

Abstract
Activation, anergy, and apoptosis are all possible outcomes of T cell receptor (TCR) engagement. The first leads to proliferation and effector function, whereas the others can lead to partial or complete immunological tolerance. Structural variants of immunizing peptide–major histocompatibility complex molecule ligands that induce selective lymphokine secretion or anergy in mature T cells in association with altered intracellular signaling events have been described. Here we describe altered ligands for mature mouse CD4+ T helper 1 cells that lead to T cell apoptosis by the selective expression of Fas ligand (FasL) and tumor necrosis factor (TNF) without concomitant IL-2, IL-3, or interferon γ production. All ligands that stimulated cell death were found to induce FasL and TNF mRNA expression and TCR aggregation (“capping”) at the cell surface, but did not elicit a common pattern of tyrosine phosphorylation of the TCR-associated signal transduction chains. Thus, TCR ligands that uniquely trigger T cell apoptosis without inducing cytokines that are normally associated with activation can be identified.
Tcell receptor (TCR) engagement can lead to several different responses in mature peripheral T lymphocytes, including activation and proliferation, anergy, and programmed cell death (apoptosis). These events are controlled by signals resulting from interaction of the TCR and CD4 or CD8 coreceptors with peptide–MHC complex ligands and contribute to the quality and extent of the T cell response. Structural variants of a TCR ligand can selectively affect individual effector functions and biochemical events in T cells (1–5). Introduction of amino acid substitutions in the peptide component of an agonist ligand for a given T cell may variously result in the formation of: (a) a nonagonist that elicits no T cell effector function; (b) a weaker or stronger agonist that elicits all T cell effector functions with a different dose response from the original ligand; (c) a partial agonist with the capacity to induce a subset of effector functions or anergy in T cells; or (d) an antagonist that diminishes the response to agonists. The discovery of these functional classes of variant TCR ligands promoted the notion that T cells can sense the “quality” as well as the “quantity” of TCR engagements (1–5). This concept has been strengthened by recent evidence for unique patterns of early tyrosine phosphorylation events induced by variant TCR ligands that cannot be seen in response to agonists (2, 3, 6).
The activation state of a T cell also dictates the outcome of interactions between the TCR and the peptide–MHC ligand. Agonist ligands stimulate lymphokine production and proliferation in resting T cells, whereas in cycling cells, they cause lymphokine production followed by death via apoptosis (7, 8). The differential signaling capacity of the TCR prompted us to investigate whether variant ligands that selectively induce T cell death without effector activation might exist. Such ligands could be useful for dissecting the signaling pathways leading to apoptosis as well as for developing effective immunotherapeutic agents. We therefore screened numerous variants of the pigeon cytochrome c (PCC)1 88–104 peptide for the ability to trigger apoptosis in an activated murine CD4+ T cell clone against PCC/I-Ek without inducing activation cytokines such as IL-2, IL-3, or IFN-γ. We have identified altered TCR ligands with selective death-inducing activity and report the initial characterization of their effects on activated T cells.
Materials and Methods
T Cell Clones and APCs.
CD4+ A.E7 Th1 cells (9) were maintained by biweekly stimulation with PCC protein (5 μM) and irradiated splenocytes from B10.A (H-2a) mice. A.E7 cells stimulated in this manner 2 d previously were ficolled and transferred to fresh medium containing IL-2 (50–100 IU/ml recombinant human IL-2 (Chiron, Emeryville, CA) or 10–15% T-StimTM (Collaborative Biomedical Products, Bedford, MA) and incubated for an additional 2–5 d to generate cycling A.E7 cells that are predisposed to apoptosis after TCR engagement (8). P13.9, L cell transfectants expressing I-Ek, intercellular adhesion molecule 1 (ICAM-1), and B7.1 (CD80) molecules, were used as APCs (3).
Peptides.
PCC (88–104) is KAERADLIAYLKQATAK. Peptides Y99, C99, R99, A99 are synthetic peptides in which the primary TCR contact residue, lysine in position 99, is changed to tyrosine, cysteine, arginine, or alanine, respectively. All peptides were synthesized by the Peptide Synthesis Facility, NIAID, National Institutes of Health [NIH], Bethesda, MD.
Cell Death Assays.
5 × 104 P13.9 cells were incubated with 5 × 104 cycling A.E7 cells in the presence of various concentrations of PCC peptides in quadruplicate in 96-well round bottomed plates. To block apoptosis, Fas-Fc and TNFR-Fc (10 μg/ml; gift from Dr. David Lynch, Immunex, Seattle, WA) were included in some assays. Cells were harvested after 24 h of incubation, two of each four wells were pooled, and 10 μg/ml of propidium iodide were added. Ungated cells were acquired for 30 s at a constant flow rate using a FACScan® equipped with CellQuest software (Becton-Dickinson, Mountain View, CA) and the number of viable A.E7 cells in each sample was calculated as previously described (10, 11). In some experiments, P13.9 cells were preloaded with 0.1 μM carboxyfluorescein diacetate-acetylester (CMFDA; Molecular Probes, Inc., Eugene, OR) to allow APCs to be excluded from the analysis by gating on fluorescein-negative cells. In other experiments, A.E7 cells were stained with fluoresceinated anti-CD4 before flow cytometry to differentiate them from APCs.
Cytokine Measurement.
Supernatants from the apoptosis assays were harvested at 24 h and assayed for cytokine production by ELISA. IL-2 and IL-3 were detected using antibodies purchased from PharMingen (San Diego, CA), essentially as per the manufacturer's instructions. IFN-γ was measured by ELISA as previously described (6). Data are expressed as the mean of quadruplicate wells calculated as a percentage of the response to 100 μM wild-type (WT) peptide.
mRNA Analysis by Semiquantitative Reverse Transcriptase PCR.
A.E7 cells were incubated with P13.9 cells prepulsed for 2 h with 100 μM of the indicated peptides and total RNA was isolated using RNAzol according to the manufacturer's instructions (Tel-test, Inc., Friendswood, TX). The reverse transcriptase (RT) reaction was performed using random hexamer primers and part of each sample was subjected to PCR amplification using β-actin primers and 32P-labeled nucleotides (12). Serial dilutions of RT products were used to ensure comparisons were in the linear range. After normalizing the input cDNA to the β-actin signal, β-actin, Fas ligand (L), TNF-α, IL-2, IFN-γ and Bcl-X primers were then used to amplify their respective cDNA under the same conditions. The PCR protocol was 95°C for 30 s, 55°C for 30 s, 72°C for 60 s, for 30 cycles. Sequences of the primers are as follows: β-actin, 5′ primer (5′GAT GAC GAT ATC GCT GCG CTG3′), 3′primer (5′GTA CGA CCA GAG GCA TAC AGG3′); mIL-2, 5′ primer (5′ATG TAC AGC ATG CAG CTC GCA TC3′), 3′ primer (5′GGC TTG TTG AGA TGA TGC TTT GAC A3′); mFas-L, 5′ primer (5′CTG GTG GCT CTG GTT GGA AT3′), 3′ primer (5′GTT TAG GGG CTG GTT GTT GC3′); mTNF-α, 5′ primer (5′ ATG AGC ACA GAA AGC ATG ATG CGC3′), 3′ primer, (5′CCA AAG TAG ACC TGC CCG GAC TC3′); Bcl-X, 5′primer, (5′CAA TGG TGG CTG AAG AGA3′), 3′ primer, (5′GGA GAG CGT TCA GTG ATC3′).
Protein Biochemistry.
2–5 × 106 P13.9 APCs were incubated for 2–4 h in 200 μl complete medium alone (no Ag) or with the indicated peptides at 100 μM. Pulsed APCs were then centrifuged together with 1–1.25 × 107 cycling A.E7 cells in Eppendorf tubes and warmed to 37°C, as described (6). After stimulation, T cells + APCs were lysed (6) and lysates were then subjected to immunoprecipitation with anti-CD3ε (500A2 mAb; PharMingen), anti–ZAP-70 (rabbit antiserum; gift from Dr. L. Samelson, NICHD, NIH, Bethesda, MD), or anti–TCR-ζ (rabbit antiserum; gift from Dr. J. Bolen, DNAX, Palo Alto, CA; 13). Immunoprecipitates were resolved by SDS-PAGE, transferred to nitrocellulose, and blotted with antiphosphotyrosine antibody (6). Blots were developed using SuperSignalTM chemiluminescent substrate (Pierce Chemical Co., Rockford, IL).
Fluorescence Microscopy.
106 P13.9 APCs were incubated for 2–4 h in 200 μl complete medium alone (NP) or with the indicated peptides at 100 μM. Pulsed APCs were then centrifuged together with 106 cycling A.E7 cells in Eppendorf tubes and warmed to 37°C for 5 min. Cells were incubated on ice for 5 min and an additional 15 min in presence of 10 μg/ml of anti-CD3 Ab (PharMingen), washed twice with cold PBS, and fixed for 30 min at room temperature in 1% PFA. For the detection of actin polymerization, A.E7 cells were similarly stimulated with APCs and different variant peptides. Cells were fixed for 30 min at room temperature in 1% PFA, washed twice in PBS 1% FCS and incubated 30 min in PBS/0.1% saponin/1% FCS/Texas red conjugated–X phalloidin (2 U/ml). Cells were washed twice with PBS and were mounted on slides using Fluoromount-GTM (Electron Microscopy Sciences, Fort Washington, PA). The images were obtained on a Zeiss Axiophot (Carl Zeiss, Inc., Thornwood, NY) with a CCD camera (Princeton Instruments, Inc., Trenton, NJ).
Results
Variant Ligands Induce Apoptosis without the Production of Activation Lymphokines such as IL-2, IL-3, or IFN-γ.
To determine whether variant ligands can selectively elicit apoptosis responses, we screened many peptides with single amino acid substitutions at the major epitopic residue (lysine 99) of the agonist 88–104 peptide of PCC (PCC 88-104) for their effect on A.E7 cells that were actively cycling under the influence of IL-2. Certain variants of PCC 88-104 with substitutions at position 99 have been previously found to induce variant signaling in unactivated, resting A.E7 cells (3). These substitutions do not affect binding to the I-Ek molecule and therefore are assumed only to alter TCR recognition of peptide–MHC ligand (3, 14). In our study of cycling A.E7 cells, we observed that the WT PCC 88–104 peptide induces both cell death and the secretion of IL-2, IL-3, and IFN-γ (Fig. 1 A). In contrast, at high concentrations, two of the variant peptides tested, Y99 and C99, induce as much cell death as the WT peptide, but fail to induce IL-2, IL-3, or IFN-γ production (Fig. 1 A). T cell death under these circumstances has the morphological and nuclear fragmentation characteristics of apoptosis (8 and data not shown). Other variant peptides, such as A99 and R99, do not stimulate either apoptosis or lymphokine secretion (Fig. 1 A). To address the possibility that the selective cell death induction by Y99 and C99 was due to weak stimulation of all effector functions, we compared dose-response curves for apoptosis and lymphokine responses. The apoptosis assay is slightly more sensitive than the cytokine ELISAs (Fig. 1 B); however, 50% maximal apoptosis requires ∼1 nM WT ligand (Fig. 1 B) or 1 μM Y99 (Fig. 1 D). Because half-maximal cytokine production in response to the WT peptide requires at most 10-fold more agonist than half-maximal apoptosis, cytokine production should be seen with 10 μM Y99 assuming the latter simply behaves as a weak agonist. Yet 100 μM of Y99 or C99 causes no production of IL-2, IL-3, or IFN-γ (Fig. 1 D and data not shown). We conclude that Y99 and C99 are true partial agonists for A.E7 cells, because they induce apoptosis but not other typical effector functions. By contrast, R99 and A99 fail to induce these responses at any dose and are therefore nonagonists with respect to the responses we have examined (Fig. 1 C and data not shown).
Figure 1.

(A) Variant ligands induce apoptosis of CD4+ Th1 cells without the accompanying production of IL-2, IL-3, or IFN-γ. (B–D) Dose-response analyses of cycling A.E7 cells stimulated by P13.9 APCs incubated with PCC (88–104) WT peptide (B), variant peptide R99 (C), or variant peptide Y99 (D). T cell apoptosis (solid square), IL-2 (diamond), IL-3 (circle), and IFN-γ (triangle) production in cycling A.E7 cells was measured after stimulation for 24 h with P13.9 APCs in the presence of the indicated concentrations of WT peptide or variant analogues. All values are expressed as a percentage of the response obtained with 100 μM of the WT peptide. The actual 100% maximal values were as follows: apoptosis, 55% dead cells at 24 h; IL-2, 1.4 ng/ml; IL-3, ⩾250 ng/ml; IFN-γ, 120 ng/ml. All error bars are shown and represent one SD from the mean.
T Cell Death Induced by Partial Agonists Uses the Fas and TNF Pathways.
Because T cell apoptosis can result from the action of Fas or TNF (15–18), we assessed the role of these molecules in cell death induced by the Y99 and C99 partial agonists. Stimulation of cycling A.E7 cells for 2 h with the apoptogenic WT, Y99, or C99 peptides induced the mRNAs for TNF and Fas-L but not for Bcl-X, a protein that prevents apoptosis (19; Fig. 2 A). There was less induction of these mRNAs with the variants compared to the WT peptide, in accordance with the latter's greater lethality at low concentrations (Fig. 1). Fas-L or TNF mRNA levels were not increased using the nonagonist peptides A99 and R99 (Fig. 2 A). Both Fas-L and TNF were functionally important in the apoptosis caused by the WT, C99, and Y99 peptides because death was potently inhibited by reagents that specifically block these cytokines (Fig. 2 B). In contrast to the WT peptide, none of the variants elicited IL-2 (Fig. 2 A) or IFN-γ mRNA (data not shown), confirming the results obtained by ELISA (Fig. 1).
Figure 2.

Induction of T cell death by partial agonists is mediated by both the Fas and TNF pathways. (Left) mRNA expression by semiquantitative RT-PCR analysis of β-actin, IL-2, Fas-L, TNF-α, and Bcl-X in cycling A.E7 cells stimulated with variant or WT ligands. Cycling A.E7 cells were stimulated for 2 h with P13.9 APCs preincubated with either medium (no Ag) or 100 μM of the WT or variant peptides as indicated. Dilutions of the RT product were pretested to ensure that the PCR reaction was in the linear range. (Right) Fas-L and TNF blocking of variant ligand-induced cell death of A.E7 cells. Cycling A.E7 cells were stimulated with APCs prepulsed with 2.5 nM WT, 100 μM C99, or 100 μM Y99 peptides, in medium alone or in the presence of either Fas-Fc (10 μg/ml), TNFR-Fc (10 μg/ml), or both Fas-Fc and TNFR-Fc as indicated. A.E7 cell death was measured as described in Materials and Methods and data are expressed as the percentage of dead cells. In control experiments, Fas-Fc and TNFR-Fc alone did not induce proliferation or apoptosis. Error bars represent one SD of the mean.
Tyrosine Phosphorylation Patterns Induced by Variant Ligands.
In resting T cells, partial agonist or antagonist ligands generate unique patterns of protein tyrosine phosphorylation (2, 3, 6). Hence, we determined whether apoptosis-inducing variant peptides produce characteristic TCR-associated phosphorylation patterns in cycling T cells. CD3-ε, ZAP-70, or TCR-ζ (Fig. 3, A, B, and C, respectively) and their associated proteins were immunoprecipitated from lysates prepared from A.E7 cells that had been briefly stimulated with WT and variant ligands. In cycling A.E7 cells, the WT peptide induced prominent tyrosine-phosphorylated species, including the three isoforms of phospho-ζ (p18, p21, and p23), phosphorylated CD3-ε, and phosphorylated ZAP-70 (Fig. 3, A and B, lane 2). This is the same pattern as that obtained when resting A.E7 cells were stimulated with the WT peptide (3). However, we found no correlation between the pattern of TCR-ζ tyrosine phosphorylation and programmed cell death. The apoptosis-inducing partial agonist, C99, and the nonagonist ligands, A99 and R99, all produced a modest increase in the p21 form of phospho-ζ (Fig. 3, A–C, lanes 3–5). However, the partial agonist Y99 surprisingly failed to induce a detectable increase in p21 phospho-ζ, even upon examination by direct anti-ζ immunoprecipitation (Fig. 3, A–C, lane 6). None of the variant ligands induced detectable levels of the other species of phospho-ζ (p18 and p23), phosphorylated CD3-ε, or phosphorylated ZAP-70 (Fig. 3, A and B, lanes 3–6).
Figure 3.
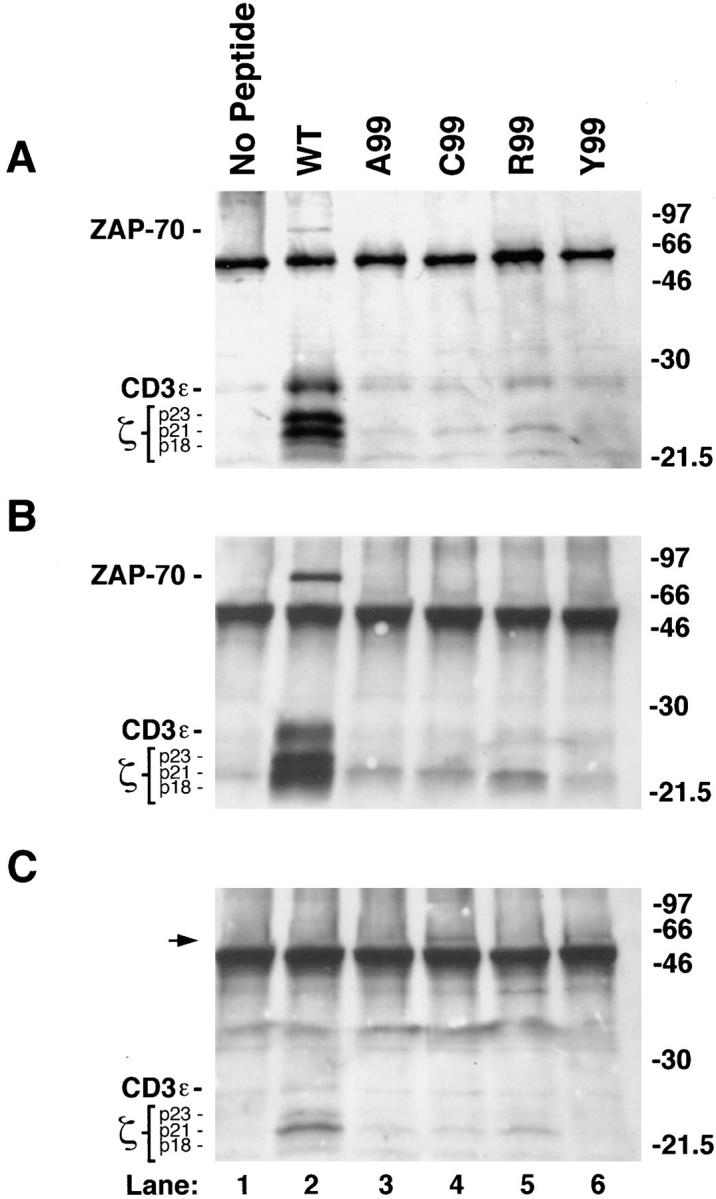
Effect of variant peptide stimulation on early tyrosine phosphorylation in cycling A.E7 cells. Tyrosine phosphorylation of major protein species (indicated on left) coprecipitating with CD3-ε (A), ZAP-70 (B), or TCR-ζ (C), revealed by immunoprecipitation followed by SDS-PAGE and phosphotyrosine immunoblotting. For each lane, 107 cycling A.E7 cells were stimulated for 10 min with 5 × 106 P13.9 APCs prepulsed with the indicated peptides. The arrowhead in C identifies a 55–60-kD phosphorylated protein coprecipitating with ζ after stimulation with C99 or Y99, the only two variants capable of inducing A.E7 cell death. The anti-ζ antiserum used here preferentially precipitates the p21 form of phospho-ζ, resulting in underdetection of the p23 species (C). SDS-PAGE was performed on a 12% gel under reducing conditions. The migration of prestained molecular weight markers is indicated on the right. B–D are from the same experiment, whereas A is from a separate experiment.
Ligands that Deliver Death-inducing Signals Induce TCR Capping.
Successful activation of resting T cells with an agonist peptide causes the redistribution of the TCR into polarized focal aggregates on the membrane, known as “capping” (20). We examined by fluorescent microscopy the ability of the variant ligands to induce TCR aggregation and actin polymerization after stimulating A.E7 cells for 5 min. We observed TCR polarization at points of contact with the APCs after stimulation with the WT peptide and the apoptosis-inducing Y99 and C99 peptides but not by the A99 peptide which, by itself, does not induce cell death. The aggregation of the TCR correlated closely with actin polymerization detected after intracellular staining of A.E7 cells + APCs with Texas red–conjugated X phalloidin (Fig. 4). However no distinct capping or TCR polarization was seen after stimulation with the variants R99 (data not shown) and A99 (Fig. 4), which fail to cause apoptosis even though they induced readily detectable ζ-chain phosphorylation.
Figure 4.
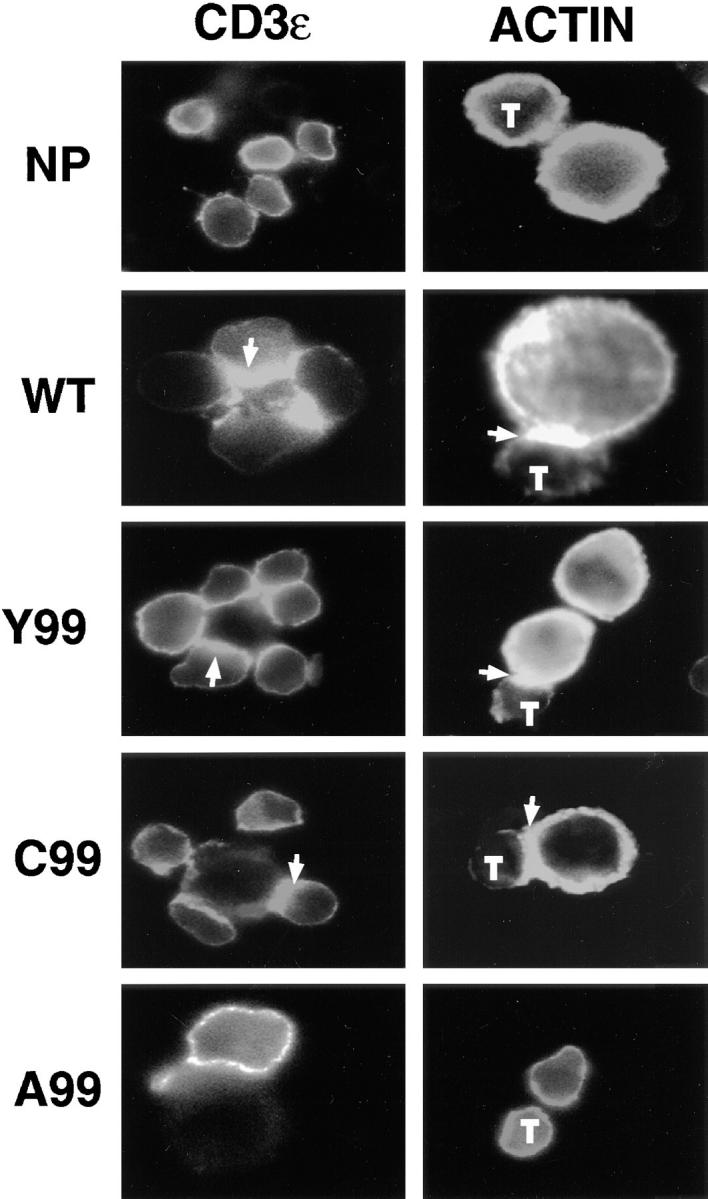
Variant TCR ligands that induce programmed cell death promote TCR aggregation. Cycling A.E7 cells were stimulated with P13.9 APCs pulsed with medium alone (NP) or with the indicated peptides at 100 μM for 5 min at 37°C. Cells were stained with fluoresceinated anti-CD3 Ab (PharMingen) (left) and Texas red conjugated–X phalloidin (right). Slides were analyzed by fluorescence microscopy. Arrows indicate TCR or actin aggregation. Small cells are A.E7 T cells (T) and larger cells are APCs.
Discussion
A key difference between resting and cycling mature T cells is that antigen will stimulate lymphokine production and proliferation in resting cells, whereas it will stimulate lymphokine production and apoptosis in cycling T cells (8). Death of cycling T lymphocytes occurs as part of a homeostatic feedback control mechanism and can be used to ameliorate the autoimmune demyelinating disease, experimental allergic encephalomyelitis, in mice (21, 22). For the first time, we have examined the biological and signaling properties of TCR interaction with variant peptide–MHC class II molecule ligands in cycling T cells. We used ligands that differ in a key TCR contact residue in an attempt to influence the quality of TCR engagement by altering the TCR–peptide–MHC conjugate. Cytochrome c peptide variants at position 99, as we have used here (Y99, C99, A99, and R99) have been shown to bind Ek with the same affinity but to bind differently to the TCR through residues in the CDR3 (14). We have discovered that Y99 and C99 represent a novel class of variant ligands that induce autoregulatory apoptosis of A.E7 cells without secretion of numerous cytokines (IL-2, IL-3, IFN-γ) whose production typically accompanies TCR signaling of Th1 clones such as A.E7. This contrasts with the WT agonist, which causes both T cell death and cytokine production. For A.E7 cells, we have not detected any ligands that induce activation cytokines without apoptosis. The discovery of TCR ligands that induce death without other effector functions leads one to consider whether other ligands could possibly elicit cytokine secretion without death. Models suggesting a hierarchy of T cell responses do not support this possibility. However, if distinct signaling pathways for each effector response exist, then novel variants that stimulate activation cytokines without apoptosis might be found (2, 3).
We observed similarities and differences in the signalling events triggered by the WT agonist and the selectively apoptogenic variant peptides, C99 and Y99. Each of the peptides that caused apoptosis shared the ability to induce TCR polarization in the membrane, as well as Fas-L and TNF gene activation. At the same time, stimulation with these ligands did not result in a consistent pattern of TCR-subunit phosphorylation, as the WT and C99 ligand induced clear-cut ζ chain phosphorylation, but the Y99 ligand failed to do so. Moreover, the C99 and Y99 partial agonists failed to induce other tyrosine phosphorylation signals involving CD3-ε and the ZAP-70 kinase, which correlated with their failure to induce IL-2, IL-3, and IFN-γ expression. However, despite the absence of these phosphorylation events, the C99 and Y99 peptides induce Fas-L and TNF at levels sufficient to cause death. Thus, the TCR signals associated with the induction of death cytokines are potentially different from the signals necessary for inducing activation lymphokines.
Our observations concerning signal transduction are surprising in the context of standard models of WT and variant ligand signaling. Most current hypotheses are based on the lower affinity of altered ligands for the TCR and propose that the variation in TCR phosphorylation seen with such rapidly dissociating ligands arises from their short time of TCR engagement (1, 4, 5, 23, 24). In these models, only a subset of signaling events and effector functions would be induced by the variant ligands because there is rapid loss of ligand–TCR contact. If cellular responses are directly linked to the sequential set of signaling events that occur over time, one would expect a consistent pattern of signaling for ligands capable of inducing similar functional responses. We do not observe a consistent pattern of phosphorylation associated with the ability of peptides to cause apoptosis, despite the shared ability of the apoptosis-inducing ligands to cause TCR aggregation and actin polymerization. However, engagement of the TCR may elicit both positive and negative regulatory feedback loops that compete kinetically to control the extent and duration of receptor phosphorylation. As such, all ligands might initiate similar early signaling events, including immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation (which is required for active polarization of the TCR), but may differ in the extent to which phosphatase recruitment has occurred at the time the cells are lysed for biochemical analysis. Alternatively, one must consider the possibility that a complex alteration in TCR signal generation arising from conformational properties of the altered ligand–TCR interaction, rather than simple kinetics, accounts for the selective induction of apoptosis.
Because mature T cell apoptosis is a powerful mechanism for establishing peripheral immune tolerance, the new class of variant ligands described here, which selectively induce apoptosis of the responding T cell, constitute a new type of potentially valuable immunomodulatory reagent. The availability of apoptosis-inducing peptides that do not elicit lymphokine secretion could permit the elimination of activated T cells without the possibility of exacerbating disease by the simultaneous release of inflammatory mediators. Likewise, selective death-inducing responses to self-ligands recognized by peripheral T cells as partial agonists, might be envisioned to play a natural role in preventing destructive autoimmune disease by eliminating self-reactive T cells without allowing them to expand or cause inflammation. Further experimentation will be needed to address these possibilities.
Acknowledgments
We thank Drs. J. Bolen, D. Lynch and L. Samelson for generously providing reagents. We thank Dr. I. Stefanova for helpful discussions and Drs. B. Lucas, J. O'Shea, R. Siegel and P. Schwartzberg for critically reading the manuscript.
Footnotes
Abbreviations used in this paper: Fas-L, Fas ligand; PCC, pigeon cytochrome c; WT, wild-type.
B. Combadière and C. Reis e Sousa contributed equally to this work.
References
- 1.Evavold BD, Sloan LJ, Allen PM. Tickling the TCR: selective T-cell functions stimulated by altered peptide ligands. Immunol Today. 1993;14:602–609. doi: 10.1016/0167-5699(93)90200-5. [DOI] [PubMed] [Google Scholar]
- 2.Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: altered phospho-zeta and lack of ZAP-70 recruitment in APL-induced T cell anergy. Cell. 1994;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 3.Madrenas J, Wange RL, Wang JL, Isakov N, Samelson LE, Germain RN. Zeta phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science. 1995;267:515–518. doi: 10.1126/science.7824949. [DOI] [PubMed] [Google Scholar]
- 4.Jameson SC, Bevan MJ. T cell receptor antagonists and partial agonists. Immunity. 1995;2:1–11. doi: 10.1016/1074-7613(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 5.Madrenas J, Germain RN. Variant TCR ligands: new insights into the molecular basis of antigen-dependent signal transduction and T cell activation. Semin Immunol. 1996;8:83–101. doi: 10.1006/smim.1996.0011. [DOI] [PubMed] [Google Scholar]
- 6.Reis e Sousa C, Levine EH, Germain RN. Partial signaling by CD8+T cells in response to antagonist ligands. J Exp Med. 1996;184:149–157. doi: 10.1084/jem.184.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lenardo MJ. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- 8.Lenardo MJ, Boehme SA, Chen L, Combadière B, Fisher G, Freedman M, McFarland H, Pelfrey C, Zheng L. Autocrine feedback death and the regulation of mature T lymphocyte antigen responses. Int Rev Immunol. 1995;13:115–134. doi: 10.3109/08830189509061742. [DOI] [PubMed] [Google Scholar]
- 9.Hecht TT, Longo DL, Matis LA. The relationship between immune interferon production and proliferation in antigen-specific, MHC-restricted T cell lines and clones. J Immunol. 1983;131:1049–1054. [PubMed] [Google Scholar]
- 10.Boehme SA, Lenardo MJ. Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur J Immunol. 1993;23:1552–1560. doi: 10.1002/eji.1830230724. [DOI] [PubMed] [Google Scholar]
- 11.Duke, R.C., J.J. Cohen, S.A. Boehme, M.J. Lenardo, C.D. Surh, H. Kishimoto, and J. Sprent. 1995. Morphological, biochemical and flow cytometric assays of apoptosis. In Current Protocols in Immunology. J.E. Coligan, A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober, editors. Wiley, New York. Chapter 3.17.
- 12.Boehme SA, Lenardo MJ. TCR-mediated death of mature T lymphocytes occurs in absence of p53. J Immunol. 1996;156:4075–4078. [PubMed] [Google Scholar]
- 13.Burkhardt AL, Stealey B, Rowley RB, Mahajan S, Prendergast J, Fargnoli J, Bolen JB. Temporal regulation of non-transmembrane protein tyrosine enzyme activity following T cell antigen receptor engagement. J Biol Chem. 1994;269:23642–23647. [PubMed] [Google Scholar]
- 14.Jorgensen JL, Esser U, Fazekas de St B, Groth, Reay PA, Davis MM. Mapping T-cell receptor–peptide contacts by variant peptide immunization of single chain trangenics. Nature. 1992;355:224–230. doi: 10.1038/355224a0. [DOI] [PubMed] [Google Scholar]
- 15.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 16.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, Green DR. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 17.Ju ST, Panka DJ, Cui H, Ettinger R, El-Khatib M, Sherr DH, Stanger BZ, Marshak-Rothstein A. Fas (CD95)/Fas-L interactions required for programmed cell death after activation. Nature. 1995;373:444–447. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 18.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 19.Boise LH, Gonzalez-Garcia M, Posteme CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CG. bcl-x, a bcl-2–related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 20.Kupfer A, Singer SJ, Janeway CA, Jr, Swain SL. Coclustering of CD4 (L3T4) molecule with the T-cell receptor is induced by specific direct interaction of helper T cells and antigen-presenting cells. Proc Natl Acad Sci USA. 1987;84:5888–5892. doi: 10.1073/pnas.84.16.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Critchfield JM, Racke MK, Zúñiga-Pflücker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 22.Lenardo MJ. Introduction: the molecular regulation of lymphocyte apoptosis. Semin Immunol. 1997;9:1–5. doi: 10.1006/smim.1996.0050. [DOI] [PubMed] [Google Scholar]
- 23.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NRJ. T-cell–receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 24.Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y-H, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]