

Abstract
Evidence has been accumulating that shows that insulin-dependent diabetes is subject to immunoregulation. To determine whether cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) is involved, we injected anti–CTLA-4 mAb into a TCR transgenic model of diabetes at different stages of disease. When injected into young mice, months before they would normally become diabetic, anti–CTLA-4 induced diabetes rapidly and essentially universally; this was not the result of a global activation of T lymphocytes, but did reflect a much more aggressive T cell infiltrate in the pancreatic islets. These effects were only observed if anti–CTLA-4 was injected during a narrow time window, before the initiation of insulitis. Thus, engagement of CTLA-4 at the time when potentially diabetogenic T cells are first activated is a pivotal event; if engagement is permitted, invasion of the islets occurs, but remains quite innocuous for months, if not, insulitis is much more aggressive, and diabetes quickly ensues.
Insulin-dependent diabetes mellitus (IDDM) is a highly regulated autoimmune disease, certainly in mice and probably in humans (1, 2). Three major phases of disease can be distinguished: the generation of T lymphocytes potentially reactive to proteins made by pancreatic islet β cells, infiltration of these cells and other leukocytes into the islets, and the terminal destruction of β cells leading to hyperglycemia. Progression from one stage to the next is neither automatic nor immediate. The nonobese diabetic (NOD) mouse has been instrumental in defining these three disease phases, but they are even more clear in the BDC2.5 TCR transgenic (tg) model of diabetes (3). BDC2.5 tgs carry the rearranged TCR genes from a CD4+, β cell–specific, diabetogenic T cell clone isolated from an NOD mouse (4). Despite their autoreactivity, T cells expressing the transgene-encoded specificity escape clonal deletion and make an exaggerated contribution to the peripheral T cell repertoire. In BDC2.5 mice on the NOD genetic background, the autoreactive cells appear inert during the animals' first weeks and begin to infiltrate the islets shortly after 2 wk of age, but do not provoke diabetes until several months later (3); in mice on the C57Bl/6 (B6) background, the delay between the onset of insulitis and diabetes is shortened to weeks (5). The nature of the cells and molecules modulating the initiation of insulitis and evolution to diabetes has been the subject of much discussion (6).
One molecule that makes an attractive candidate as a diabetes regulator is cytotoxic T lymphocyte–associated antigen 4 (CTLA-4; CD152; 7–9). This Ig superfamily member shares many features with the CD28 molecule; the two have marked structural homology, their expression is restricted to T cells, they both bind to B7-1 and B7-2. CD28 is the prototypical T cell costimulator, transmitting a positive signal that synergizes with the signal delivered through the TCR to promote optimum activation. Some reports (10, 11) have suggested that CTLA-4 has an analogous function as a secondary costimulator; however, others have argued for an opposing role, as a dampener of T cell activation. CTLA-4 blockade enhances T cell responses in vitro (12–15) and in vivo (16, 17), augments antitumor immunity (18), and exacerbates an induced autoimmune disease (19, 20). In addition, mice unable to express CTLA-4 due to an engineered null mutation show massive accumulation of activated T cells in the peripheral lymphoid organs and leukocyte infiltration into a variety of tissues (21, 22). Earlier studies emphasized a role for CTLA-4 in downmodulating an ongoing T cell response, but more recent experiments point to an alternative or additional influence on the initial character of the response (8, 9). A nice example of an early role is the recent report that anti–CTLA-4 mAb can prevent tolerance induction and promote an abortive response if administered together with a normally tolerogenic stimulus (23).
Clearly, then, CTLA-4 is an important regulator of the immune response, exerting its influence on reactivity to both foreign and self antigens. There are two hints that it might be involved in regulating the progression of insulin-dependent diabetes mellitus. First, one of the human diabetes susceptibility loci encompasses the CD28 and CTLA-4 genes (24), as does one of the loci modulating diabetes in BDC2.5 TCR tg mice (5). Second, reagents that block B7-1 and/or B7-2 had clear, though complicated, effects on disease in NOD mice (25). Unfortunately, in neither of these cases was it possible to distinguish an effect promoted by CD28 from one mediated by CTLA-4.
Our strategy to evaluate the influence of CTLA-4 on the unfolding of diabetes was to inject an anti–CTLA-4 mAb into BDC2.5 TCR tg mice at different ages. Such an approach was appealing for a number of reasons. The major advantages of generating a CTLA-4 deficiency by means of an mAb rather than a null mutation were that we could intervene at will during the different phases of disease, and were more likely to avoid physiological adaptations to the absence of CTLA-4. Among the advantages of studying the TCR tg model rather than standard NOD mice were that the three disease phases are more precisely defined and synchronized in the former, and that it would be easier to visualize effects on the actual pathogenic T cells. This strategy revealed an important role for CTLA-4 in controlling diabetes progression.
Materials and Methods
Diabetes and Insulitis.
BDC2.5 TCR tg mice (3) backcrossed onto the NOD genetic background for 16 or 17 generations were housed in our conventional facility. Mice were followed for diabetes daily for 2 wk after the last mAb injection, twice a week for another 2 wk, and weekly thereafter, quantitating urine glucose levels and confirming positives by blood glucose measurements (3). Hematoxylin–eosin staining of thin sections from Bouin's solution–fixed, paraffin-embedded pancreata was performed as described (26). Multiple sections from each animal were scored for insulitis, at least 40 islets per individual.
Antibodies and Flow Cytometry.
For the injections, the anti– CTLA-4 mAbs 9H10 (13) and UC10-4F10-11 (reference 12; PharMingen, San Diego, CA), the anti-CD28 mAb 37N51.1 (27), and, as controls, the irrelevant hamster antibodies F536 (13) or G235-2356 (anti-TNP) (PharMingen) were used. The following mAbs were used for T cell analysis: phycoerythrin-conjugated anti-CD4 (Caltag Labs., South San Francisco, CA); biotin-conjugated anti-CD8 (Caltag Labs.); FITC-conjugated anti-CD69 (PharMingen); IM7, specific for CD44; Mel-14, specific for CD62L; and PC61, specific for CD25 (3). Biotinylated antibody was revealed with Cy5-labeled streptavidin (Jackson ImmunoResearch, West Grove, PA), and the other antibodies either with Texas red– labeled anti–rat IgG (Jackson ImmunoResearch) or FITC-labeled anti–rat IgG (Caltag Labs.).
Immunostainings and analyses were performed essentially as described (28). Pancreatic T cells were extracted by gently teasing the pancreas in a small volume of medium on ice (after the pancreatic lymph nodes were carefully removed); due to the young age of the mice and the aggressive nature of the insulitis, it was not possible to isolate islets.
Results
Treatment with Anti–CTLA-4, but Not Anti-CD28, Precipitates Diabetes in Young BDC2.5/NOD tgs.
We began by probing CTLA-4 function at disease onset in BDC2.5 TCR tg mice carried on the NOD genetic background. The first sign of disease in these animals is an impressive insulitis beginning after about 15 d of age (3). We injected anti–CTLA-4, anti-CD28, or control mAb on days 9, 12, and 15, a protocol analogous to one that was effective at modulating antitumor responses (18). As indicated in Fig. 1, diabetes was rapidly precipitated in almost all of the tgs treated with anti–CTLA-4. Compared with unmanipulated BDC2.5/ NOD mice, this represents a striking increase in both the time of onset (in comparison with the usual 5–6 mo) and incidence (compared with the typical 15–30%). Diabetes was not provoked prematurely by treatment with either the anti-CD28 or control mAbs.
Figure 1.

Treatment with anti–CTLA-4 mAb provokes a rapid onset of diabetes. BDC2.5/NOD mice were injected intraperitoneally with anti–CTLA-4, anti-CD28, or control mAb at 9, 12, and 15 d of age (total dose: 250–300 μg).
Anti–CTLA-4 mAb Is Diabetogenic only when Present before the Initiation of Insulitis.
Next, we explored the influence of CTLA-4 on disease progression by varying the time of anti–CTLA-4 mAb injection. As illustrated in Fig. 2 A, only those animals that got anti–CTLA-4 on or before day 12 exhibited premature diabetes. Interestingly, no matter when the first dose of mAb was administered, anywhere from days 2 to 12, hyperglycemia was not detectable until 17 d of age. When the initial anti–CTLA-4 injection was given on day 17 or later, diabetes was not induced. Initiating the treatment at much later times, e.g., at 10 wk of age, also had no effect (data not shown).
Figure 2.

Anti–CTLA-4 treatment works only when first administered before the initiation of insulitis. (A) BDC2.5/NOD mice were treated with anti–CTLA-4 mAb using a variety of protocols. All received a total dose of 250–300 μg, except those injected only on day 12, which got 200 μg. The day on which the mAb was first administered is indicated. Carrots, individual injections; stippling, the presence of anti–CTLA-4; solid black, the cumulative frequency of diabetes. The numbers to the right represent the final frequency of diabetics. All experiments included anti-CD28 and/or irrelevant mAb controls. In several experiments, paired littermates received earlier (before 12 d) or later (after 17 d) injections. (B) Groups of BDC2.5/NOD mice were killed at different ages, and insulitis quantitated. Each bar represents the average proportion of infiltrated islets (groups of 5–12 mice). The horizontal scale (days of age) matches that of A.
As mentioned above, one advantage of working with the BDC2.5 TCR tg model is that early disease events are more synchronous than in NOD mice, in particular, the initiation of insulitis, which occurs over about a week in individual BDC2.5 tgs but can be scattered over many weeks in NOD animals. Thus, we could more precisely correlate the window of anti–CTLA-4 efficacity with the development of insulitis. A comparison of A and B of Fig. 2 reveals that anti–CTLA-4 mAb had to be present before the initiation of insulitis to provoke premature diabetes.
Anti–CTLA-4 Treatment Dramatically Alters the Aggressiveness, but Not the Timing, of Insulitis.
We also looked for more immediate alterations in the phenotype or behavior of T cells as a result of the anti–CTLA-4 treatment. Fig. 3 A compares the expression of activation markers on CD4+ T cells from 17-d-old mice administered control mAb or anti–CTLA-4 before the initiation of insulitis. Few cells from the peripheral lymphoid organs of control mAb-injected animals expressed the early activation markers CD69 and CD25 (the latter not shown); however, a significant fraction displayed late activation markers indicative of chronically activated or memory cells, i.e., low levels of CD62L and high levels of CD44 (the latter not shown); many more of the cells isolated from the pancreatic lesion expressed these early and late activation markers. The profiles from the different tissues of animals injected with anti– CTLA-4 were analogous, notably the differences between the pancreas and the lymphoid tissues. By this measure, then, the anti–CTLA-4 treatment did not alter the activation state of the bulk of CD4+ T cells, either systemically or locally, within the pancreas. Nor did the treatment provoke a generalized inflammatory response (reflecting the phenotype of the CTLA-4–null mice; references 21, 22). We observed no infiltrates in the heart, lung, liver, kidney, brain, gut, or spleen on day 17 of age after having injected the mAb according to diverse protocols (data not shown).
Figure 3.

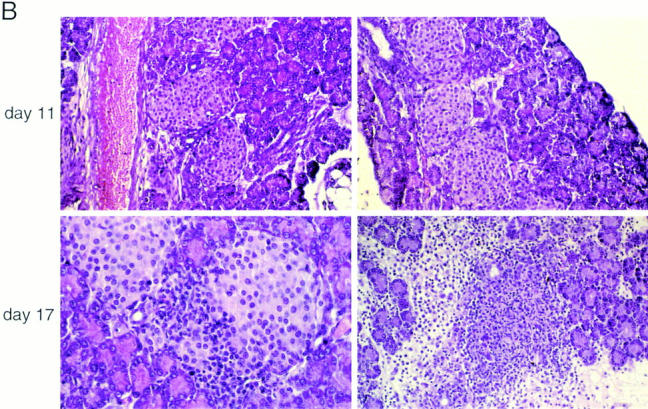
Treatment with anti–CTLA-4 does not provoke global T cell activation but enhances the aggressiveness of insulitis. (A) Lymphocytes from the spleen, mesenteric lymph nodes, or pancreas of 17-d-old BDC2.5/NOD mice, after injection with either control mAb (left) or anti–CTLA-4 (right) on day 12, were stained for CD4, CD8, and CD69 or CD62L. The profiles depict cells gated for CD4 positivity. The bars indicate positive staining. (B) Representative pancreas sections from BDC2.5/NOD transgenics treated with control mAb (left) or anti–CTLA-4 (right). (Top) Injections on days 2, 5, and 8 and pancreas removal on day 11. (Bottom) Injections on days 9, 12, and 15 and pancreas removal on day 17.
Fig. 3 B compares T cell invasiveness in BDC2.5 tgs administered control mAb or anti–CTLA-4 well before insulitis initiation. The kinetics of insulitis in the two types of animals appeared the same; there was no infiltrate seen at 11 d and a substantial one at 17 d. However, the nature of the insulitis was strikingly different in the two cases. In control mAb-injected mice, the invading cells penetrated the islets, but rested at the outskirts, quite segregated from the β cell mass. This picture is typical of insulitis in BDC2.5/NOD tgs. The mice injected with anti–CTLA-4 exhibited a much more aggressive infiltrate, with complete intermingling of the invading leukocytes and the β cells, even spilling out into the exocrine tissue, and marked edema. This aggressive image is reminiscent of insulitis in BDC2.5/NOD tgs just after cyclophosphamide injection (6), young BDC2.5/B6 mice (5), and neonatal NOD mice transferred with activated BDC2.5 T cells (29).
Discussion
Our data indicate that CTLA-4 plays a pivotal role at disease onset in the BDC2.5/NOD TCR tg model of diabetes. Whether or not CTLA-4 can be engaged on the T cells of animals 12 d old or less conditions the nature of the insulitis that ensues, how aggressive it looks and how rapidly it evolves to full-blown diabetes. We do not know precisely what happens to BDC2.5 T cells during the animals' first days of life other than that they are generated in the thymus and populate the peripheral lymphoid organs. Presumably, they must encounter the relevant β cell antigen, become activated, and migrate to the islets, but the order and details of these events have not yet been established. Whatever the precise scenario turns out to be, it must include an opportunity for CTLA-4 to be engaged by its ligand and to exert its influence on the nature of insulitis.
CTLA-4's role in BDC2.5/NOD mice appeared to be confined to disease onset. Anti–CTLA-4 mAb precipitated diabetes when administered before insulitis began, but never afterwards. The opposite is true with cyclophosphamide; this drug rapidly induces diabetes in BDC2.5/NOD animals, but only in those which already exhibit insulitis (6). The effect of cyclophosphamide indicates that the nondestructive form of insulitis is certainly not refractory to incitement. It seems, then, that preempting the engagement of CTLA-4 can alter disease course in the BDC2.5 model, but annulling the engagement cannot. CTLA-4 seems to control entry into a state of stable and balanced autoimmunity; once this state is established, mechanisms independent of CTLA-4 must maintain it. Such a restricted window of influence would not have been expected according to the original idea of CTLA-4 function, i.e., that it is critical for the downmodulation of an ongoing T cell response. However, it fits very well with the newer notion that CTLA-4 can be important in setting, at the time of initiation, the timbre of a response (15, 30).
How might CTLA-4 engagement when BDC2.5 T cells first encounter their antigen influence the nature of the insulitis that develops? It is well established that CD28 transmits a supplementary positive signal when engaged together with the TCR; such costimulation augments the production of IL-2, expression of the IL-2 receptor, and proliferation, and is required for most, though not all, T cell responses to reach fruition (8, 9). There is now strong evidence that engagement of CTLA-4, along with CD28 and the TCR, relays a negative signal (31), dampening T cell responses by reducing levels of IL-2, IL-2 receptor, and proliferation (14, 15, 23). The ultimate result can be functional nonresponsiveness, even if some signs of activation are still evident (23). In the case of BDC2.5 tgs, CTLA-4 engagement permits activation and mobilization of the β cell–reactive T cells, as evidenced by the strong islet infiltrate that develops, but it clearly alters the quality of the response since the insulitis appears less aggressive and does not provoke the destruction of β cells until after a long delay. The exact phenotypic alterations that CTLA-4 engagement induces in responding to BDC2.5 T cells remain to be identified. It is possible that the production of inflammatory cytokines is attenuated, that the ratio of T helper phenotypes is skewed against cells that destroy β cells or towards protective cells, or that other effector functions are compromised. Functional nonresponsiveness is also a possibility, but it is unlikely to be as profound as that described previously (23), given the active division of T cells in the islets of BDC2.5/NOD tgs (3 and our unpublished data).
It is necessary to place our results in the context of previous findings on the influence of CD28/CTLA-4–B7-1/2 interactions on the progression of disease in NOD mice. In their initial study, Lenschow et al. reported perplexing results on the effects of anti-B7 reagents (25): mAbs that block B7-1 or both B7-1 and B7-2 greatly accelerated diabetes onset if treatment was begun in the first 2–3 wk of life and continued for several weeks thereafter. An anti– B7-2 mAb or a soluble CTLA-4 fusion protein capable of blocking both B7-1 and B7-2 inhibited disease when given according to the same protocol. None of the anti-B7 reagents had an effect when administered after 10 wk of age. Matters became even more confusing with a subsequent report on tg mice expressing the soluble B7-1/2 blocker in the blood from birth and “knockout” animals genetically incapable of expressing CD28 (32); in both cases, diabetes was greatly accelerated. It is difficult to amalgamate these findings into a coherent picture, and even harder to integrate them with our results. Clearly, the complexity of the CD28/CTLA-4–B7-1/2 system, with its variable patterns of inducible and constitutive expression and widely ranging affinities of interaction, demands punctual interventions with specific reagents during discrete time windows.
The striking results we obtained no doubt reflect the simplicity of the BDC2.5 model, in particular, the relative synchrony of early disease events. Demonstrating an analogous role for CTLA-4 at disease onset in standard NOD mice and eventually humans, with their more variable disease courses, will be significantly more challenging. Preliminary results on NOD mice indicate that diabetes can be induced by injection of anti–CTLA-4 mAb at early ages but that, as expected, the results are less consistent (Lühder, F., D. Mathis, and C. Benoist, unpublished data).
Acknowledgments
We would like to thank Dr. Dana Leach, C. Waltzinger, T. Ding, F. Fischer, V. Louerat, P. Michel, J. Hergueux, P. Gerber and C. Ebel for assistance with various aspects of this study.
This work was supported by grants to CB and DM from the Juvenile Diabetes Foundation International (196078) and to JPA from the NIH (US) (CA40041), and by funds from the Institut National de la Santé et de la Recherche Médicale, the Centre National de la Recherche Scientifique, the Centre Hospitalier Universitaire Régional and Bristol-Myers Squibb. FL was supported by the Deutsche Forschungsgemeinschaft (lu 634/1-1) and the European Economic Community and PH by the Wenner-Gren Foundation of Sweden.
References
- 1.Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev. 1994;15:516–542. doi: 10.1210/edrv-15-4-516. [DOI] [PubMed] [Google Scholar]
- 2.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 3.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 4.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+islet-specific T-cell clone. Science. 1990;249:1433–1436. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D. Genetic control of diabetes progression. Immunity. 1997;7:873–883. doi: 10.1016/s1074-7613(00)80405-7. [DOI] [PubMed] [Google Scholar]
- 6.André I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: Lessons from diabetes models. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tivol EA, Schweitzer AN, Sharpe AH. Costimulation and autoimmunity. Curr Opin Immunol. 1996;8:822–830. doi: 10.1016/s0952-7915(96)80011-2. [DOI] [PubMed] [Google Scholar]
- 8.Chambers CA, Allison JP. Co-stimulation in T cell responses. Curr Opin Immunol. 1997;9:396–404. doi: 10.1016/s0952-7915(97)80087-8. [DOI] [PubMed] [Google Scholar]
- 9.Bluestone JA. Is CTLA-4 a master switch for peripheral T cell tolerance? . J Immunol. 1997;158:1989–1993. [PubMed] [Google Scholar]
- 10.Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, Damle NK. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Y, Guo Y, Huang A, Zheng P, Liu Y. CTLA-4–B7 interaction is sufficient to costimulate T cell clonal expansion. J Exp Med. 1997;185:1327–1335. doi: 10.1084/jem.185.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 13.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kearney ER, Walunas TL, Karr RW, Morton PA, Loh DY, Bluestone JA, Jenkins MK. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J Immunol. 1995;155:1032–1036. [PubMed] [Google Scholar]
- 17.Krummel MF, Sullivan TJ, Allison JP. Superantigen responses and co-stimulation: CD28 and CTLA-4 have opposing effects on T cell expansion in vitro and in vivo. Int Immunol. 1996;8:519–523. doi: 10.1093/intimm/8.4.519. [DOI] [PubMed] [Google Scholar]
- 18.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 19.Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184:783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333–1336. [PubMed] [Google Scholar]
- 21.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–989. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 22.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 23.Perez VL, Parijs LV, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- 24.Nistico L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, Larrad MT, Rios MS, Chow CC, Cockram CS, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Hum Mol Genet. 1996;5:1075–1080. doi: 10.1093/hmg/5.7.1075. [DOI] [PubMed] [Google Scholar]
- 25.Lenschow DJ, Ho SC, Sattar H, Rhee L, Gray G, Nabavi N, Herold KC, Bluestone JA. Differential effects of anti–B7-1 and anti–B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med. 1995;181:1145–1155. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Böhme J, Schuhbaur B, Kanagawa O, Benoist C, Mathis D. MHC-linked protection from diabetes dissociated from clonal deletion of T cells. Science. 1990;249:293–295. doi: 10.1126/science.2115690. [DOI] [PubMed] [Google Scholar]
- 27.Gross JA, Callas E, Allison JP. Identification and distribution of the costimulatory receptor CD28 in the mouse. J Immunol. 1992;149:380–388. [PubMed] [Google Scholar]
- 28.Cosgrove D, Gray D, Dierich A, Kaufman J, Lemeur M, Benoist C, Mathis D. Mice lacking MHC class II molecules. Cell. 1991;66:1051–1066. doi: 10.1016/0092-8674(91)90448-8. [DOI] [PubMed] [Google Scholar]
- 29.Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin-dependent diabetes. Science. 1995;268:1185–1188. doi: 10.1126/science.7761837. [DOI] [PubMed] [Google Scholar]
- 30.Chambers CA, Krummel MF, Boitel B, Hurwitz A, Sullivan TJ, Fournier S, Cassell D, Brunner M, Allison JP. The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol Rev. 1996;153:27–46. doi: 10.1111/j.1600-065x.1996.tb00919.x. [DOI] [PubMed] [Google Scholar]
- 31.Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science. 1996;272:1170–1173. doi: 10.1126/science.272.5265.1170. [DOI] [PubMed] [Google Scholar]
- 32.Lenschow DJ, Herold K, Rhee L, Patel B, Koons A, Qin H-Y, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]