-cell–specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature. 1996;383:542–547. doi: 10.1038/383542a0. [DOI] [PubMed] [Google Scholar]
- 72.Rothe J, Lesslauer W, Lötscher H, Lang Y, Koebel P, Köntgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. . Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 73.Grilli M, Chiu J-S, Lenardo MJ. NF-κB and Rel-participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 74.Lin S-C, Stavnezer J. Activation of NF-κB/Rel by CD40 engagement induces the mouse germ line immunoglobulin Cγ1 promoter. Mol Cell Biol. 1996;16:4591–4603. doi: 10.1128/mcb.16.9.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berberich I, Shu GL, Clark EA. Cross-linking CD40 on B cells rapidly activates nuclear factor-κB. J Immunol. 1994;153:4357–4366. [PubMed] [Google Scholar]