

Abstract
It has been suggested that Fas ligand–Fas receptor interactions are involved in the regulation of eosinophil apoptosis and that dysfunctions in this system could contribute to the accumulation of these cells in allergic and asthmatic diseases. Here, we demonstrate that nitric oxide (NO) specifically prevents Fas receptor–mediated apoptosis in freshly isolated human eosinophils. In contrast, rapid acceleration of eosinophil apoptosis by activation of the Fas receptor occurs in the presence of eosinophil hematopoietins. Analysis of the intracellular mechanisms revealed that NO disrupts Fas receptor–mediated signaling events at the level of, or proximal to, Jun kinase (JNK), but distal to sphingomyelinase (SMase) activation and ceramide generation. In addition, activation of SMase occurs downstream of an interleukin 1 converting enzyme–like (ICE-like) protease(s) that is not blocked by NO. However, NO prevents activation of a protease that targets lamin B1. These findings suggest a role for an additional NO-sensitive apoptotic signaling pathway that amplifies the proteolytic cascade initialized by activation of the Fas receptor. Therefore, NO concentrations within allergic inflammatory sites may be important in determining whether an eosinophil survives or undergoes apoptosis upon Fas ligand stimulation.
Inhibition of eosinophil apoptosis has been proposed as a key mechanism for the development of blood and tissue eosinophilia in diseases such as bronchial asthma and other allergic disorders (1, 2). The delay of eosinophil death might be due, at least in part, to overproduction of T cell–derived cytokines (2–9). Besides cytokines, eosinophil apoptosis also seems to be regulated by at least one member of the TNF/ nerve growth factor (NGF) receptor superfamily, namely the Fas receptor (CD95/APO-1) (10–13). Cross-linking of the Fas receptor is associated with the induction of apoptosis in eosinophils from normal individuals. In contrast, blood and tissue eosinophils derived from eosinophilic donors often do not undergo cell death after Fas receptor cross-linking, although Fas protein is normally expressed in these cells, suggesting that receptor activity is regulated as previously observed in other systems (13). Fas receptor susceptibility does not seem to be regulated by cytokines that promote eosinophil survival (10, 11).
Patients with bronchial asthma and allergic rhinitis show an increased level of nitric oxide (NO)1 in exhaled air (14, 15). NO originates from the biotransformation of l-arginine to l-citrulline by an enzyme called NO synthase (NOS; 16). There is evidence that the inducible isoform of NOS (iNOS) is expressed in the bronchial mucosa of patients with bronchial asthma, but not of normal control individuals (17), suggesting that increased levels of NO may result from increased NO production by iNOS. Recently, it has been demonstrated that eosinophils themselves are a source of NO production in eosinophilic inflammation (18).
The pathophysiological consequences of increased NO production in allergic diseases are not yet known. However, it is clear that NO has effects on immune responses. For example, NO inhibits both the proliferation of Th1s and their production of IL-2 and IFN-γ, as demonstrated in several infectious disease models (19, 20). In contrast, Th2s are not affected by NO (21). These data suggest that increased amounts of NO may contribute to a preferential Th2 response in allergic diseases of the respiratory tract (22).
Consequently, we were interested in whether NO may also play a role in later events of Th2 responses such as inhibition of eosinophil apoptosis (1, 2). A possible involvement of NO in defective Fas ligand–Fas receptor interactions was concluded from studies performed in mice where NO protected liver cells from TNF-induced apoptosis (23). Since at least one signaling cascade in the induction of cell death is common to both TNF and Fas receptors (24), we hypothesized that NO, at least in some cellular systems, may also counterregulate Fas receptor–mediated apoptosis. In this study, we demonstrate that NO mediates a functional defect in the Fas receptor signal transduction cascade in human eosinophils.
Materials and Methods
Reagents and Antibodies
All cell cultures were performed using complete culture medium, which was RPMI 1640 supplemented with 10% fetal calf serum (both Life Technologies, Basel, Switzerland). SNAP (S-nitroso- N-acetylpenicillamine), dibutyryl-cGMP, dibutyryl-cAMP, C2-ceramide, C6-ceramide, C2-dihydroceramide, DAG (1,2-dioctanoyl-sn-glycerol), IBMX (3-isobutyl-1-methylxanthine), LY 83583 (6-anilinoquinoline-5,8-quinone), and GST-fusion protein of c-Jun (GST-c-Jun, 1–79) were from Biomol (Plymouth Meeting, PA). Azide and hydroxylamine hydrochloride were obtained from Fluka (Buchs, Switzerland). The IL-1 converting enzyme (ICE) inhibitor II (N-acetyl-Tyr-Val-Ala-Asp-chloromethylketone; Ac-YVAD-cmk) was obtained from Bachem (Bubendorf, Switzerland). NG-monomethyl-l-arginine (NMMA) was from Alexis Corp. (Läufelfingen, Switzerland). The concentrations used for all these reagents are indicated in the text and figure legends. Anti-CD16 mAb microbeads were from Miltenyi Biotec (Bergisch-Gladbach, Germany). Unconjugated (clone CH-11, IgM) and FITC-conjugated (clone UB2, IgG1) anti-CD95 mAb were from Immunotech (Marseille, France). For functional assays, anti-Fas mAb (IgM) was used at 1 μg/ml. Anti–Jun kinase (JNK) Ab (C-17) was from Santa Cruz Biotechnology (Santa Cruz, CA). Anti–human lamin B1 mAb (clone 101-B7, IgG1) was from Matritech Inc. (Cambridge, MA). Neutralizing anti–GM-CSF mAb was purchased from R&D Sys. Inc. (Minneapolis, MN). LPS was from Sigma Chemical Co. (Buchs, Switzerland). IL-5 and IFN-γ were obtained from Genzyme (Cambridge, MA). Unless stated otherwise, all other reagents were obtained from Sigma Chemical Co.
iNOS Messenger RNA Expression in Nasal Polyp Tissues
RNA was isolated from fresh nasal polyp and inferior turbinate tissues by using an RNA purification kit (Stratagene, Heidelberg, Germany). The inferior turbinate tissues were obtained from two individuals undergoing nasal surgery for deviation of the septum and used as noninflamed control tissues. First strand cDNA synthesis and PCR amplification was conducted as described (2, 9, 13, 25). Primers used for PCR amplification were oligonucleotides recognizing sequences in iNOS (Clontech, Palo Alto, CA).
NO Production in Eosinophilic Inflammation (Nasal Polyposis)
NO was measured in the nasal cavity of 20 patients with nasal polyps and 20 normal control individuals. Both groups were age and sex matched. Analysis was performed with a chemiluminescence analyzer (Eco Physics, Dürnten, Switzerland), which measures emitted light caused by the reaction NO + O3 = NO2 + O2. One nasal olive was closed using an occlusive nasal olive from a rhinomanometer (Homoth, Hamburg, Germany), and connected to a syringe that had access to the nasal vestibulum. The contralateral nostril was open. Subjects were asked to keep the mouth closed. Thus, breathing was carried out through the open nostril only. This allowed analysis of the local NO concentration with free flow of air from one nostril to the other via the nasopharynx. Using the syringe, 20 ml of nasal air was aspirated over a time period of 15 s. The aspirated air was injected into the analyzer. NO concentrations were recorded by an on-line microcomputer and given in parts per million. The analyzer was calibrated before each experiment using certified NO mixtures. NO concentrations in the room were <5 parts per billion.
Eosinophil Purification
Eosinophils were purified as previously described (7–9, 13, 25, 26). The resulting cell populations contained 99% eosinophils as controlled by staining with Diff-Quik (Baxter, Düdingen, Switzerland) and light microscopy.
Coculture of Fas Receptor–activated Eosinophils with U937 Cells
The human monocyte-like cell line U937 was received from American Type Culture Collection (Rockville, MD). U937 cells were stimulated with 10 ng/ml LPS and 100 U/ml IFN-γ for 24 h before coculture to induce iNOS expression. Freshly purified eosinophils were incubated with 1 μg/ml anti-Fas mAb (IgM) for 1 h before coculture. Pretreated U937 cells and eosinophils were cocultured at a 1:1 ratio for 24 h at 37°C in 5% CO2 and 95% air in a humidified atmosphere. In additional experiments, the NOS inhibitor NMMA (1 mM) was included at the start of U937 stimulation. Furthermore, to block possible mediators, an inhibitor of soluble guanylyl cyclase (sGC), LY 83583 (10 μM; reference 27), and a neutralizing anti–GM-CSF mAb (20 μg/ml) were added to the coculture. To determine eosinophil apoptosis in this system, cells were morphologically examined. To this end, cytospin preparations were made, stained with Diff-Quik, and photographed under a Zeiss Axioscope microscope at 1,000 magnifications.
Determination of Cell Death
Eosinophils (106/ml) were cultured in the presence or absence of anti-Fas mAb or ceramides in the indicated conditions and for the indicated times. Cell death of eosinophils was assessed by uptake of 1 mM ethidium bromide and flow cytometric analysis (EPICS XL; Coulter, Hialeah, FL) as previously described (7–9, 13).
Determination of Apoptosis
Determination of DNA Fragmentation.
Oligonucleosomal DNA fragmentation, a characteristic feature of cells undergoing apoptosis, was assessed by flow cytometry as previously described (13).
Analysis of Phosphatidylserine Redistribution.
Phosphatidylserine (PS) is normally confined to the inner plasma membrane leaflet. In contrast, PS appears on the external leaflet in apoptotic cells. Annexin V is a PS-binding protein and can be used to detect apoptotic cells (28). The appearance of PS on the eosinophil cell surface in response to the various indicated culture conditions was assessed by flow cytometry (EPICS XL) using a commercial apoptosis detection kit according to the manufacturer's instructions (R&D Sys., Inc.).
Determination of Acidic Sphingomyelinase Activity
Purified eosinophils were cultured in the presence or absence of anti-Fas mAb in the indicated conditions and for the indicated times. The ICE inhibitor II Ac-YVAD-cmk (1 μM) was added 2 h before anti-Fas mAb stimulation. Sphingomyelinase (SMase) activity in eosinophils was determined with an in vitro assay as previously described (29).
Determination of JNK Activation
Eosinophils were cultured in the presence or absence of C2-ceramide (C2) in the indicated conditions for 60 min. JNK activation was measured as previously described (30).
Detection of Total Intracellular Proteolytic Activity
Fluorescamine Assay.
Protein fragmentation occuring during Fas receptor–mediated apoptosis in eosinophils was first determined by the fluorescamine assay as previously described (31). In brief, purified eosinophils were cultured in the presence or absence of anti-Fas mAb in the indicated conditions and for the indicated times. Cells (106) were washed twice with 1 ml homogenization buffer (10 mM Hepes, 1 mM EGTA, pH 7.4, and 10 mM PMSF), and then resuspended in 100 μl of this buffer. Cells were lysed by sonication. 500 μl of fluorescamine solution (0.3 mg/ml in acetone) were added to a quartz cuvette (Hellma, Basel, Switzerland) containing 1 ml of a 1:100 dilution of cell lysates in homogenization buffer. Fluorescence was measured at excitation and emission wavelengths of 390 nm and 480 nm, respectively, using a FluoroMax spectrophotometer (Spex Industries Inc., Edison, NJ). Intracellular proteolytic activity was determined as fluorescence intensity (cps)/μg protein cell lysate added to the cuvette. Protein concentrations in the cell lysates were measured by a Bradford protein assay (BioRad Labs., München, Germany).
Zymogram Gels.
To further confirm the data obtained using the fluorescamine assay, we used Zymogram gels (Novex, San Diego, CA). These 0.1% gelatin-containing and standardized 10% SDS-polyacrylamide gels can be used to detect a wide variety of proteases that can use gelatin as a substrate (32). After cell culture, eosinophils (5 × 105) were washed with cold PBS, resuspended in 50 μl of sample buffer (0.06 M Tris-HCl, pH 6.8, 2% SDS, 0.0025% bromophenol blue, and 10% glycerol), and lysed by sonication. Electrophoresis was conducted under standard conditions according to the manufacturer's instructions. Gels were incubated in renaturing buffer (0.25% Triton X-100) for 30 min, and then briefly washed with developing buffer (5 mM Tris-HCl, pH 7.6, 20 mM NaCl, 0.5 mM CaCl2, and 0.002% Brij 35). The gels were stained in 0.5% Coomassie Blue R 250, and partially destained in destaining solution (10% CH3COOH, and 40% CH3OH) to make digested gelatin spots visible.
Immunoblotting
We measured expression and/or degradation of lamin B1 in eosinophils that had been cultured in the presence or absence of anti-Fas mAb in the indicated conditions and for the indicated times. Eosinophils (106) were washed, resuspended in Lämmli buffer, and lysed by sonication. Equal sample volumes of whole cell lysates were applied to an 8% SDS polyacrylamide gel, and separated proteins were electrotransferred onto a nitrocellulose filter (Hybond–enhanced chemiluminescence [ECL]; Amersham Intl., Buckinghamshire, UK). The blots were blocked at room temperature for 1 h in blocking solution (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20, and 5% BSA). Filters were incubated in 1% BSA blocking solution containing 0.5 μg/ml anti–lamin B1 mAb at room temperature for 1–2 h, followed by incubation with horseradish peroxidase (HRP)-conjugated anti– murine IgG1 mAb (1:2,000; Amersham Intl.). Blots were developed by an ECL technique (ECL kit; Amersham Intl.) according to the manufacturer's instructions.
Results
Increased NO Production in Nasal Polyp Tissues.
We have previously reported that eosinophils in nasal polyp tissues often have a decreased susceptibility to undergo apoptosis after Fas receptor activation (13). Recently published work suggests that NO may protect liver cells from TNF receptor–mediated apoptosis (23). To verify a potential role of NO for Fas receptor resistance in eosinophils, we measured NO concentrations in the nasal cavity in 20 healthy subjects and 20 patients with nasal polyps. As shown in Fig. 1 A, NO concentrations were increased by fourfold in patients with nasal polyps, suggesting increased production of NO in this model of eosinophilic inflammation.
Figure 1.
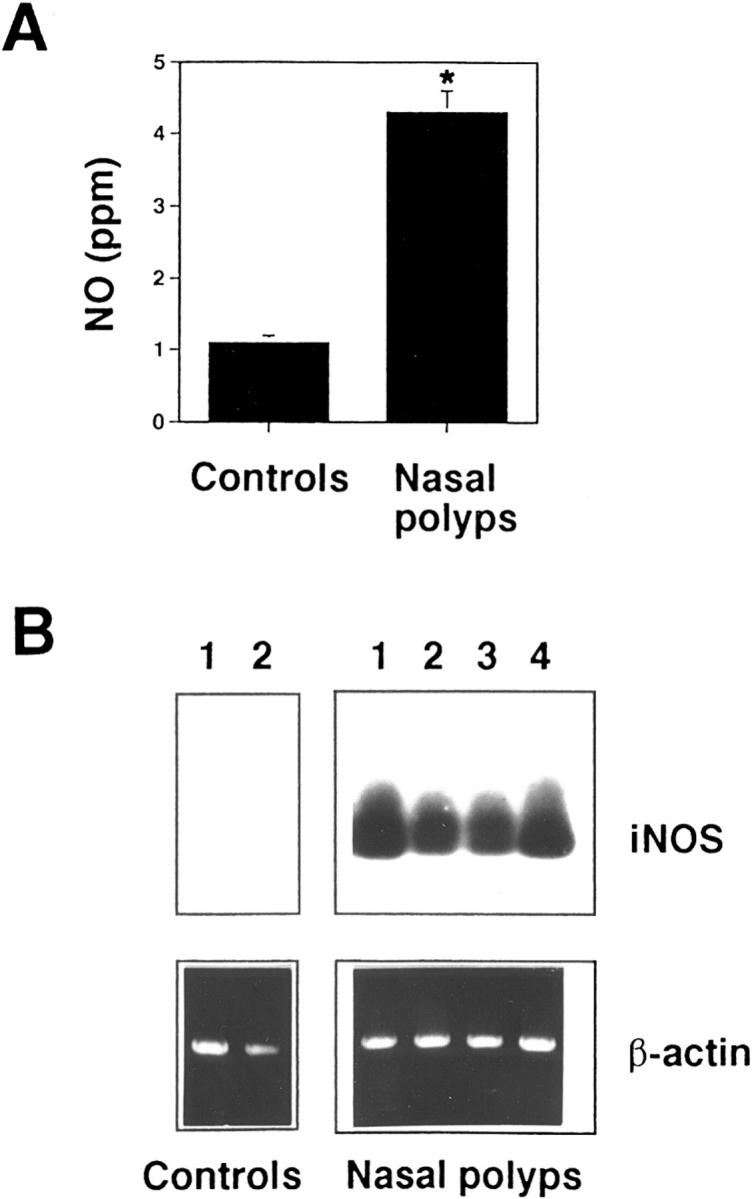
Role of NO in eosinophilic inflammation. (A) Increased NO concentrations in the nasal cavity of patients with nasal polyps compared to normal control individuals. Measurements were performed with a chemiluminescence analyzer as described in Materials and Methods. Values are means ± SEM. This figure includes results from 20 different individuals in each group (*, P < 0.001). (B) iNOS mRNA is expressed in nasal polyp tissues. In contrast, under the same experimental conditions, control nasal tissues expressed little or no iNOS mRNA. RNA was isolated, and first-strand synthesis was performed. Human iNOS cDNA was amplified by PCR using specific iNOS primers. The PCR products were electrophoresed on a 1% agarose gel, transferred onto a nitrocellulose filter, and hybridized with a random-primed fluorescein-12-dUTP–labeled iNOS probe. As a control, human β-actin cDNA was amplified and PCR products were stained by ethidium bromide in an agarose gel. Data from four different patients with nasal polyps and two control individuals are presented.
Increased NO production is often achieved by activation of the iNOS gene (22). Consequently, we investigated the pattern of iNOS gene expression by reverse transcriptase PCR in nasal polyp tissues. The results are shown in Fig. 1 B. Nasal polyp, but not control nasal tissues, expressed detectable iNOS messenger RNA (mRNA), as indicated by the presence of a PCR product with the expected size of 257 bp. iNOS specificity of this PCR product was demonstrated by hybridization to an iNOS-specific probe. β-actin–specific amplification products were of similar intensity between all samples, suggesting equality of the RNA populations. Thus, it is likely that the expression of iNOS in nasal polyp tissues accounts for the increased NO production measured in the nasal cavities of these patients.
NO Inhibits Fas Receptor–induced Apoptosis of Eosinophils.
To more directly determine a possible role of NO for the reduction of eosinophil sensitivity to Fas receptor– mediated apoptosis, we cocultured stimulated monocytic-like U937 cells with anti–Fas receptor–activated freshly purified eosinophils. U937 cells were stimulated with LPS and IFN-γ to induce iNOS expression and NO production. As shown in Fig. 2, eosinophils undergo normal Fas receptor– mediated apoptosis under conditions where U937 cells were not stimulated. In contrast, LPS– and IFN-γ–stimulated U937 cells prevented eosinophil apoptosis in this coculture system. This suggests that activated U937 cells produce a factor that counterregulates Fas receptor–mediated death. Moreover, addition of 1 mM NMMA (NOS inhibitor) or 10 μM LY 83583 (sGC inhibitor; NO signals via sGC; reference 27) abolished the effect of stimulated U937 cells on Fas receptor–mediated eosinophil apoptosis (Fig. 2). In addition, since activated U937 cells may also produce eosinophil survival factors such as GM-CSF, we also used a neutralizing anti–human GM-CSF mAb in this system, and observed no effect on Fas receptor–mediated eosinophil apoptosis (not presented). These data suggest that NO, but not GM-CSF, is involved in U937-mediated Fas receptor resistance of eosinophils.
Figure 2.
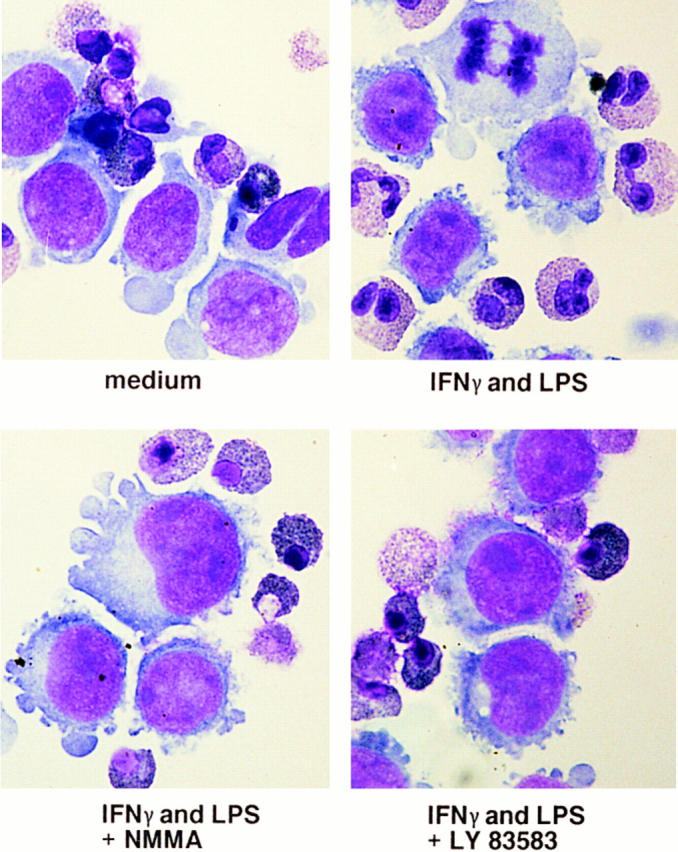
Activated U937 cells prevent Fas receptor–mediated apoptosis in eosinophils. Purified eosinophils were stimulated with anti-Fas mAb, and cocultured with U937 cells for 24 h. LPS– and IFN-γ–stimulated U937 cells produce a factor that prevents eosinophils apoptosis. In contrast, unstimulated U937 cells or cocultures incubated with the NOS inhibitor NMMA or the sGC inhibitor LY 83583 did not prevent eosinophil apoptosis (original magnification: 1,000). Neutralizing anti–GM-CSF mAb did not counteract the effects of the factor that prevents eosinophil apoptosis in this system (not presented).
To complement these studies and to establish a functional approach without the need of a coculture system, we generated NO in pure eosinophil cultures either by endogenous catalase from azide or hydroxylamine or by hydrolysis from SNAP (33, 34). As shown in Fig. 3 A, cell viability of purified eosinophils from individuals with normal or slightly increased eosinophil numbers averaged 63 ± 2% after 24 h of in vitro culture. Anti-Fas mAb treatment accelerated eosinophil death by twofold (31 ± 3%). Azide, in the range of 0.02 to 1.2 mM, hydroxylamine, in the range of 0.3 to 1.2 mM, and SNAP, in the range of 0.001 to 1 mM, significantly inhibited Fas receptor–mediated eosinophil death. At optimal concentrations, the rescue effect was 95% for azide (1.2 mM), 89% for hydroxylamine (1.2 mM), and 100% for SNAP (1 mM) (Fig. 3 B). Similar to the coculture system, LY 83583 abrogated the protective effect of 1 mM SNAP on anti-Fas mAb-induced eosinophil death. Moreover, the specific eosinophil survival factor IL-5 (3, 4) had only little rescue effects (17%) in this system (Fig. 3 B). In addition, similar to IL-5, IFN-γ, IL-3, and GM-CSF were unable to significantly protect eosinophils from Fas receptor–mediated death (not presented). Furthermore, and in contrast to the strong rescue effect of NO on Fas receptor– activated cells, it is important to note that all NO donors only slightly inhibited spontaneous eosinophil death (16% for azide, 18% for hydroxylamine, and 5% for SNAP).
Figure 3.

Fas receptor–mediated death is prevented by NO, but not by IL-5 in freshly purified eosinophils. (A) Three different NO donors (azide and hydroxylamine release NO due to endogenous catalase activity; SNAP spontaneously releases NO) inhibited, in a dose-dependent manner, Fas receptor–mediated eosinophil death. Horizontal lines, cell viability of untreated eosinophils. Eosinophils were cultured for 24 h in the presence of anti-Fas mAb and NO donors at the indicated concentrations. Cell viability was assessed by uptake of 1 μM ethidium bromide and flow cytometric analysis. Values are means ± SEM of three independent experiments. (B) In contrast to NO, optimal concentration of IL-5 (50 ng/ ml) did not inhibit Fas receptor–mediated eosinophil death. LY 83583 was used to demonstrate the specificity of NO-mediated effects. Experiments were performed as described in A. The percentage of inhibition of Fas receptor–mediated eosinophil death was calculated. Values are means ± SEM of three independent experiments.
Using a DNA fragmentation assay and flow cytometric analysis of surface PS expression, we demonstrated that the anti-Fas mAb triggered apoptotic cell death. This apoptotic cell death was inhibited by NO. We found 35 ± 2% DNA fragmentation in nontreated purified eosinophils after 16 h of in vitro culture (Fig. 4 A a). Anti-Fas mAb treatment of eosinophils increased the levels of DNA fragmentation to 64 ± 4% DNA (Fig. 4 A b). In this system, SNAP inhibited Fas receptor–mediated apoptosis in a dose-dependent manner (Fig. 4 A, c, and d and right). Similar results were obtained using azide and hydroxylamine as NO donors (not presented). Moreover, anti-Fas mAb-induced externalization of PS, an early marker of apoptosis (28), was inhibited in the presence of 1 mM SNAP, further suggesting that NO inhibits typical apoptotic processes induced by Fas receptor activation in human eosinophils (Fig. 4 B).
Figure 4.

NO prevents Fas receptor–mediated apoptosis in freshly purified eosinophils. (A) SNAP inhibited, in a dose-dependent manner, Fas receptor–mediated DNA fragmentation. Eosinophils were cultured for 16 h. DNA fragmentation (black) was determined by DNA staining with propidium iodide and flow cytometric analysis. (Left) a, untreated; b, anti-Fas mAb; c, anti-Fas mAb plus 0.5 mM SNAP; d, anti-Fas mAb plus 1 mM SNAP. (Right) Values are means ± SEM of three independent experiments. (B) SNAP inhibited Fas receptor–mediated redistribution of PS. Eosinophils were cultured for the indicated times. FITC-conjugated annexin V binding (black) was assessed by flow cytometric analysis. (Left, measurements after 8 h) a, untreated; b, anti-Fas mAb; c, anti-Fas mAb plus 0.5 mM SNAP; d, anti-Fas mAb plus 1 mM SNAP. (Right) Values are means ± SEM of three independent experiments.
NO Causes Reduced Fas Receptor Sensitivity via sGC in Eosinophils.
sGC is generally accepted as being the main molecular target of NO leading to the production of cGMP. The subsequent regulation of cGMP-dependent protein kinases, nucleotide phosphodiesterases, and ion channels mediates most of the physiological NO functions (35). Therefore, we mimicked intracellular cGMP increases after sGC activation by adding cell membrane-permeable dibutyryl (db)-cGMP to the in vitro cultures. Similarly to SNAP, azide, or hydroxylamine, db-cGMP reduced anti-Fas mAb-induced death in purified eosinophils in a dose-dependent manner (Fig. 5 A). The inhibition of cell death was 100% at 50 mM db-cGMP. In contrast, DAG, an important second messenger molecule in other signaling systems, had no effect on Fas receptor–mediated eosinophil death (not presented), indicating specificity of the observed db-cGMP effects.
Figure 5.

Fas receptor–mediated death is prevented by NO-induced second messengers in freshly purified eosinophils. (A) cGMP and cAMP inhibited, in a dose-dependent manner, Fas receptor–mediated death. In contrast, DAG had no effect in this system (not presented). The dashed line represents cell viability of untreated eosinophils. Eosinophils were cultured for 24 h. Cell viability was assessed by uptake of 1 μM ethidium bromide and flow cytometric analysis. Values are means ± SEM of three independent experiments. (B) Inhibition of phosphodiesterase activity by IBMX enhanced low-dose NO effects. 0.5 mM, but not 0.1 mM, SNAP inhibited Fas receptor–mediated eosinophil death. However, 0.1 mM SNAP could completely block death when IBMX was added. Experiments and analysis were performed as described in A. Values are means ± SEM of three independent experiments.
The generation of cGMP subsequently inhibits a cGMP-regulated cyclic nucleotide phosphodiesterase (35) and, consequently, increases intracellular cAMP. Therefore, membrane-permeable db-cAMP was added to our system to mimic increases in cAMP levels. Similarly to db-cGMP, db-cAMP was an effective blocker of Fas receptor–mediated eosinophil death (Fig. 5 A). In fact, the db-cAMP rescue effect was, at concentrations between 1 and 50 mM, even more potent than db-cGMP.
cGMP and cAMP are both targets of regulatory phosphodiesterases that cleave these molecules to 5′-GMP and 5′-AMP, respectively. To confirm the roles for cGMP and cAMP in the reduction of eosinophil sensitivity to Fas receptor–mediated apoptosis, we next inhibited phosphodiesterases by using IBMX to increase intracellular cGMP and cAMP levels. Previously published work has demonstrated that inhibition of phosphodiesterase activity enhances NO effects (36). Therefore, we performed experiments using suboptimal concentrations of the NO donor SNAP to investigate whether IBMX is able to increase SNAP-mediated protection from cell death. Inhibition of phosphodiesterase activity by IBMX and suboptimal concentrations of SNAP blocked Fas receptor–mediated death almost completely, whereas IBMX had no significant effect when optimal concentrations of SNAP were used (Fig. 5 B). We also performed DNA fragmentation assays and observed similar results (not presented). Together, these data suggest that NO exerts its potent inhibitory effect on Fas receptor– mediated apoptosis through activation of sGC and consequent enhancement of intracellular cGMP and cAMP levels.
NO Inhibits Fas Receptor Signaling Events Distal to Acidic SMase Activition in Eosinophils.
In preliminary experiments, we excluded the possibility of Fas receptor downregulation as a mechanism to reduce eosinophil sensitivity to Fas receptor–mediated apoptosis by NO (not presented). Rather, this implied that the reduced sensitivity of NO-treated eosinophils to Fas receptor–mediated apoptosis reflected a functional defect to transduce signals via this receptor. Previously published work suggested that acidic SMase-generated ceramide is involved in the Fas receptor signaling pathway leading to apoptosis (29, 37, 38). Therefore, the effect of anti-Fas mAb treatment on SMase activity was studied in the absence and presence of optimal concentrations of SNAP. Enzyme activity was determined by degradation of radioactively labeled sphingomyelin (SM) in an in vitro assay. As shown in Fig. 6 A, anti-Fas mAb treatment was followed by induction of SMase activity in both SNAP-untreated and -treated eosinophils, indicating that NO may act distally from SM breakdown. In contrast, the ICE inhibitor II Ac-YVAD-cmk abrogated Fas receptor– mediated activation of SMase (Fig. 6 A), suggesting that SM breakdown is distal to early ICE protease activation in the Fas receptor signaling pathway.
Figure 6.

NO inhibits Fas receptor signaling events at the level of, or proximal to, JNK, but distal to acidic SMase activation in freshly purified eosinophils. (A) The ICE inhibitor Ac-YVAD-cmk (1 μM), but not SNAP (1 mM), blocked acidic SMase activation. Eosinophils were stimulated, as indicated, for 20 min. SMase activity was assessed in cell lysates by a radioactive in vitro assay using 14C-SM as a substrate. SMase activity is presented as percent degraded substrate calculated from densitometry measurements after autoradiography. Values are means ± SEM of three independent experiments. (B) The ceramide analogues C2- and C6-ceramide induced, in a dose-dependent manner, eosinophil death. In contrast, biologically inactive C2-dihydroceramide (DH-C2) had no effect in this system. Eosinophils were cultured for 24 h. Cell viability was assessed by uptake of 1 μM ethidium bromide and flow cytometric analysis. Values are means ± SEM of three independent experiments. (C) SNAP (1 mM), cGMP (50 mM), and cAMP (5 mM) inhibited C2-ceramide–mediated eosinophil death. Experiments and analysis were performed as described in B. Values are means ± SEM of three independent experiments. (D) cGMP (50 mM) and cAMP (5 mM) abrogated anti-Fas mAb- or ceramide-mediated phosphorylation of c-Jun. Eosinophils were stimulated for 60 min. The cell lysates were immunoprecipitated with an anti-JNK Ab, and phosphorylation of GST-c-Jun (1–79) was observed by in vitro kinase assay.
We next investigated whether SMase-generated ceramide could induce apoptosis in eosinophils as it has been reported in other systems (29, 37, 38). Indeed, 10–50 μM C2- or C6-ceramide induced, in a dose-dependent manner, eosinophil death (Fig. 6 B). In contrast, the biologically inactive structural analogue of C2-ceramide, C2-dihydroceramide, had no effect on cell death in the same concentration range. Higher ceramide concentrations were found to damage the cell membrane. To determine whether NO can counterregulate ceramide-induced apoptosis, untreated and C2-ceramide–treated eosinophils were incubated in the absence and presence of optimal concentrations of SNAP, db-cGMP, and db-cAMP. SNAP, db-cGMP, and db-cAMP inhibited C2-ceramide–induced cell death (Fig. 6 C) and DNA fragmentation (not presented). These data suggest that NO, as well as its second messengers, counterregulate Fas receptor signaling distal to ceramide generation.
NO Inhibits Fas Receptor–induced JNK Activation.
Recently, it has been demonstrated that both Fas receptor activation as well as the presence of C2-ceramide results in activation of an alternative mitogen-activated protein (MAP) kinase pathway leading to JNK activation followed by apoptosis in human T and B cells (30, 39). Activated JNK phosphorylates c-Jun in many systems (40–42). Therefore, phosphorylation of GST-c-Jun (1–79) was used to measure JNK activity by an immune complex kinase assay. As shown in Fig. 6 D, C2-ceramide stimulation for 60 min induced phosphorylation of c-Jun. In contrast, these increases were abolished by the second messengers of NO cGMP and cAMP. These data suggest that NO interrupts the Fas receptor signaling pathway at the level of, or proximal to, JNK activation.
NO Inhibits Fas Receptor–induced Proteolytic Activity in Eosinophils.
Since proteases are believed to be the central components of the cell death machinery (43), we analyzed proteolytic activity in Fas receptor–activated eosinophils and its modulation by NO. Total protease activation was first assessed using a fluorescence assay to measure the amounts of degraded proteins by conjugating terminal amino groups to the fluorochrome Fluram. Anti-Fas mAb stimulation of eosinophils for 16 h resulted in an increase in specific fluorescence intensity in the Fluram assay by a factor of 2.3, indicating increased proteolytic activity. This increase was not detected in the presence of optimal SNAP concentrations (Fig. 7 A). Intracellular proteolytic activity was also analyzed by separating and incubating cell lysates on a Zymogram gel that contained 0.1% gelatin. Again, Fas receptor– activated eosinophils demonstrated elevated proteolytic activity compared to unstimulated cells (Fig. 7 B). Increased proteolytic activity after Fas receptor activation involved proteases with molecular masses of 49, 58, 84, and between 90 and 95 kD beginning 7 h after stimulation. In contrast, additional treatment of eosinophils with 50 mM db-cGMP resulted in reduced proteolytic acitivity in these cells. Moreover, the pattern of protease activation did not differ between spontaneously and anti-Fas mAb-mediated dying cells. These data suggest that NO reduces activation of proteases within the Fas receptor signaling pathway, and thereby prevents Fas receptor–mediated apoptosis in eosinophils.
Figure 7.
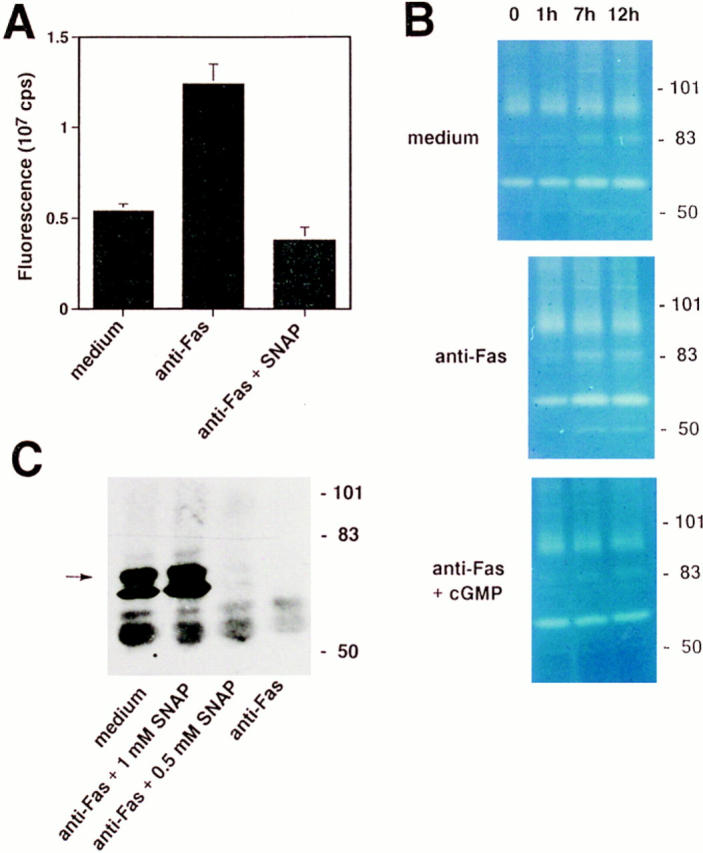
NO and NO-mediated second messengers inhibit activation of intracellular proteases in freshly purified eosinophils. (A) SNAP (1 mM) inhibited anti–Fas receptor–mediated activation of intracellular proteases. Intracellular proteolytic activity was assessed by a fluorescence assay; levels of terminal amino groups were measured by binding of Fluram and fluorescence spectrometry. Eosinophils were cultured for 16 h. Values are means ± SEM of three independent experiments. (B) cGMP (50 mM) inhibited anti–Fas receptor–mediated activation of intracellular proteases. A time course of intracellular proteolytic activity was assessed by Zymogram electrophoresis. Spontaneous and anti–Fas receptor–mediated eosinophil apoptosis was associated with the activation of proteases with molecular masses of 49, 58, 84, and between 90 and 95 kD. (Right) The positions of molecular size standards. (C) SNAP inhibited, in a dose-dependent manner, Fas receptor–mediated proteolysis of lamin B1. Eosinophils were cultured for 4 h. Immunoblot analysis of cell lysates was performed with anti–lamin B1 mAb. The full-length lamin B1 is indicated (arrow). (Right) The positions of molecular size standards.
Previous studies have identified important targets of proteases that execute cell death. One of these targets is a nuclear 74-kD protein, lamin B1. In contrast to the slow turnover of lamins in nonapoptotic cells (t 1/2 > 24 h), apoptotic cells are characterized by a rapid decrease in levels of intact lamins (44). Lamin cleavage seems to be required for packaging of the condensed chromatin into apoptotic bodies (45). Therefore, we characterized cleavage of lamin B1 in Fas receptor–activated eosinophils in the presence or absence of SNAP by Western blotting. As shown in Fig. 7 C, complete cleavage of lamin B1 occurred within 4 h after anti-Fas mAb treatment. In contrast, optimal concentrations of SNAP completely inhibited Fas receptor–mediated cleavage of lamin B1, whereas a suboptimal SNAP concentration was only partially protective. In addition, a second protein with a molecular mass of ∼70 kD reacted with the antibody, probably representing an early degradation product of lamin B1. These data suggest that NO blocks the proteolytic action of proteases on specific target proteins that are important for the apoptotic process, such as lamin B1.
Discussion
Allergic diseases of the respiratory tract are associated with a marked eosinophilic inflammation (46) and enhanced cytokine production (47). Some of these cytokines inhibit eosinophil apoptosis and may, therefore, contribute to tissue eosinophil accumulation (1, 2). In addition to these effects, there is evidence to suggest upregulation of iNOS in eosinophilic inflammation airways (17). Consequently, measurements of NO in exhaled air have demonstrated elevated levels in asthma and seasonal allergic rhinitis patients (14, 15, 22). In agreement with these studies, we measured increased NO levels within the nasal cavity of patients with nasal polyps. We also provided evidence for increased iNOS mRNA expression in polyp tissues. Together with the previously published work (14, 15, 22), these data suggest that increased NO production may represent a general feature of eosinophilic inflammation.
Fas receptors are broadly expressed on many different cell types. Activation of the Fas receptors leads to induction of apoptosis in many, but not all systems (48). For example, hematopoietic and nonhematopoietic tumor cells resistant to Fas receptor–mediated apoptosis have been found, although they express the receptor on the cell surface (49, 50). Therefore, the mechanisms of Fas receptor resistance have generated great interest. The results presented in this study suggest a novel NO-mediated mechanism causing nonfunctional Fas receptors by disruption of the death signaling pathway in human eosinophils.
Extensive analysis of the signaling pathways associated with Fas receptor–mediated apoptosis has revealed that oligomerization of the receptor induces conformational changes of the intracellular domains leading to binding of cytoplasmic proteins and formation of the death-inducing signaling complex (DISC; 51). One of the DISC proteins is an ICE-like protease (24, 52). Thus, perhaps there is a direct physical connection between initialization of the death signal at the cell membrane and the death machinery (48). However, other cell signaling events involving tyrosine phosphorylation (53), SMase-ceramide (29, 37, 38), and Ras/Raf/ MAP kinase pathways (39, 40) have also been reported to be involved in cell death. The contribution of all of these pathways to the induction of apoptosis as well as possible interactions between different pathways have not yet been clearly elucidated.
Ceramide, generated by activated SMase, triggers apoptosis in response to Fas receptor activation (29, 37, 38) and many other death stimuli (54). Thus, ceramide production appears to be a pleiotropic activator of apoptosis. Therefore, we first analyzed activation of SMase after Fas receptor activation in the presence and absence of NO or second messengers of NO. This strategy allowed us to determine whether the possible disruption of the Fas receptor signaling pathway by NO occurs proximal or distal to SMase activation. We observed a Fas receptor–mediated activation of SMase in both NO-untreated and -treated eosinophils, suggesting that NO may block the death signal distal to SMase. The possibility to inhibit Fas receptor–initiated signaling events downstream of SMase activation has been previously demonstrated in cells overexpressing Abl tyrosine kinases (55). In contrast to the effects observed with NO donors, activation of SMase was abrogated when the tetrapeptide YVAD was used to block activation of the ICE protease. In agreement with a previously published work (56), these data demonstrate that activation of ICE proteases is required for SMase activation. Moreover, these data indicate that NO did not disrupt DISC formation in Fas receptor–activated eosinophils.
There has been some progress in the identification of ceramide targets. The first reported target for its activity was a serine/threonine protein kinase, termed ceramide-activated protein kinase, that was only recently identified (57). The target of this kinase appears to be the protooncogene Raf (57). Moreover, Fas receptor activation and ceramide induce activation of another, alternative MAP kinase pathway resulting in JNK stimulation. JNK activation has been shown to be critical for induction of apoptosis in many systems (30, 39). To examine the possibility of a functional JNK inactivation by NO, we monitored kinase activity after anti-Fas mAb or ceramide treatment of eosinophils in the presence and absence of NO or second messengers of NO. The results suggest that JNK activation is also necessary for Fas receptor–mediated apoptosis in eosinophils. Moreover, since second messengers of NO prevent c-Jun phosphorylation, NO may act at the level of, or proximal to, JNK activation to prevent death.
It is now clear that proteases play a key role in apoptosis (43, 48). This study provides evidence that NO prevents Fas receptor–mediated proteolysis. However, the observation that it is possible to block activation of SMase by using an inhibitor of ICE suggests that there is some protease activation even in the presence of NO. It has recently been demonstrated that activation of a protease can lead to further activation of other proteases within the apoptotic process (58), generating a protease cascade (43, 48). We hypothesize at this point that the generation of ceramide and subsequent JNK activation may represent a signaling event responsible for amplification of the proteolytic cascade. Therefore, disruption of ceramide-induced signals prevents further proteolysis. This idea is further supported by very recent reports demonstrating that the central ICE-like protease CPP32 (Yama/Apopain) is not only a target of ICE (58) but, at least in a later phase of the apoptotic death process, also of MAP kinase and JNK signaling pathways (59). Thus, it is possible that, even in the presence of NO, activation of eosinophils via the Fas receptor leads to an immediate but limited activation of proteases able to degrade only a limited number of substrates. These can then be replaced without any damage to the cell. In contrast, in the absence of NO, ceramide-mediated amplification of the proteolytic cascade takes place and the apoptotic process initialized via the Fas receptor proceeds, causing irreversible damage to the cell.
In summary, the data reported here indicate that NO, a secretory product released in increased amounts within chronic eosinophilic inflammatory responses such as bronchial asthma and other chronic-allergic disorders, disrupts Fas receptor–mediated apoptosis in eosinophils. Therefore, we have identified a mechanism of Fas receptor resistance that might contribute to the eosinophilia associated with these diseases. In this context, it is tempting to speculate that glucocorticoids, which are known to suppress NO concentration in asthmatic patients (15), decrease eosinophil numbers, besides other possible mechanisms, by sensitization of the Fas receptor. Moreover, we have localized the disruption of the Fas signaling pathway by NO at the level of, or proximal to, JNK, but distal to SMase activation. We also provide evidence that the ceramide-induced apoptotic response may serve as an amplification step within the proteolytic cascade of the apoptotic process that, on the other hand, can be counterregulated by additional signals.
Acknowledgments
We wish to thank M. Roth for densitometry measurements. We also thank G.P. Anderson, C. Borner, G.B. Mills, and M.E. Peter for critical reading and discussion of the manuscript.
This work was supported by grant 32-49210.96 from the Swiss National Science Foundation (to H.-U. Simon).
Footnotes
Abbreviations used in this paper: DAG, 1,2-dioctanoyl-sn-glycerol; db, dibutyryl; DISC, death-inducing signaling complex; ECL, enhanced chemiluminescence; IBMX, 3-isobutyl-1-methylxanthine; ICE, IL-1 converting enzyme; iNOS, inducible isoform of NOS; JNK, Jun kinase; LY 83583, 6-anilinoquinoline-5,8-quinone; MAP, mitogen-activated protein; mRNA, messenger RNA; NMMA, NG-monomethyl-l-arginine; NO, nitric oxide; NOS, NO synthase; PS, phosphatidylserine; sGC, soluble guanylyl cyclase; SM, sphingomyelin; SMase, sphingomyelinase; SNAP, S-nitroso-N-acetylpenicillamine.
References
- 1.Simon H-U, Blaser K. Inhibition of programmed eosinophil death: a key pathogenic event for eosinophilia? . Immunol Today. 1995;16:53–55. doi: 10.1016/0167-5699(95)80086-7. [DOI] [PubMed] [Google Scholar]
- 2.Simon H-U, Yousefi S, Schranz C, Schapowal A, Bachert C, Blaser K. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J Immunol. 1997;158:3902–3908. [PubMed] [Google Scholar]
- 3.Rothenberg ME, Stevens RL, Silberstein DS, Soberman R, Austen KF, Owen WF. IL-5 promotes long-term culture and enhances functional properties of human eosinophils. J Immunol. 1989;143:2311–2316. [PubMed] [Google Scholar]
- 4.Yamaguchi Y, Suda T, Ohta S, Tominaga K, Miura Y, Kasahara T. Analysis of the survival of mature human eosinophils: interleukin-5 prevents apoptosis in mature human eosinophils. Blood. 1991;78:2542–2547. [PubMed] [Google Scholar]
- 5.Walker C, Virchow JC, Bruijnzeel PLB, Blaser K. T cell subsets and their soluble products regulate eosinophilia in allergic and nonallergic asthma. J Immunol. 1991;146:1829–1835. [PubMed] [Google Scholar]
- 6.Stern M, Meagher L, Savill J, Haslett C. Apoptosis in human eosinophils. Programmed cell death in the eosinophil leads to phagocytosis by macrophages and is modulated by IL-5. J Immunol. 1992;148:3543–3549. [PubMed] [Google Scholar]
- 7.Yousefi S, Green DR, Blaser K, Simon H-U. Protein-tyrosine phosphorylation regulates apoptosis in human eosinophils and neutrophils. Proc Natl Acad Sci USA. 1994;91:10868–10872. doi: 10.1073/pnas.91.23.10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yousefi S, Hoessli DC, Blaser K, Mills GB, Simon H-U. Requirement of Lyn and Syk tyrosine kinases for the prevention of apoptosis by cytokines in human eosinophils. J Exp Med. 1996;183:1407–1414. doi: 10.1084/jem.183.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon H-U, Yousefi S, Dommann-Scherrer CC, Zimmermann DR, Bauer S, Barandun J, Blaser K. Expansion of cytokine-producing CD4−CD8−T cells associated with abnormal Fas expression and hypereosinophilia. J Exp Med. 1996;183:1071–1082. doi: 10.1084/jem.183.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto K, Schleimer RP, Saito H, Iikura Y, Bochner BS. Induction of apoptosis in human eosinophils by anti-Fas antibody treatment in vitro. Blood. 1995;86:1437–1443. [PubMed] [Google Scholar]
- 11.Tsuyuki S, Bertrand C, Erard F, Trifilieff A, Tsuyuki J, Wesp M, Anderson GP, Coyle AJ. Activation of the Fas receptor on lung eosinophils leads to apoptosis and the resolution of eosinophilic inflammation of the airways. J Clin Invest. 1995;96:2924–2931. doi: 10.1172/JCI118364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Druilhe A, Cai Z, Hailé S, Chonaib S, Petrolani M. Fas-mediated apoptosis in cultured human eosinophils. Blood. 1996;87:2822–2830. [PubMed] [Google Scholar]
- 13.Hebestreit H, Yousefi S, Balatti I, Weber M, Crameri R, Simon D, Hartung K, Schapowal A, Blaser K, Simon H-U. Expression and function of the Fas receptor on human blood and tissue eosinophils. Eur J Immunol. 1996;26:1775–1780. doi: 10.1002/eji.1830260817. [DOI] [PubMed] [Google Scholar]
- 14.Alving KE, Weitzberg E, Lundberg JM. Increased amount of nitric oxide in exhaled air of asthmatics. Eur Respir J. 1993;6:1268–1370. [PubMed] [Google Scholar]
- 15.Kharitonov SA, Rajakulasingam K, O'Connor B, Durham SR, Barnes PJ. Nasal nitric oxide is increased in patients with asthma and allergic rhinitis and may be modulated by nasal glucocorticoids. J Allergy Clin Immunol. 1997;99:58–64. doi: 10.1016/s0091-6749(97)70301-4. [DOI] [PubMed] [Google Scholar]
- 16.Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from l-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 17.Hamid Q, Springall DR, Riveros-Moreno V, Chanez P, Howarth P, Redington A, Bousquet J, Godard P, Holgate S, Polak JM. Induction of nitric oxide synthase in asthma. Lancet. 1993;342:1510–1513. doi: 10.1016/s0140-6736(05)80083-2. [DOI] [PubMed] [Google Scholar]
- 18.del Pozo V, de Arruda-Chaves E, de Andrés B, Cardaba B, Lopez-Farré A, Gallardo S, Cortegano I, Vidarte L, Jurado A, Sastre J, et al. Eosinophils transcribe and translate messenger RNA for inducible nitric oxide synthase. J Immunol. 1997;158:859–864. [PubMed] [Google Scholar]
- 19.Abrahamsohn IS, Coffman RL. Cytokine and nitric oxide regulation of the immunosuppression in trypanosoma cruziinfection. J Immunol. 1995;155:3955–3963. [PubMed] [Google Scholar]
- 20.Sternberg JM, Mabbott NA. Nitric oxide–mediated suppression of T cell responses during trypanosoma brucei infection: soluble trypanosome products and interferon-γ are synergistic inducers of nitric oxide synthase. Eur J Immunol. 1996;26:539–543. doi: 10.1002/eji.1830260306. [DOI] [PubMed] [Google Scholar]
- 21.Taylor-Robinson AW, Liew FY, Severn A, Xu D, McSorley SJ, Garside P, Padron J, Phillips RS. Regulation of the immune response by nitric oxide differentially produced by T helper type 1 and T helper type 2 cells. Eur J Immunol. 1994;24:980–984. doi: 10.1002/eji.1830240430. [DOI] [PubMed] [Google Scholar]
- 22.Barnes PJ, Liew FY. Nitric oxide and asthmatic inflammation. Immunol Today. 1995;16:128–130. doi: 10.1016/0167-5699(95)80128-6. [DOI] [PubMed] [Google Scholar]
- 23.Bohlinger I, Leist M, Barsig J, Uhlig S, Tiegs G, Wendel A. Interleukin-1 and nitric oxide protect against tumor necrosis factor α–induced liver injury through distinct pathways. Hepatology. 1995;22:1829–1837. [PubMed] [Google Scholar]
- 24.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1– and TNF receptor– induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 25.Yousefi S, Hemmann S, Weber M, Hölzer C, Hartung K, Simon H-U. IL-8 is expressed by human peripheral blood eosinophils. Evidence for increased secretion in asthma. J Immunol. 1995;154:5481–5490. [PubMed] [Google Scholar]
- 26.Simon H-U, Tsao PW, Siminovitch KA, Mills GB, Blaser K. Functional platelet-activating factor receptors are expressed by monocytes and granulocytes but not by resting or activated T and B lymphocytes from normal individuals or patients with asthma. J Immunol. 1994;153:364–377. [PubMed] [Google Scholar]
- 27.Moro MA, Russel RJ, Cellek S, Lizasoain I, Su Y, Darley-Usmar VM, Radomski MW, Moncada S. cGMP mediates the vascular and platelet actions of nitric oxide: confirmation using an inhibitor of the soluble guanylyl cyclase. Proc Natl Acad Sci USA. 1996;93:1480–1485. doi: 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin SJ, Reutelingsperger CPM, McGahon AJ, Rader JA, van Schie RCAA, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cifone MG, De Maria R, Roncaioli P, Rippo MR, Azuma M, Lanier LL, Santoni A, Testi R. Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J Exp Med. 1993;177:1547–1552. doi: 10.1084/jem.180.4.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson DJ, Fortner KA, Lynch DH, Mattingly RR, Macara IG, Posada JA, Budd RC. JNK, but not MAPK, activation is associated with Fas-mediated apoptosis in human T cells. Eur J Immunol. 1996;26:989–994. doi: 10.1002/eji.1830260505. [DOI] [PubMed] [Google Scholar]
- 31.Pacifici RE, Davies KJA. Protein degradation as an index of oxidative stress. Methods Enzymol. 1990;186:485–502. doi: 10.1016/0076-6879(90)86143-j. [DOI] [PubMed] [Google Scholar]
- 32.Brown PD, Levy AT, Margulies IM, Liotta LA, Stetler-Stevenson WG. Independent expression and cellular processing of Mr 72.000 type IV collagenase and interstitial collagenase in human tumorigenic cell lines. Cancer Res. 1990;50:6184–6191. [PubMed] [Google Scholar]
- 33.Nicholls P. The reactions of azide with catalase and their significance. Biochem J. 1964;90:331–343. doi: 10.1042/bj0900331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, Gruetter CA. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 35.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 36.Thusu KG, Morin FC, Russell JA, Steinhorn RH. The cGMP phosphodiesterase inhibitor Zaprinast enhances the effect of nitric oxid. Am J Respir Crit Care Med. 1995;152:1605–1610. doi: 10.1164/ajrccm.152.5.7582302. [DOI] [PubMed] [Google Scholar]
- 37.Gulbins E, Bissonnette R, Mahboubi A, Martin S, Nishioka W, Brunner T, Baier G, Baier-Bitterlich G, Byrd C, Lang F, et al. Fas-induced apoptosis is mediated via a ceramide-initiated RAS signaling pathway. Immunity. 1995;2:341–351. doi: 10.1016/1074-7613(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 38.Tepper CG, Jayadev S, Liu B, Bielawska A, Wolff R, Yonehara S, Hannun YA, Seldin MF. Role of ceramide as an endogenous mediator of Fas-induced cytotoxicity. Proc Natl Acad Sci USA. 1995;92:8443–8447. doi: 10.1073/pnas.92.18.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cahill MA, Peter ME, Kischkel FC, Chinnaiyan AM, Dixit VM, Krammer PH, Nordheim A. CD95 (APO-1/Fas) induces activation of SAP kinases downstream of ICE-like proteases. Oncogene. 1996;13:2087–2096. [PubMed] [Google Scholar]
- 40.Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G, Robertson M, Kufe DW, Anderson KC. Interleukin-6 inhibits Fas-induced apoptosis and stress-activated protein kinase activation in multiple myeloma cells. Blood. 1997;89:227–234. [PubMed] [Google Scholar]
- 41.Dérijard B, Hibi M, Wu I, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 42.Liu Z-G, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JYJ. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- 43.Martin SJ, Green DR. Protease acitvation during apoptosis: Death by a thousand cuts. Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 44.Oberhammer FA, Hochegger K, Froschl G, Tiefenbacher R, Pavelka M. Chromatin condensation during apoptosis is accompanied by degradation of lamin A + B, without enhanced activation of cdc2 kinase. J Cell Biol. 1994;126:827–837. doi: 10.1083/jcb.126.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazebnik Y, Takahashi A, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bousquet J, Chanez P, Lacoste JY, Barnéon G, Ghavanian N, Enander I, Venge P, Ahlstedt S, Simony-Lafontaine J, Godard P, Michel F-B. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 47.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med. 1996;183:1–5. doi: 10.1084/jem.183.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 49.Mapara MY, Bargou R, Zugck C, Dohner H, Ustaoglu F, Jonker RR, Krammer PH, Dörken B. APO-1 mediated apoptosis or proliferation in human chronic B lymphocytic leukemia: correlation with bcl-2 oncogen expression. Eur J Immunol. 1993;23:702–708. doi: 10.1002/eji.1830230320. [DOI] [PubMed] [Google Scholar]
- 50.Owen-Schaub, L.B., R. Radinski, E. Kruzel, K. Berry, and S. Yonehara. 1994. Anti-Fas on non-hematopoietic tumors: levels of Fas/APO-1 and bcl-2 are not predictive of biological responsiveness. Cancer Res. 54:1580–1586. 158:1912– 1918. [PubMed]
- 51.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO (Eur Mol Biol Organ) J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/ CED-3–like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 53.Eischen CM, Dick CJ, Leibson PJ. Tyrosine kinase activation provides an early and requisite signal for Fas-induced apoptosis. J Immunol. 1994;153:1947–1954. [PubMed] [Google Scholar]
- 54.Haimovitz-Friedman A, Kan C, Ehleiter D, Persaud R, McLoghlin M, Fuks Z, Kolesnick RN. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. J Exp Med. 1994;180:525–535. doi: 10.1084/jem.180.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGahon AJ, Nishioka WK, Martin SJ, Mahboubi A, Cotter TG, Green DR. Regulation of the Fas apoptotic cell death pathway by Abl. J Biol Chem. 1995;270:22625–22631. doi: 10.1074/jbc.270.38.22625. [DOI] [PubMed] [Google Scholar]
- 56.Pronk GJ, Ramer K, Amiri P, Williams LT. Requirement of an ICE-like protease for induction of apoptosis and ceramide generation by REAPER. Science. 1996;271:808–810. doi: 10.1126/science.271.5250.808. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Yao B, Delikat S, Bayoumy S, Lin X-H, Basu S, McGinley M, Chan-Hui P-Y, Lichenstein H, Kolesnick R. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell. 1997;89:63–72. doi: 10.1016/s0092-8674(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 58.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 59.Huang S, Jiang Y, Li Z, Nishida E, Mathias P, Lin S, Ulevitch RJ, Nemerow GR, Han J. Apoptosis signaling pathway in T cells is composed of ICE/Ced-3 family proteases and MAP kinase kinase 6b. Immunity. 1997;6:739–749. doi: 10.1016/s1074-7613(00)80449-5. [DOI] [PubMed] [Google Scholar]