

Abstract
CD40 activates nuclear factor kappa B (NFκB) and the mitogen-activated protein kinase (MAPK) subfamily, including extracellular signal–regulated kinase (ERK). The CD40 cytoplasmic tail interacts with tumor necrosis factor receptor–associated factor (TRAF)2, TRAF3, TRAF5, and TRAF6. These TRAF proteins, with the exception of TRAF3, are required for NFκB activation. Here we report that transient expression of TRAF6 stimulated both ERK and NFκB activity in the 293 cell line. Coexpression of the dominant-negative H-Ras did not affect TRAF6-mediated ERK activity, suggesting that TRAF6 may activate ERK along a Ras-independent pathway. The deletion mutant of TRAF6 lacking the NH2-terminal domain acted as a dominant-negative mutant to suppress ERK activation by full-length CD40 and suppress prominently ERK activation by a deletion mutant of CD40 only containing the binding site for TRAF6 in the cytoplasmic tail (CD40Δ246). Transient expression of the dominant-negative H-Ras significantly suppressed ERK activation by full-length CD40, but marginally suppressed ERK activation by CD40Δ246, compatible with the possibility that TRAF6 is a major transducer of ERK activation by CD40Δ246, whose activity is mediated by a Ras-independent pathway. These results suggest that CD40 activates ERK by both a Ras-dependent pathway and a Ras-independent pathway in which TRAF6 could be involved.
CD40 is a cell-surface glycoprotein on B lymphocytes, dendritic cells, follicular dendritic cells, and thymic epithelial cells (1), and a member of the TNF-α receptor superfamily that includes 55- and 75-kD TNF receptors (TNFR1 and TNFR2, respectively), CD30 receptor, low-affinity nerve growth factor receptor, lymphotoxin β receptor (LTβR), and Fas antigen (1).
The cytoplasmic domain of the TNFR superfamily members lacks sequences indicative of catalytic activity, but is associated with a signal transducer, TNFR-associated factor (TRAF; reference 2). The cytoplasmic domain of human CD40 consists of 62 amino acids at positions 196–257 and is associated with TRAF2, TRAF3, TRAF5, and TRAF6 (3–9) and Janus kinase (Jak)3 (10); the membrane proximal region of the cytoplasmic tail of CD40 contains a proline-rich region at positions 202–209 that is crucial for Jak3 binding (10). TRAF6 binds to the NH2-terminal cytoplasmic tail of CD40 at positions 210–225, although the possibility can not be excluded that full association of TRAF6 with CD40 may also require the COOH-terminal part at positions 226–249 (9). TRAF2, TRAF3, and TRAF5 bind to the COOH-terminal CD40 cytoplasmic domain at positions 226–249 (9), containing a minimum element, designated TIMct, responsible for TRAF2 and TRAF3 binding and signal transduction mediating nuclear factor kappa B (NFκB) activation (7).
Stimulation of CD40 results in activation of protein tyrosine kinases (PTKs), NFκB, the mitogen-activated protein kinase (MAPK), and Jak3/signal transducers and activators of transcription (STAT)3 (10–18), and it mediates critical biological effects in B cell growth, survival, and differentiation (19–27). It is known that TRAF2 and TRAF5 play a role in NFκB activation in signaling through CD40, as well as TNFR1, TNFR2, CD30, and lymphotoxin β receptor (6–8, 28–32). TRAF6 participates in NFκB activation signaled by CD40 and IL-1 receptor (9, 33). The TRAF family is characterized by a homologous COOH-terminal TRAF-COOH (TRAF-C) domain, an α-helical TRAF-NH2 (TRAF-N) domain, and an NH2-terminal RING finger with the exception of TRAF1 (2–6, 8, 9, 30, 33). The effector function of TRAF2 and TRAF5 toward NFκB activation is mediated by its NH2-terminal RING finger domain (6, 8, 30), whereas that of TRAF6 is mediated by the RING finger and zinc fingers (9, 33).
It has been reported that TRAF2 stimulates c-jun NH2-terminal kinase (JNK) activity in TNFR1 signaling (34–36), leading to the idea that TRAF2 may also play a role in JNK activation by CD40 (15, 16). However, the signaling pathway coupling CD40 to extracellular signal-regulated kinase (ERK) activation has remained unknown. To investigate which TRAF proteins might participate in ERK activation, we have performed transient transfection experiments in the human embryonic kidney 293 cell line. In the present study, we demonstrate that TRAF6 plays a role as a signal transducer in ERK activation by CD40, probably along a Ras-independent pathway.
Materials and Methods
Cell Culture.
Human embryonic kidney cell line 293 was maintained in DME supplemented with 10% FCS, 200 mM l-glutamine, and penicillin/streptomycin.
Plasmid Construction.
TRAF2, TRAF3, and human CD40 cDNA were obtained by the reverse transcription PCR (RT-PCR) by use of messenger RNA purified from B cell lymphomas, WEHI231 and Raji, with primers flanking the entire coding region, and then cloned into pMIKHygB, a gift from Dr. K. Maruyama (Tokyo Medical and Dental University, Tokyo, Japan). Deletion mutants of TRAF2 that lack a RING finger motif (amino acids [aa] 87–501) and the TRAF-C domain (aa 352–501) were constructed by PCR. Isolation of TRAF5 and TRAF6 cDNAs and construction of the expression vectors coding full-length TRAF5, full-length TRAF6, and the TRAF-C of TRAF6 were reported previously (9, 30). A deletion mutant of human CD40, designated CD40Δ246, that removes 32 aa at positions 226–257 from the cytoplasmic tail was previously described (9). Dominant-negative Raf-1 (aa 1–258) was constructed by reverse transcription PCR, according to Schaap et al. (37). Dominant-negative N17Ras (38) and glutathione-S-transferase (GST) fusion ERK2 (pGSTM1) were provided by Dr. G.M. Cooper (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA) and Dr. K. Takishima (National Defense Medical College, Saitama, Japan), respectively. Construction of a dominant-negative mutant of MAPK/ERK-activating kinase (MEK)1, designated MEK1DN, and a constitutively activated mutant of MEK1, designated MEK1EE, was previously described (39). All cDNA fragments were subcloned into pMIKHygB.
ERK-dependent activator and reporter constructs were prepared according to Seth et al. (40, 41). In brief, the fusion of the GAL4 DNA-binding domain and the nuclear localization signal (aa 1–147) and NH2-terminal transcriptional activation region of c-Myc (aa 2–103) were constructed in vector pGBT9 (Clontech, Palo Alto, CA), and its HindIII-SalI fragment was digested and subcloned into pMIKHygB, designated pGAL4MycN. A firefly luciferase reporter containing two copies of the GAL4 binding site adjacent to a thymidine kinase promoter was constructed into pGL2 (Promega, Madison, WI), designated pGAL4tkLuc. A previous study by Seth et al. (41) showed that, in cotransfected cells with the GAL4–c-Myc fusion construct and luciferase reporter, the phosphorylation state of the NH2-terminal c-Myc transactivation domain was increased in an ERK-dependent manner, resulting in an increase in the activity of the luciferase reporter gene (41). Therefore, this ERK-dependent activator and reporter system allows one to monitor ERK activity by measuring the luciferase reporter activity. NFκB-dependent luciferase constructs containing three copies of an NFκB binding site from immunogloblin κ light chain enhancer and the thymidine kinase promoter was constructed into pGL2, designed pkBtkLuc. All cDNAs and constructs were confirmed by DNA sequencing.
In Vitro MAPK Assay.
An in vitro kinase assay for endogenous ERK activity was performed as previously described (17). For estimating exogenous ERK2 kinase activity, 293 cells were cotransfected with 1 μg of GST-ERK2 and various amounts of plasmid constructs by the calcium phosphate coprecipitation method. The total amount of DNA was adjusted with empty vector at 10 μg. When 293 cells were cotransfected with 1 μg of CD40 cDNA, the cells were stimulated with anti-CD40 mAb (2 μg/ml) 24 h after transfection. In some experiments, cells were analyzed for the expression of CD40 on the surface by FACScan® (Becton Dickinson, Mountain View, CA) with FITC-conjugated anti–human CD40 mAb (5C3; PharMingen, San Diego, CA). The cell lysates were incubated with glutathione-coupled Sepharose 4B (Pharmacia, Uppsala, Sweden) at 4°C for 1 h. The Sepharose beads were pelleted down by centrifugation and washed twice with lysis buffer and with Tris-buffered saline (10 mM Tris-HCl, pH 7.2, 100 mM NaCl) supplemented with 5 mM benzamidine and 1 mM Na3VO4. The GST-ERK2 immobilized on Sepharose was directly subjected to a kinase reaction as previously described (17).
Luciferase Assay.
Cells were cotransfected with either full-length CD40 or CD40Δ246 and ERK-dependent activator and reporter plasmids consisted of 500 ng of pGAL4tkLuc and 100 ng of pGAL4MycN, or NFκB-dependent plasmid of 20 ng of pkBtkLuc, 1 μg of β-actin β-galactosidase (β-gal), and various amounts of expression plasmids by the calcium phosphate coprecipitation method. Total amounts of DNA were adjusted with empty vector at 10 μg. The cells were stimulated with anti-CD40 mAb 24 h after transfection. The cells were lysed in PicaGeneTM Cell Culture Lysis Reagent LCβ (Toyo Ink, Tokyo, Japan) 24 h after incubation, and luciferase activity was measured by use of a luciferase assay kit, PicaGeneTM (Toyo Ink), followed by normalization of transfection efficiency by β-gal activity.
Results and Discussion
TRAF6 Is a Signal Transducer of Both ERK and NFκB Activation.
Overexpression of TRAF2, TRAF5, and TRAF6 results in its aggregation and causes activation of the NFκB signaling pathway, which could be similar to that induced by ligand-triggered receptor aggregation (6, 8, 9, 30, 33). Therefore, to investigate which TRAF proteins might participate in ERK activation, we cotransfected human embryonic kidney 293 cells with various TRAF expression vectors and an GST-ERK2 fusion construct. After transfection, the GST–ERK2 fusion protein was precipitated by use of glutatione Sepharose 4B from the cell lysate, and the ERK activity was analyzed by an in vitro kinase assay, with myelin basic protein (MBP) used as an exogenous substrate. As shown in Fig. 1 A, transient expression of full-length TRAF6, but not TRAF2, TRAF3, or TRAF5, stimulated exogenous ERK activity approximately fivefold above the vector control, suggesting that transient expression of TRAF6 stimulates ERK activity in the 293 cell line.
Figure 1.
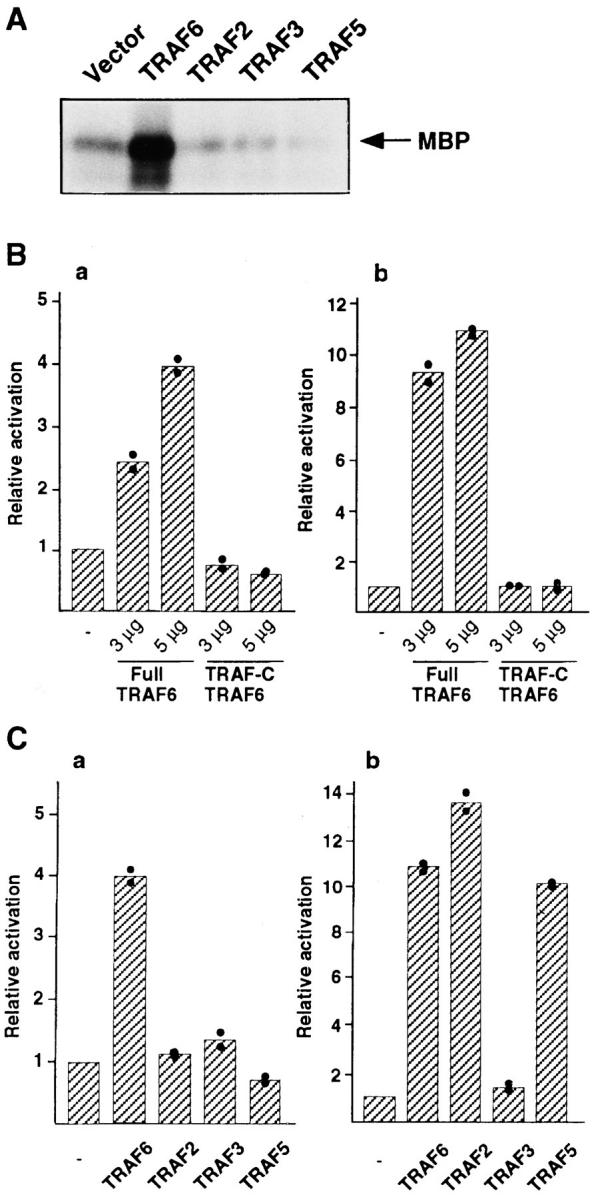
Transient expression of TRAF6-stimulated ERK activity in 293 cells. (A) Cells (106) were transiently transfected with expression plasmid of TRAF proteins (5 μg each) together with GST-ERK2. GST-ERK2 fusion protein was precipitated by use of glutathione Sepharose 4B from cell lysates 48 h after transfection and subjected to in vitro kinase assay using MBP as a substrate. Arrow, phosphorylated MBP in the autoradiogram. (B) Cells (106) were transiently transfected with 100 ng of ERK-dependent activator pGAL4MycN and 500 ng of the reporter pGAL4tkLuc (a) or 20 ng of the NFκB-dependent luciferase reporter pkBtkLuc (b), together with empty vector, full-length TRAF6 (3 or 5 μg), or the TRAF-C of TRAF6 (3 or 5 μg). Total amount of DNA was adjusted with empty vector at 10 μg. Luciferase activities were measured 48 h after transfection and normalized by β-gal expression. The extent of stimulation (relative activation) was calculated by comparison between luciferase activity given by the cells cotransfected with each TRAF expression plasmid and that of cells transfected with empty vector. (C) Cells (106) were transiently cotransfected with either TRAF2, TRAF3, or TRAF5 (5 μg each) and ERK– (a) or NFκB–dependent luciferase reporter (b). Cell lysates were subjected to luciferase assay 48 h after transfection. The results shown in B and C were obtained from parallel experiments. For comparison, the extent of stimulation in ERK and NFκB by transient expression of TRAF6 (5 μg) in B is also shown in C. The mean value of two independent experiments (column) was calculated from a mean value of a single experiment in triplicate (closed circles).
ERK is known to phosphorylate target protein within the cytosol (42), whereas, under certain conditions, ERK translocates to the nucleus to phosphorylate the nuclear substrate (41, 43–45). To investigate the effect of various TRAF proteins on ERK activity in the nucleus, we used an ERK- dependent activator and reporter system, established by Davis and coworkers (40, 41, and see Materials and Methods). Human 293 cells were transiently transfected with various TRAF expression vectors, together with an ERK-dependent activator, a fusion construct of the GAL4 DNA-binding domain and the nuclear localization signal/NH2-terminal transactivation domain of c-Myc, and a luciferase reporter plasmid containing GAL4 binding sites adjacent to a thymidine kinase promoter, as an ERK-dependent reporter (40, 41). In addition, to confirm the activity of TRAF proteins, we cotransfected 293 cells with various TRAF expression vectors and the NFκB-dependent luciferase reporter.
As shown in Fig. 1 B a, transient expression of full-length TRAF6 triggered activation of the cotransfected ERK-dependent reporter in a dose-dependent manner to a maximum of approximately fourfold above the vector control, whereas a deletion of the RING finger and zinc fingers of TRAF6 (TRAF-C) abolished the activity of ERK activation. Consistent with previous reports (9, 33), transient expression of full-length TRAF6, but not TRAF-C of TRAF6, caused NFκB activation in the 293 cell line (Fig. 1 B b), suggesting that TRAF6 stimulates both ERK and NFκB activity through its NH2-terminal region containing the RING finger and zinc fingers. Parallel experiments showed that transient expression of full-length TRAF2 and TRAF5 had no effect on ERK activity, whereas it stimulated NFκB activity efficiently (Fig. 1 C; 6, 8, 30–32). In this context, previous reports also ruled out the role of TRAF2 in ERK activation (34, 36). Transient expression of TRAF3 stimulated neither ERK nor NFκB activity, in agreement with previous reports (7, 9, 34). Taken together, these results suggest that TRAF6, but not TRAF2, TRAF3, or TRAF5, is a transducer of both ERK and NFκB activation.
TRAF6 Stimulates ERK That Is Largely Independent of Ras.
The Ras-dependent protein kinase cascade leading from various receptors to ERK is dependent on the protein kinase Raf-1, which activates a dual-specificity kinase, MEK (42). To investigate the role of Ras, Raf-1, and MEK1 in TRAF6-mediated ERK activation, we cotransfected 293 cells with a full-length TRAF6 and ERK-dependent activator and reporter, or GST-ERK, together with a dominant-negative mutant of H-Ras (N17Ras; 38), c-Raf-1, which contains only the NH2-terminal regulatory domain (DNRaf; 37), or MEK1, in which Asp208 was replaced with Ala (MEK1DN; 39). Some experiments were performed in parallel with the experiments depicted in Fig. 4. As shown in Fig. 2, coexpression of N17Ras had no effect on the activity of the cotransfected ERK-dependent reporter (Fig. 2 A) or on the exogenous ERK kinase activity (Fig. 2 B), which were increased five- to sixfold above the vector control by transient expression of TRAF6. The results suggest that TRAF6 may stimulate ERK activity along a Ras-independent pathway.
Figure 4.

ERK activation by cross-linking of full-length, but not that by CD40Δ246, was prominently suppressed by dominant negative mutant of Ras. Cells (106) were cotransfected with ERK– (A) or NFκB–dependent reporter (B) and either 1 μg of full-length CD40 (a) or CD40Δ246 (b), together with 5 μg of N17Ras, DNRaf, and MEKDN. Total amount of DNA was adjusted with empty vector at 10 μg. Cells were stimulated with anti-CD40 mAb 24 h after transfection. After 24 h incubation, cell lysates were subjected to luciferase assay. Relative activation of ERK and NFκB in each transfection was calculated as described above. The mean value of two to three independent experiments (column) was calculated from a mean value of a single experiment in triplicate (closed circle).
Figure 2.
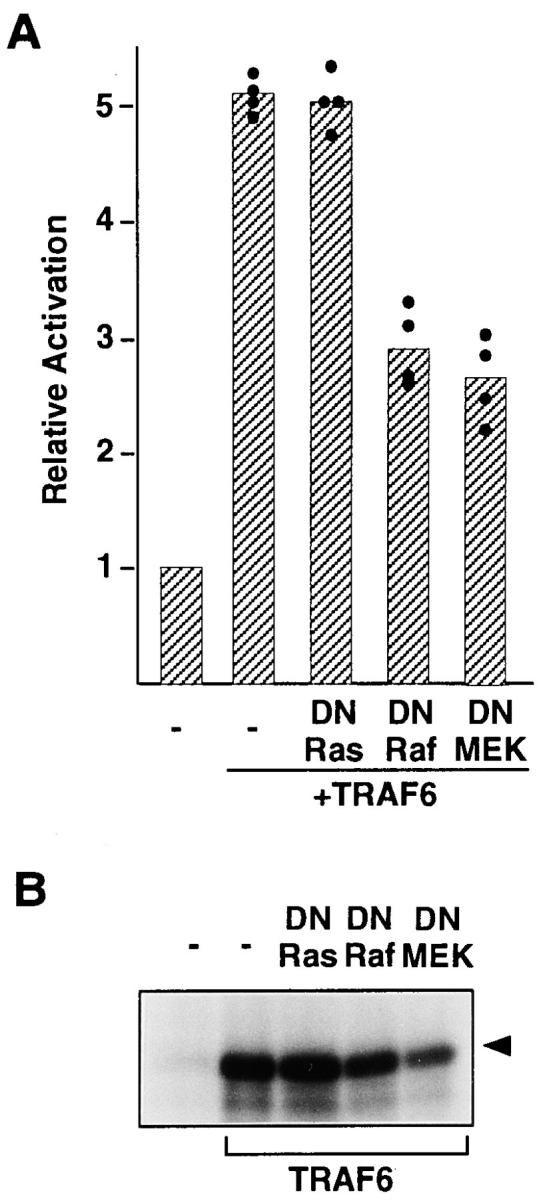
Inhibition of TRAF6-mediated ERK activity by dominant-negative mutant of Raf and MEK1, but not by Ras. Cells (106) were cotransfected with ERK-dependent activator and reporter (A) or 500 ng of GST-ERK2 (B) and full-length TRAF6 (5 μg), together with 5 μg of N17Ras (DN Ras), DNRaf, or MEK1DN (DN MEK). Cell lysates were prepared 48 h after transfection and subjected to luciferase assay (A) or in vitro kinase assay (B). The mean value of relative activity of ERK- dependent luciferase reporter (column) in four independent experiments was calculated from a mean value of a single experiment in triplicate (closed circles). Arrowhead, phosphorylated MBP in the autoradiogram.
The dominant-negative DNRaf and MEK1DN suppressed TRAF6-mediated ERK activity in the 293 cell line, but not entirely (Fig. 2, A and B). Although ERK activation is essentially induced by growth factors via a Ras-mediated pathway (42), the result could be interpreted as evidence that the diversified signal cascades regulate ERK activity, sometimes in a cell-type–specific manner, via c-Raf-1 and other possible MAPK kinase kinase in a Ras-independent fashion (46–51).
TRAF6 Is Involved in ERK Activation in CD40 Signaling.
The truncated mutant of TRAF proteins that lack the RING finger or the RING finger and zinc fingers acts as a transdominant-negative mutant to suppress NFκB activation in signaling through TNFR1, TNFR2, IL-1R, CD30, and CD40 receptor (6, 8, 9, 28, 30, 31, 33), in which the dominant-negative mutant may act specifically by replacing the corresponding endogenous wild-type TRAF protein.
To investigate whether TRAF6 could be involved in ERK activation by CD40, we cotransfected 293 cells with the TRAF-C of TRAF6 and ERK-dependent activator and reporter, or GST-ERK, together with full-length CD40 cDNA. Because a previous report suggested that the NH2-terminal CD40 cytoplasmic tail at positions 210–225 could be essential for TRAF6 binding and could function for NFκB activation (9), the involvement of TRAF6 in ERK activation by CD40 was also investigated by transient transfection of the TRAF-C of TRAF6 and ERK-dependent activator and reporter, or GST-ERK, into 293 cells coexpressing a deletion mutant of CD40, CD40Δ246 (9), that removes the COOH-terminal cytoplasmic tail at positions 226–257. Previous results showed that TRAF6 interacts with the CD40Δ246 construct in vivo (9).
As shown in Fig. 3, A and B, cross-linking of full-length CD40 and CD40Δ246 caused ERK activation approximately four- to sixfold above the level of unstimulated cells, comparable to the magnitude of the endogenous ERK activity that was transiently increased in 293 transfectants which constitutively expressed CD40 after stimulation with anti-CD40 mAb (data not shown). As shown in Fig. 3, A (a) and B both ERK-dependent reporter activity (Fig. 3 A) and the exogenous ERK kinase activity (Fig. 3 B) mediated by full-length CD40 were suppressed by coexpression of TRAF-C of TRAF6 in a dose-dependent manner to a maximum of 30–35% of inhibition, compatible with the notion that TRAF6-dependent and -independent pathways could be involved in ERK activation by CD40 (see below). Coexpression of the TRAF-C of TRAF6 caused prominent suppression of ERK activity mediated by cross-linking of CD40Δ246 in a dose-dependent manner, suggesting a major role of TRAF6 in ERK activation by CD40Δ246. It is not likely that the transdominant-negative mutant of TRAF6 nonspecifically diminished the activity of the ERK-dependent activator and reporter, because activation of the ERK-dependent reporter mediated by coexpression of catalytically active MEK1 (39) in the 293 cell line was not prevented by coexpression of TRAF-C of TRAF6 (data not shown).
Figure 3.
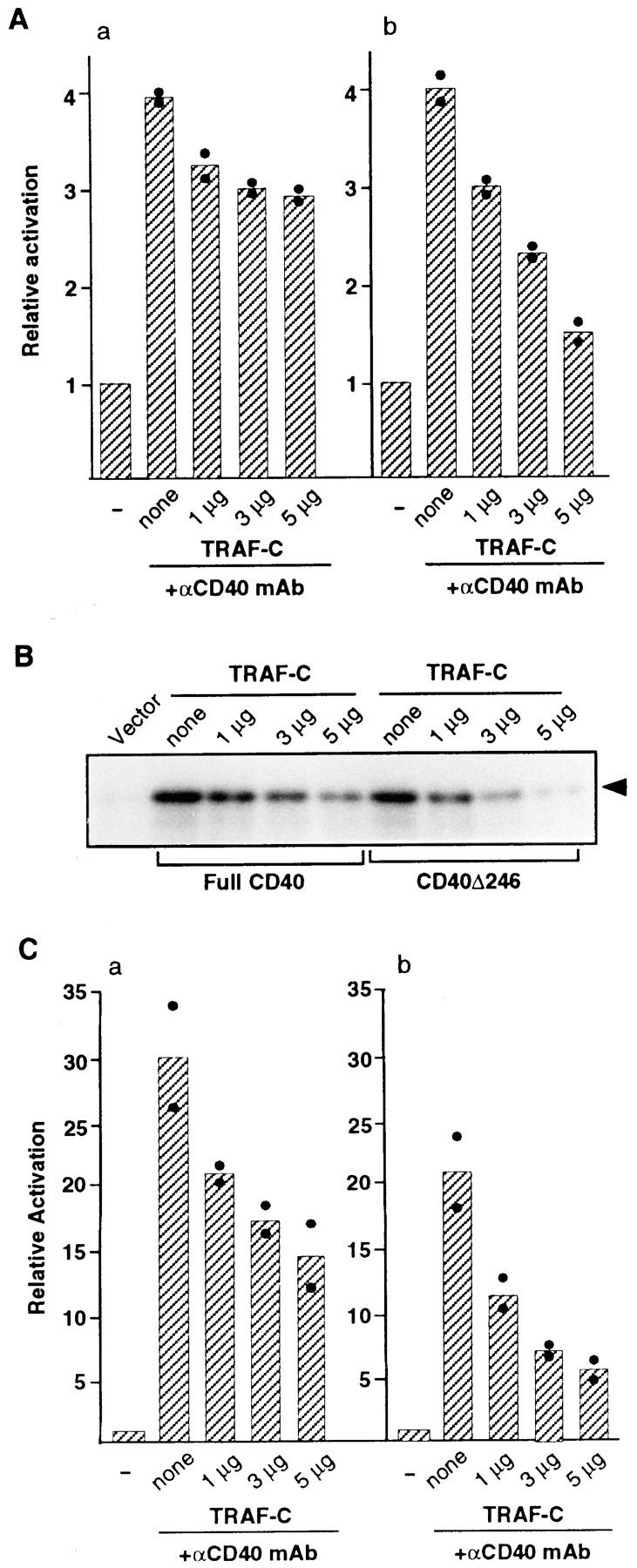
Transient expression of TRAF-C of TRAF6 caused inhibition of ERK and NFκB activity mediated by cross-linking of CD40. Cells (106) were transiently cotransfected with ERK-dependent activator and reporter (A), GST-ERK2 fusion construct (B), or NFκB-dependent reporter (C) and either l μg of full-length CD40 (a) or CD40Δ246 (b), together with 1, 3, or 5 μg of TRAF-C domain of TRAF6. Total amount of DNA was adjusted with empty vector at 10 μg. Cells were stimulated with anti-CD40 mAb 24 h after transfection. After 24 h of incubation, ERK activity was measured by luciferase assay (A) or in vitro kinase assay (B). Arrowhead, phosphorylated MBP in the autoradiogram is indicated by an arrowhead. NFκB activity was measured by luciferase assay (C). Relative activation of ERK and NFκB in 293 cells at each transfection was calculated as described above. The mean value of two independent experiments (column) was calculated from a mean value of a single experiment in triplicate (closed circles).
In control experiments, transient expression of the TRAF-C of TRAF6 caused prominent suppression of the activity of cotransfected NFκB-dependent reporter mediated by CD40Δ246 in the 293 cell line (Fig. 3 C b), consistent with previous reports (9). In contrast to ERK activation, the activation of NFκB activity by full-length CD40 was greater than that by CD40Δ246, and ∼50% of the activity was suppressed by coexpression of the TRAF-C of TRAF6 (Fig. 3 C a). Because of a similar frequency and the level of expression of full-length CD40 and CD40Δ246 in the 293 cell line in transient transfection assays (data not shown), the results seem compatible with the notion that distinct regions of the CD40 cytoplasmic domain could be required for full activation of NFκB in the 293 cell line.
CD40 Activates ERK by the Ras-dependent Pathway and Ras-independent Pathway in which TRAF6 Could Be Involved.
To investigate the role of Ras, Raf-1, and MEK in ERK activation by CD40, we cotransfected 293 cells with the ERK-dependent activator and reporter, or NFκB-dependent reporter as a control, and with N17Ras, DNRaf, or MEK1DN, together with either full-length CD40 or CD40Δ246. The experiments were performed in parallel with some of the experiments depicted in Fig. 2 A. Cotransfection of N17Ras, DNRaf, and MEK1DN caused ∼70% inhibition of ERK activity mediated by cross-linking of full-length CD40 (Fig. 4 A a). In contrast, coexpression of N17Ras and MEK1DN did not affect NFκB activation by both full-length CD40 and CD40Δ246, whereas DNRaf marginally suppressed NFκB activation (Fig. 4 B). In conjunction with the previous observation indicating activation of the Ras-mediated pathway by CD40 stimulation in B cells (52), the results support the possibility that the Ras-Raf-MEK–mediated pathway could be involved in ERK activation, but not NFκB, by cross-linking of CD40. Transient expression of N17Ras resulted in ∼30% inhibition of ERK activity mediated by CD40Δ246, compatible with the notion that TRAF6 is a major transducer of ERK activation by CD40Δ246, whose activity is mediated by a Ras-independent pathway.
Taken together, these results support the possibility that CD40 may activate ERK via Ras-dependent and -independent pathways in which TRAF6 could be involved. The inhibition by a dominant-negative mutant of MEK1 in ERK activity by full-length CD40 was more prominent than in ERK activity mediated by TRAF6 and by CD40Δ246, in which TRAF6 may play a major role. Because the MEK isoforms MEK1 and MEK2 are an ERK-specific kinase (53), these results might imply that preferential usage of MEK isoforms would differ in TRAF6-dependent and -independent pathways in CD40 signaling. In this context, it has been suggested that the signaling pathways leading to activation of MEK isoforms appear to differ, and that each MEK isoform could be preferentially used in a particular external stimulus in a certain type of cell (54–56). Further study is needed to clarify the signaling pathway coupling TRAF6 to ERK activation.
Given that full ERK activity is stimulated through TRAF6-dependent and -independent pathways in CD40 signaling, a similar extent of ERK activation by full-length CD40 and CD40Δ246 in the 293 cell line led to the idea that ERK activity could be regulated in full-length CD40 signaling in 293 cells, probably via activation of MAPK phosphatase or downregulation of MEK activity (57–60). In contrast to ERK activation in murine splenic resting B cells (17, 18), it was reported that CD40 stimulation did not cause ERK activation in human tonsil B cells (15, 16). Whether ERK activation is regulated in CD40 signaling, depending on the maturation stage of B cells, would be worth analyzing.
Acknowledgments
We are grateful to Drs. G.M. Cooper (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA), K. Maruyama (Tokyo Medical and Dental University, Tokyo, Japan), and K. Takishima (National Defense Medical College, Saitama, Japan) for reagents. We are also grateful to Drs. Tsunetsugu-Yokota and Z. Matsuda (NIID, Tokyo, Japan) for helpful discussions and reading of the manuscript, and to Mr. H. Shimizu, Ms. M. Takizawa, and K. Ishii for technical help.
This work was supported by a grant from the Agency of Technology and Science of the Japanese government and partly from the Ministry of Education and Science and Japanese Human Sciences Foundation to T. Takemori.
Footnotes
Abbreviations used in this paper: β-gal, β-galactosidase; aa, amino acids; DN, dominant negative; ERK, extracellular signal–regulated kinase; GST, glutathione-S-transferase; Jak, Janus kinase; JNK, c-jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MBP, myelin basic protein; MEK, MAPK/ERK-activating kinase; NFκB, nuclear factor kappa B; TRAF, TNFR-associated factor; TRAF-C, TRAF-COOH; TRAF-N, TRAF-NH2.
References
- 1.Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, van Kooten C, Liu YJ, Rousset F, Saeland S. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 2.Rothe M, Wong SC, Henzel WJ, Goeddel DV. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 3.Hu HM, O'Rourke K, Boguski MS, Dixit VM. A novel RING finger protein interacts with the cytoplasmic domain of CD40. J Biol Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- 4.Sato T, Irie S, Reed JC. A novel member of the TRAF family of putative signal transducing proteins binds to the cytosolic domain of CD40. FEBS Lett. 1995;358:113–118. doi: 10.1016/0014-5793(94)01406-q. [DOI] [PubMed] [Google Scholar]
- 5.Cheng G, Cleary AM, Ye ZS, Hong DI, Lederman S, Baltimore D. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- 6.Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 7.Cheng G, Baltimore D. TANK, a co-inducer with TRAF2 of TNF- and CD40L-mediated NF-kappaB activation. Genes Dev. 1996;10:963–973. doi: 10.1101/gad.10.8.963. [DOI] [PubMed] [Google Scholar]
- 8.Ishida TK, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J. TRAF5, a novel tumor necrosis factor receptor–associated factor family protein, mediates CD40 signaling. Proc Natl Acad Sci USA. 1996;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishida T, Mizushima SI, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, et al. Identification of TRAF6, a novel tumor necrosis factor receptor–associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. 1996;271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]
- 10.Hanissian SH, Geha RS. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity. 1997;6:379–387. doi: 10.1016/s1074-7613(00)80281-2. [DOI] [PubMed] [Google Scholar]
- 11.Lalmanach GA, Chiles TC, Parker DC, Rothstein TL. T cell–dependent induction of NF-kappa B in B cells. J Exp Med. 1993;177:1215–1219. doi: 10.1084/jem.177.4.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren CL, Morio T, Fu SM, Geha RS. Signal transduction via CD40 involves activation of lyn kinase and phosphatidylinositol-3-kinase, and phosphorylation of phospholipase C gamma 2. J Exp Med. 1994;179:673–680. doi: 10.1084/jem.179.2.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faris M, Gaskin F, Parsons JT, Fu SM. CD40 signaling pathway: anti-CD40 monoclonal antibody induces rapid dephosphorylation and phosphorylation of tyrosine-phosphorylated proteins including protein tyrosine kinase Lyn, Fyn, and Syk and the appearance of a 28-kD tyrosine phosphorylated protein. J Exp Med. 1994;179:1923–1931. doi: 10.1084/jem.179.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berberich I, Shu GL, Clark EA. Cross-linking CD40 on B cells rapidly activates nuclear factor–kappa B. J Immunol. 1994;153:4357–4366. [PubMed] [Google Scholar]
- 15.Sakata N, Patel HR, Terada N, Aruffo A, Johnson GL, Gelfand EW. Selective activation of c-Jun kinase mitogen-activated protein kinase by CD40 on human B cells. J Biol Chem. 1995;270:30823–30828. doi: 10.1074/jbc.270.51.30823. [DOI] [PubMed] [Google Scholar]
- 16.Berberich I, Shu G, Siebelt F, Woodgett JR, Kyriakis JM, Clark EA. Cross-linking CD40 on B cells preferentially induces stress-activated protein kinases rather than mitogen-activated protein kinases. EMBO (Eur Mol Biol Organ) J. 1996;15:92–101. [PMC free article] [PubMed] [Google Scholar]
- 17.Kashiwada M, Kaneko Y, Yagita H, Okumura K, Takemori T. Activation of mitogen-activated protein kinases via CD40 is distinct from that stimulated by surface IgM on B cells. Eur J Immunol. 1996;26:1451–1458. doi: 10.1002/eji.1830260708. [DOI] [PubMed] [Google Scholar]
- 18.Li YY, Baccam M, Waters SB, Pessin JE, Bishop GA, Koretzky GA. CD40 ligation results in protein kinase C-independent activation of ERK and JNK in resting murine splenic B cells. J Immunol. 1996;157:1440–1447. [PubMed] [Google Scholar]
- 19.Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, Bedell MA, Edelhoff S, Disteche CM, Simoneaux DK, et al. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science. 1993;259:990–993. doi: 10.1126/science.7679801. [DOI] [PubMed] [Google Scholar]
- 20.Aruffo A, Farrington M, Hollenbaugh D, Li X, Milatovich A, Nonoyama S, Bajorath J, Grosmaire LS, Stenkamp R, Neubauer M, et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell. 1993;72:291–300. doi: 10.1016/0092-8674(93)90668-g. [DOI] [PubMed] [Google Scholar]
- 21.Korthauer U, Graf D, Mages HW, Briere F, Padayachee M, Malcolm S, Ugazio AG, Notarangelo LD, Levinsky RJ, Kroczek RA. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:539–541. doi: 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- 22.DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, de Saint G, Basile CD40 ligand mutations in X-linked immunodeficiency with hyper-IgM. Nature. 1993;361:541–543. doi: 10.1038/361541a0. [DOI] [PubMed] [Google Scholar]
- 23.Fuleihan R, Ramesh N, Loh R, Jabara H, Rosen RS, Chatila T, Fu SM, Stamenkovic I, Geha RS. Defective expression of the CD40 ligand in X chromosome-linked immunoglobulin deficiency with normal or elevated IgM. Proc Natl Acad Sci USA. 1993;90:2170–2173. doi: 10.1073/pnas.90.6.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawabe T, Naka T, Yoshida K, Tanaka T, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T, Kikutani H. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167–178. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 25.Castigli E, Alt FW, Davidson L, Bottaro A, Mizoguchi E, Bhan AK, Geha RS. CD40-deficient mice generated by recombination-activating gene-2–deficient blastocyst complementation. Proc Natl Acad Sci USA. 1994;91:12135–12139. doi: 10.1073/pnas.91.25.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Foy TM, Laman JD, Elliott EA, Dunn JJ, Waldschmidt TJ, Elsemore J, Noelle RJ, Flavell RA. Mice deficient for the CD40 ligand (published erratum 1:613) Immunity. 1994;1:423–431. doi: 10.1016/1074-7613(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 27.Clark EA, Ledbetter JA. How B and T cells talk to each other. Nature. 1994;367:425–428. doi: 10.1038/367425a0. [DOI] [PubMed] [Google Scholar]
- 28.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 29.Gedrich RW, Gilfillan MC, Duckett CS, Van Dongen DJ, Thompson CB. CD30 contains two binding sites with different specificities for members of the tumor necrosis factor receptor–associated factor family of signal transducing proteins. J Biol Chem. 1996;271:12852–12858. doi: 10.1074/jbc.271.22.12852. [DOI] [PubMed] [Google Scholar]
- 30.Nakano H, Oshima H, Chung W, Williams AL, Ware CF, Yagita H, Okumura K. TRAF5, an activator of NF-kappaB and putative signal transducer for the lymphotoxin-beta receptor. J Biol Chem. 1996;271:14661–14664. doi: 10.1074/jbc.271.25.14661. [DOI] [PubMed] [Google Scholar]
- 31.Lee SY, Lee SY, Kandala G, Liou ML, Liou HC, Choi Y. CD30/TNF receptor–associated factor interaction: NF-kappa B activation and binding specificity. Proc Natl Acad Sci USA. 1996;93:9699–9703. doi: 10.1073/pnas.93.18.9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aizawa S, Nakano H, Ishida T, Horie R, Nagai M, Ito K, Yagita H, Okumura K, Inoue J, Watanabe T. Tumor necrosis factor receptor–associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediated NFkappaB activation. J Biol Chem. 1997;272:2042–2045. doi: 10.1074/jbc.272.4.2042. [DOI] [PubMed] [Google Scholar]
- 33.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 34.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 35.Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, Levrero M. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science. 1997;275:200–203. doi: 10.1126/science.275.5297.200. [DOI] [PubMed] [Google Scholar]
- 36.Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor α–induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO (Eur Mol Biol Organ) J. 1997;16:1080–1092. doi: 10.1093/emboj/16.5.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaap D, van der Wal J, Howe LR, Marshall CJ, van Blitterswijk WJ. A dominant-negative mutant of raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic p21ras. J Biol Chem. 1993;268:20232–20236. [PubMed] [Google Scholar]
- 38.Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okazaki K, Sagata N. MAP kinase activation is essential for oncogenic transformation of NIH3T3 cells by Mos. Oncogene. 1995;10:1149–1157. [PubMed] [Google Scholar]
- 40.Seth A, Alvarez E, Gupta S, Davis RJ. A phosphorylation site located in the NH2-terminal domain of c-Myc increases transactivation of gene expression. J Biol Chem. 1991;266:23521–23524. [PubMed] [Google Scholar]
- 41.Seth A, Gonzalez FA, Gupta S, Raden DL, Davis RJ. Signal transduction within the nucleus by mitogen-activated protein kinase. J Biol Chem. 1992;267:24796–24804. [PubMed] [Google Scholar]
- 42.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 43.Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lenormand P, Sardet C, Pages G, L'Allemain G, Brunet A, Pouyssegur J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukuda M, Gotoh Y, Nishida E. Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO Eur Mol Bio Organ) J. 1997;16:1901–1908. doi: 10.1093/emboj/16.8.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burgering BM, de Vries-Smits AMM, Medema RH, van Weeren PC, Tertoolen LG, Bos JL. Epidermal growth factor induces phosphorylation of extracellular signal-regulated kinase 2 via multiple pathways. Mol Cell Biol. 1993;13:7248–7256. doi: 10.1128/mcb.13.12.7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buscher D, Hipskind RA, Krautwald S, Reimann T, Baccarini M. Ras-dependent and -independent pathways target the mitogen-activated protein kinase network in macrophages. Mol Cell Biol. 1995;15:466–475. doi: 10.1128/mcb.15.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reuter CW, Catling AD, Jelinek T, Weber MJ. Biochemical analysis of MEK activation in NIH3T3 fibroblasts. Identification of B-Raf and other activators. J Biol Chem. 1995;270:7644–7655. doi: 10.1074/jbc.270.13.7644. [DOI] [PubMed] [Google Scholar]
- 49.Winston BW, Lange CC, Gardner AM, Johnson GL, Riches DW. Tumor necrosis factor alpha rapidly activates the mitogen-activated protein kinase (MAPK) cascade in a MAPK kinase kinase-dependent, c-Raf-1–independent fashion in mouse macrophages. Proc Natl Acad Sci USA. 1995;92:1614–1618. doi: 10.1073/pnas.92.5.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Q, Lin TH, Der CJ, Juliano RL. Integrin-mediated activation of MEK and mitogen-activated protein kinase is independent of Ras. J Biol Chem. 1996;271:18122–18127. doi: 10.1074/jbc.271.30.18122. [DOI] [PubMed] [Google Scholar]
- 51.Cowen DS, Sowers RS, Manning DR. Activation of a mitogen-activated protein kinase (ERK2) by the 5-hydroxytryptamine 1A receptor is sensitive not only to inhibitors of phosphatidylinositol 3–kinase, but to an inhibitor of phosphatidylcholine hydrolysis. J Biol Chem. 1996;271:22297–22300. doi: 10.1074/jbc.271.37.22297. [DOI] [PubMed] [Google Scholar]
- 52.Gulbins E, Brenner B, Schlottmann K, Koppenhoefer U, Linderkamp O, Coggeshall KM, Lang F. Activation of the Ras signaling pathway by the CD40 receptor. J Immunol. 1996;157:2844–2850. [PubMed] [Google Scholar]
- 53.Zheng CF, Guan KL. Properties of MEKs, the kinases that phosphorylate and activate the extracellular signal–regulated kinases. J Biol Chem. 1993;268:23933–23939. [PubMed] [Google Scholar]
- 54.Jelinek T, Catling AD, Reuter CW, Moodie SA, Wolfman A, Weber MJ. RAS and RAF-1 form a signalling complex with MEK-1 but not MEK-2. Mol Cell Biol. 1994;14:8212–8218. doi: 10.1128/mcb.14.12.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Downey GP, Butler JR, Brumell J, Borregaard N, Kjeldsen L, Sue A, Quan AK, Grinstein S. Chemotactic peptide-induced activation of MEK-2, the predominant isoform in human neutrophils. Inhibition by wortmannin. J Biol Chem. 1996;271:21005–21011. doi: 10.1074/jbc.271.35.21005. [DOI] [PubMed] [Google Scholar]
- 56.Winston BW, Remigio LK, Riches DW. Preferential involvement of MEK1 in the tumor necrosis factor-alpha–induced activation of p42mapk/erk2 in mouse macrophages. J Biol Chem. 1995;270:27391–27394. doi: 10.1074/jbc.270.46.27391. [DOI] [PubMed] [Google Scholar]
- 57.Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P. Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- 58.Xu S, Robbins D, Frost J, Dang A, Lange CC, Cobb MH. MEKK1 phosphorylates MEK1 and MEK2 but does not cause activation of mitogen-activated protein kinase. Proc Natl Acad Sci USA. 1995;92:6808–6812. doi: 10.1073/pnas.92.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bokemeyer D, Sorokin A, Yan M, Ahn NG, Templeton DJ, Dunn MJ. Induction of mitogen-activated protein kinase phosphatase 1 by the stress-activated protein kinase signaling pathway but not by extracellular signal-regulated kinase in fibroblasts. J Biol Chem. 1996;271:639–642. doi: 10.1074/jbc.271.2.639. [DOI] [PubMed] [Google Scholar]
- 60.Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO (Eur Mol Biol Organ) J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]