

Abstract
Although it has been reported that activated platelets can adhere to intact endothelium, the receptors involved have not been fully characterized. Also, it is not clear whether activated platelets bind primarily to matrix proteins at sites of endothelial cell denudation or directly to endothelial cells. Thus, this study was designed to further clarify the mechanisms of activated platelet adhesion to endothelium. Unstimulated human umbilical vein endothelial cell (HUVEC) monolayers were incubated with washed, stained, and thrombin-activated human platelets. To exclude matrix involvement, HUVEC were harvested mechanically and platelet binding was measured by flow cytometry. Before the adhesion assay, platelets or HUVEC were treated with different receptor antagonists. Whereas blockade of platelet β1 integrins, GPIbα, GPIV, P-selectin, and platelet-endothelial cell adhesion molecule (PECAM)-1 did not reduce platelet adhesion to HUVEC, blockade of platelet GPIIbIIIa by antibodies or Arg-Gly-Asp (RGD) peptides markedly decreased adhesion. Moreover, when platelets were treated with blocking antibodies to GPIIbIIIa-binding adhesive proteins, including fibrinogen and fibronectin, and von Willebrand factor (vWF), platelet binding was also reduced markedly. Addition of fibrinogen, fibronectin, or vWF further increased platelet adhesion, indicating that both endogenous platelet-exposed and exogenous adhesive proteins can participate in the binding process. Evaluation of the HUVEC receptors revealed predominant involvement of intercellular adhesion molecule (ICAM)-1 and αvβ3 integrin. Blockade of these two receptors by antibodies decreased platelet binding significantly. Also, there was evidence that a component of platelet adhesion was mediated by endothelial GPIbα. Blockade of β1 integrins, E-selectin, P-selectin, PECAM-1, vascular cell adhesion molecule (VCAM)-1 and different matrix proteins on HUVEC did not affect platelet adhesion. In conclusion, we show that activated platelet binding to HUVEC monolayers is mediated by a GPIIbIIIa-dependent bridging mechanism involving platelet-bound adhesive proteins and the endothelial cell receptors ICAM-1, αvβ3 integrin, and, to a lesser extent, GPIbα.
Although the pathophysiologic consequences of activated platelets in circulation are not yet fully understood, it is well established that increased platelet activation is associated with an enhanced risk of thrombotic complications in different clinical disorders, such as diabetes, preeclampsia, unstable angina, peripheral vascular disease, and stroke and after angioplastic and fibrinolytic therapy (1). Because activated, but not resting, platelets have been shown to adhere to intact endothelium, it has been suggested that platelet thrombi may also occur in the absence of endothelial cell denudation, particularly in the microvasculature (2–5). However, while the platelet receptors involved in aggregate formation and matrix adhesion have been studied extensively, the pathways responsible for the interaction of platelets and the endothelium are not well characterized.
So far, three different platelet receptors have been reported to be involved in the binding to endothelium. Rolling of activated platelets on high endothelial venules was found to depend primarily on platelet P-selectin (αIIbβ3; CD62P; 6), whereas firm adhesion to human saphenous vein endothelial cells was inhibited by anti-GPIIbIIIa (CD41a/ CD61) antibodies and RGD peptides (7). Furthermore, it has been shown that platelet-sialylated glycoproteins may, at least in part, be responsible for the increased adhesion of platelets from diabetics to bovine valvular endothelial cells (8).
Likewise, several distinct endothelial cell molecules have been reported to be involved in the binding of resting and activated platelets. Both endothelial-sialylated glycoproteins (6), as well as P-selectin on activated endothelium (9), have been proposed to mediate platelet rolling. With human umbilical vein endothelial cells (HUVEC)1 infected with herpes virus or stimulated with IL-1 or plasma containing chemotherapeutic drugs, platelet adhesion was effectively inhibited by antibodies to endothelial von Willebrand factor (vWF) and αvβ3 integrin (CD51/CD61), respectively (10–12). Moreover, a recent in vivo study has presented evidence that platelet–endothelial cell adhesion molecule-1 (PECAM-1; CD31) on endothelial cells may contribute to platelet adhesion and aggregation at a site of injured but not denuded endothelium (13).
Thus, this study was designed to further clarify the role of the different receptors that have been implicated in the adherence interaction of platelets with endothelial cells. Because both resting and activated platelets adhere primarily to matrix proteins, rather than to endothelial cells, many investigators have used fixed endothelial cells in the adhesion assay in an attempt to maintain complete confluence. However, fixation can alter the receptor function and does not exclude the involvement of matrix proteins exposed by small intercellular gaps or expressed on the endothelial cells themselves. Hence, to avoid this problem, platelet binding to HUVEC was determined in suspension using flow cytometry. Our results show that thrombin-activated platelets bind to HUVEC by a GPIIbIIIa-dependent bridging mechanism involving platelet-bound adhesive proteins, including fibrinogen, fibronectin, and vWF. Importantly, activated platelet binding did not involve endothelial cell–associated adhesive proteins such as collagen IV, fibronectin, and vWF, but instead used intercellular adhesion molecule-1 (ICAM-1; CD54) and αvβ3 integrin. In addition, we also found evidence for the involvement of endothelial GPIbα (CD42b). Thus, these endothelial adhesion molecules may contribute to the recruitment of activated platelets to intact endothelium and, consequently, to the formation of intravascular platelet aggregates, thereby promoting thrombotic processes.
Materials and Methods
Endothelial Cell Culture.
HUVEC were obtained by collagenase treatment of umbilical cord veins as previously described (14). Cells were cultured on gelatin-coated dishes and propagated in RPMI 1640 medium (BioWhittaker, Walkersville, MD) supplemented with 20% bovine calf serum (Hyclone Laboratories Inc., Logan, UT), 90 μg/ml heparin (Sigma Chemical Co., St. Louis, MO), and 50 μg/ml endothelial cell growth factor prepared from bovine hypothalamus (15). For flow cytometry assays, HUVEC derived from passages two or three were allowed to grow to confluence in 12-well dishes. For assays with HUVEC matrix, cells were cultivated in 48-well plates and harvested with trypsin after at least 4 d. Before the adhesion assay, HUVEC were washed twice with RPMI medium and incubated with receptor antagonists in phenol red–free RPMI medium for 30 min at 37°C.
Platelet Preparation.
Blood was obtained by venipuncture from healthy adult volunteers according to a protocol approved by the Human Subjects Division of the University of Washington (Seattle, WA). The volunteers did not take any drugs for the previous 10 d. Isolation of platelets was performed as described by Baenziger and Majerus (16). In brief, blood was drawn into polypropylene syringes containing one-tenth volume of 0.11 M sodium citrate and centrifuged at 1,000 g for 4 min to obtain platelet-rich plasma. Platelets were sedimented by centrifugation at 2,000 g for 10 min and washed twice with 10 ml of Hepes buffer (10 mM Hepes, 0.5 mM MgCl2, 130 mM NaCl, 4 mM KCl, 1 mM CaCl2, and 5 mM glucose, pH 7.4). To pellet erythrocytes selectively, platelets were resuspended in the same buffer and centrifuged twice at 120 g for 3 min. Subsequently, the content of erythrocytes was <1% as calculated using a hemocytometer. Platelets were stained with 2.5 μM calcein–acetoxymethyl ester (Molecular Probes Inc., Eugene, OR) in the dark for 15 min. To avoid platelet activation, all centrifugations were done at room temperature and in the presence of 1 μM prostaglandin E1 (Alprostadil, Prostin VR Pediatric®; The Upjohn Co., Kalamazoo, MI). After washing, platelets were adjusted to a final concentration of 2 × 109/ml and activated with 0.5 U/ml human thrombin (Sigma Chemical Co.) for 10 min. Thrombin was inactivated with 2 U/ml hirudin (Sigma Chemical Co.) for 10 min. Platelets were incubated with receptor antagonists for 30 min at room temperature.
Antibodies.
To block β1 integrins (CD29), the mAb P4C10 (IgG1; GIBCO BRL, Gaithersburg, MD) was used. This mAb blocks cell–cell as well as cell–matrix adhesion (17). The anti–human αvβ3 integrin mAb LM609 (IgG1; Chemicon International, Inc., Temecula, CA), blocks the RGD-binding site, whereas the anti– human αvβ3 integrin mAb LM142 (IgG1; Chemicon International Inc.) is a nonblocking mAb (18). Platelet GPIIbIIIa was blocked by the mAb P2 (IgG1; Immunotech Inc., Westbrook, ME) that inhibits platelet binding to fibrinogen and thrombin-induced platelet aggregation (19). Both anti–human GPIbα mAbs, SZ2 (IgG1; Immunotech Inc.) and 2E4 (provided by Dr. G.J. Roth, Veterans Affairs Medical Center, Seattle, WA) are blocking mAbs that inhibit ristocetin-dependent binding of vWF to platelets and ristocetin-induced platelet aggregation (reference 20 and our unpublished observation). The anti–human GPIX (CD42a) mAb BL-H6 (IgG1; Sigma Chemical Co.) was used as a negative control for the GPIb–IX complex on platelets. Platelet GPIV (CD36) was inhibited by the blocking mAb FA6.152 (IgG1; Immunotech Inc.). ICAM-1 was tested by four different blocking mAbs. mAb R6.5 (provided by Dr. R. Rothlein, Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT) inhibits adhesion of neutrophils to ICAM-1 by blocking the β2 integrin–binding site (CD18; reference 21). The mAbs 6E6 and 2D5 (provided by Dr. D.C. Altieri, The Boyer Center for Molecular Medicine, Yale University School of Medicine, New Haven, CT) block the β2 integrin- and the fibrinogen-binding site of ICAM-1, respectively (22), whereas the mAb VF27 (IgG1; Endogen Inc., Woburn, MA) inhibits adhesion of rhinoviruses to endothelial cells by blocking the first Ig domain of ICAM-1 (23). P-selectin was tested by mAb WAPS12.2 (IgG1; Endogen Inc.) and mAb G1 (IgG1; Biosource Intl., Camarillo, CA) that block platelet rolling on endothelium and platelet adhesion to neutrophils, respectively (6, 24). To block E-selectin (CD62E), HUVEC were treated with the mAbs 1.2B6 (IgG1; Endogen) and H18-7 (IgG2; Becton Dickinson, San Jose, CA) that both block leukocyte adhesion to stimulated endothelial cells (25, 26). Vascular cell adhesion molecule-1 (VCAM-1; CD106) was blocked by the mAbs E1-6 (IgG1; Becton Dickinson) and 1G11 (IgG1; Immunotech Inc.). Both mAbs inhibit leukocyte binding to stimulated endothelial cells (26, 27). PECAM-1 was tested by the mAbs JC70A (IgG1; Accurate Chemicals Co., Westbury, NY) and M89D3 (IgG1; provided by Dr. M.R. Zocchi, Laboratory of Adoptive Immunotherapy, Milan, Italy). To block fibrinogen binding to GPIIbIIIa, platelets were treated with the anti–human fibrinogen mAb D1G10VL2 (IgG1; Immunotech Inc.) that is directed to the GPIIbIIIa-binding D fragment. The anti–human fibronectin mAb type 2 (Calbiochem-Novabiochem Corp., La Jolla, CA) reacts with the cell attachment site of fibronectin and inhibits ADP-induced platelet aggregation (28). All of these mAbs were used at a final concentration of 40 μg/ml. The anti–human collagen IV mAb COL-94 (IgG1) and the anti–human vWF rabbit polyclonal antibody (IgG fraction; both Sigma Chemical Co.) were used at a final dilution of 1:250. A nonspecific isotype control was performed with a mouse IgG1κ mAb (Sigma Chemical Co.).
Other Receptor Antagonists.
Gly-Arg-Gly-Asp-Ser (GRGDS) and Gly-Arg-Gly-Glu-Ser (GRGES) peptides (Peninsula Laboratories, Belmont, CA) and recombinant annexin V (provided by Dr. J.F. Tait, University of Washington, Seattle, WA) were used at final concentrations of 50 μg/ml and 5 μM, respectively. Treatment with neuraminidase from Vibrio cholerae (Calbiochem-Novabiochem Corp.) was performed at a final concentration of 0.5 U/ ml for 4 h (platelets 60 min). For divalent cation depletion assays, cells were incubated with 1 mM EDTA (Sigma Chemical Co.) for 30 min. Adhesive proteins were added to HUVEC immediately before addition of platelets. Purified human vWF (American Diagnostica Inc., Greenwich, CT), fibrinogen (Sigma Chemical Co.), fibronectin (Calbiochem-Novabiochem Corp.), and bovine albumin (Sigma Chemical Co.) were used at concentrations as indicated in the figure legends. Ristocetin (Sigma Chemical Co.) was added together with vWF at concentrations as indicated in the figure legends.
Determination of HUVEC-associated Adhesive Proteins.
Washed , suspended HUVEC were incubated with antibodies to either collagen IV, fibronectin, or vWF (same antibodies as described above) for 30 min. After washing twice, the secondary FITC-conjugated antibody, either goat anti–mouse IgG (Caltag Lab., San Francisco, CA) or mouse anti–rabbit IgG (Sigma Chemical Co.), was incubated for 30 min (final dilution of 1:200). At least 10,000 cells were then analyzed by a FACScan® (Becton Dickinson). Cells treated with FITC-conjugated isotype-specific IgG were used as negative controls.
Adherence Assay (Flow Cytometry).
HUVEC were washed twice with RPMI medium and incubated with 600 μl of Hepes buffer (same buffer as described above, but with 2 mM CaCl2 added) and 40 μl of the suspension of activated and stained platelets for 30 min at 37°C. The final platelet concentration was 1.25 × 108/ ml. After removing unbound platelets followed by two washes, HUVEC were harvested mechanically by vigorous pipetting, washed once, and then immediately fixed with 80% ethanol on ice for 30 min. To differentiate HUVEC from residual unbound platelets, HUVEC were stained with the DNA-binding dye propidium iodide. The cells were resuspended in 200 μl PBS buffer, pH 7.4, and 0.1% Triton X-100 containing 5 μg/ml propidium iodide and 50 μg/ml ribonuclease A (R-6513; all Sigma Chemical Co.). Subsequently, specimens were analyzed by a FACScan®. At least 10,000 cells that stained positive for propidium iodide (FL-2-H) were evaluated. Platelet adhesion to HUVEC was expressed by the median fluorescence (FL-1-H) of the entire HUVEC population. Based on the distribution of the DNA content (FL-2-H), HUVEC were routinely tested for homotypic aggregate formation. On average, ∼5% of the cells exhibited more than tretraploid DNA content indicating that they were clumped. Statistical significance was determined using Student's t test.
Adherence Assay (Fluorescence Plate Reader).
Assays of platelet adhesion to HUVEC matrix were performed in 48-well dishes. Platelets were isolated, stained, and activated as described above. After harvesting the HUVEC with EDTA, 2.5 × 107 platelets per well were allowed to adhere to the matrix for 30 min at 37°C. Adherence was then assessed in a Cytofluor Series 4000 fluorescence plate reader (PerSeptive Biosystems, Framingham, MA). Plates were scanned before and after washing for total and adherent cells, respectively. Adherence was expressed as a percentage of the total fluorescence.
Transmission Electron Microscopy.
Adhesion assays were performed as described above. HUVEC were then harvested, washed, and fixed with 3% glutaraldehyde in PBS buffer. After postfixation with 2% osmium tetroxide for 60 min, cells were washed and dehydrated with increasing concentrations of ethanol and finally propylene oxide. Fragments of the cell pellet were infiltrated with 100% Medcast in vacuum for 4 h and subsequently embedded in 100% fresh (Ted Pella, Reading, CA) Medcast. After polymerization for 48 h at 60°C, specimens were visualized by a JEOL 1200-EX11 (JEOL USA Inc., Peabody, MA).
Results
Time Course of Platelet Binding.
Over a time period of 30 min, the number of HUVEC with bound resting platelets was quite low (Fig. 1). However, with thrombin-activated platelets HUVEC exhibited a significant increase in fluorescence after only 5 min. Maximal binding of activated platelets was found after 20–30 min. As determined by an arbitrary gate, ∼10 and 65% of HUVEC bound resting and activated platelets, respectively. Using flow cytometry, it was not possible to determine whether the increased fluorescence of HUVEC was due to multiple single platelets bound to HUVEC or to a few large platelet aggregates attached to HUVEC. However, large platelet aggregate formation on HUVEC was found to be a rare event (see below), suggesting that the adhesive interaction was platelet– HUVEC rather than platelet–platelet.
Figure 1.
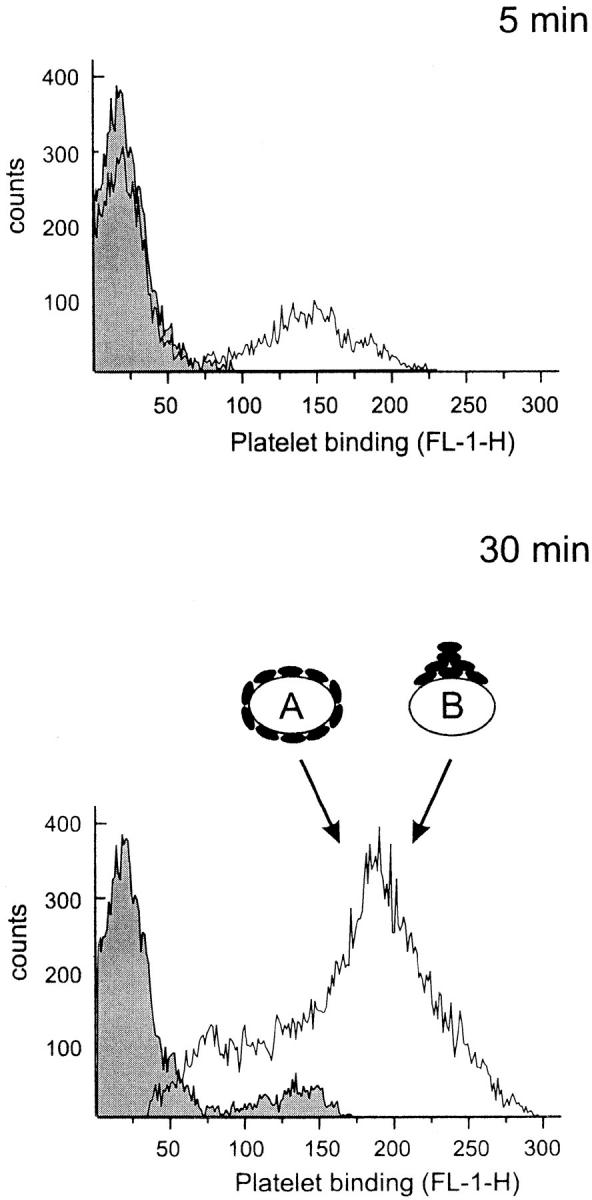
Time course of platelet binding to HUVEC monolayers. Washed and calcein-loaded resting or thrombin-activated platelets were allowed to adhere to a HUVEC monolayer for 5 or 30 min. HUVEC were then harvested and assayed by flow cytometry to determine platelet binding that is expressed as Platelet binding (FL-1-H) of the entire HUVEC population. The shaded and open curves represent HUVEC incubated with resting and activated platelets, respectively. When estimated by an arbitrary gate, HUVEC bound resting and activated platelets by ∼10 and 65% after 30 min, respectively. As depicted by the symbols A and B, the increased fluorescence of HUVEC incubated with activated platelets may be due to either numerous single platelets (symbol A) or only one large aggregate (symbol B). Using flow cytometry, it is not possible to distinguish between these two mechanisms. A representative experiment of three performed is shown.
HUVEC Become Coated Mainly by Single Platelets.
The striking difference in the fluorescence intensity between HUVEC incubated with either resting or activated platelets was also evident by transmission electron microscopy (TEM). As shown in Fig. 2 A, resting platelets bound minimally to HUVEC. However, activated platelets bound avidly to HUVEC with single or clumped platelets coating the endothelial cells (Fig. 2 B). Occasionally, large platelet aggregates adherent to HUVEC could also be found (Fig. 2 C). However, on average, we observed only one large aggregate in every 10–12 samples.
Figure 2.
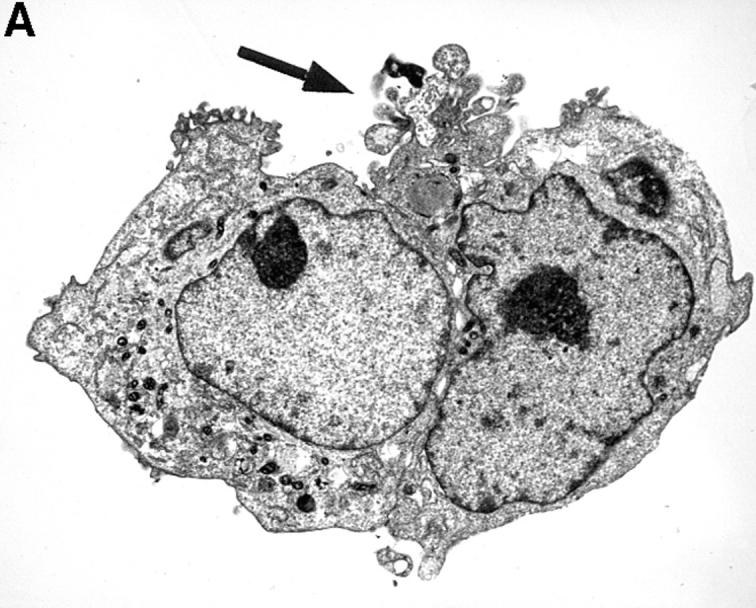

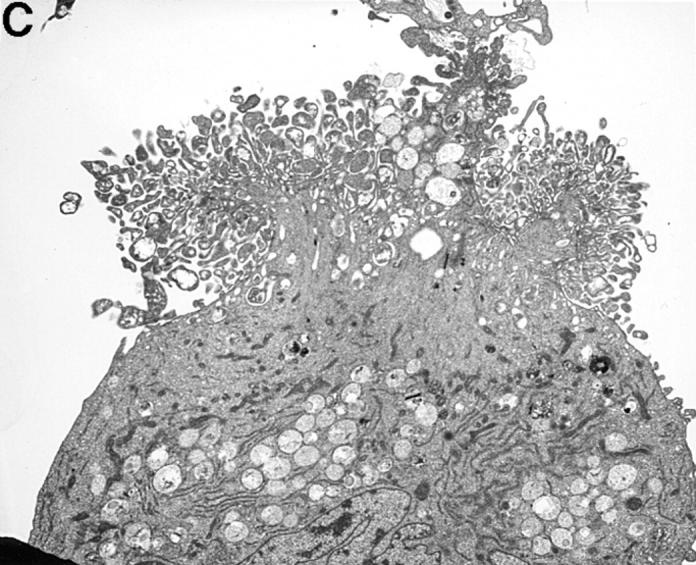
TEM of HUVEC monolayers incubated with platelets. A illustrates two adherent HUVEC that were incubated with resting platelets. On the entire circumference of the cell surfaces, there is only one area (black arrow) exhibiting adherent platelets (A). However, when incubated with activated platelets HUVEC were covered completely with single platelets (black arrow, B). Occasionally, large platelet aggregates were found (C). The magnification is 1:5,000–6,000.
Activated Platelets Bind to Different HUVEC Adhesion Molecules.
Treatment of HUVEC with annexin V, neuraminidase, and the blocking anti-β1 mAb P4C10 did not affect platelet adhesion (Fig. 3 A). When HUVEC were treated with the anti–PECAM-1 mAb M89D3, platelet adhesion decreased by ∼10%. However, this decrease was not significant. Although HUVEC were not stimulated by any exogenous agents before the assay, we also tested stimulation-dependent HUVEC receptors, assuming that platelets might be able to activate HUVEC. Blocking mAbs to VCAM-1, E-selectin, or P-selectin did not inhibit platelet binding. However, a significant decrease in platelet binding was observed when HUVEC were treated with mAbs to ICAM-1, αvβ3 integrin, or GPIbα. All four mAbs to ICAM-1 exhibited a blocking effect. HUVEC treated with anti–ICAM-1 mAb R6.5 or 6E6, which both block the β2 integrin–binding site of ICAM-1, decreased platelet binding by ∼26%. Similarly, the mAb VF27, which has been found to block adhesion of rhinoviruses to endothelial ICAM-1, also reduced platelet binding by ∼32%. However, the greatest inhibition (50%) was found when HUVEC were treated with the mAb 2D5, which blocks the fibrinogen-binding site of ICAM-1 (22). Involvement of αvβ3 integrin was evident when platelet adhesion decreased by ∼38% after treating HUVEC with the anti-αvβ3 integrin mAb LM609, whereas the nonblocking anti-αvβ3 integrin mAb LM142 had no effect. Interestingly, the anti-GPIbα mAb SZ2 also exhibited an inhibitory effect, reducing platelet binding significantly by ∼22%. A second blocking anti-GPIbα mAb 2E4 inhibited platelet adhesion by ∼12%, which, however, was not statistically significant. Incubation with RGD peptides and EDTA decreased platelet binding by 60 and 71%, respectively. A combined treatment with mAbs to ICAM-1 (2D5), GPIbα (SZ2), and αvβ3 integrin (LM609) decreased platelet adhesion by ∼59%, but still did not reduce binding to the level of resting platelets.
Figure 3.

Platelet adhesion to HUVEC monolayers after blockade of HUVEC receptors. Thrombin-activated platelets were allowed to adhere to a HUVEC monolayer that was treated with either antagonists of different endothelial cell adhesion molecules (A) or antibodies to adhesive proteins (B). Platelet binding was then determined by flow cytometry as described above. The following concentrations were used: 5 μM annexin V, 0.5 U/ml neuraminidase, 50 μg/ml GRGES and GRGDS peptides, 1 mM EDTA, 40 μg/ml mAb, and a dilution of 1:250 for polyclonal antibodies. Results are expressed as the mean ± SD of the median fluorescence (FL-1-H) of at least four experiments. *P <0.01 and **P <0.05 by Student's t test, compared with activated platelet adhesion to HUVEC treated with the isotype-specific control mAb.
Activated Platelets Do Not Bind to HUVEC-associated Matrix Proteins.
Platelets are well known to bind rapidly to the adhesive proteins that are exposed by an injured vessel wall. Thus, HUVEC were tested for the exposure of matrix proteins known to be important for platelet adhesion, such as collagen IV, fibronectin, and vWF. When analyzed by flow cytometry, all three of these proteins were found to be exposed on detached HUVEC with vWF staining greater than fibronectin and collagen IV. Incubation of HUVEC monolayers with the same antibodies to collagen IV, fibronectin, and vWF did not affect platelet adhesion (Fig. 3 B). However, each of the three antibodies was found to inhibit platelet adhesion to a matrix of the corresponding purified protein such as collagen IV, vWF, and fibronectin (data not shown). This suggests that the HUVEC monolayers do not express these adhesive proteins on the luminal surface to a degree sufficient to contribute to platelet binding. Indeed, when platelets were allowed to adhere to HUVEC in suspension, treatment of HUVEC with the antifibronectin and anti-vWF mAb markedly decreased platelet binding (data not shown). Thus, matrix proteins relevant for platelet binding, such as fibronectin and vWF, seem to be expressed primarily on the abluminal side of endothelial cells.
Activated Platelets Bind to HUVEC Matrix via β1 Integrins.
To clarify further the role of matrix proteins in the adhesion of activated platelets to HUVEC, we tested whether the platelet receptors involved in the binding to HUVEC were the same as those involved in the binding to the HUVEC matrix. Measured by a fluorescence plate reader, activated platelets bound exclusively by β1 integrins to the HUVEC matrix (Fig. 4). Blockade of other receptors including GPIbα, GPIV, and GPIIbIIIa did not significantly affect platelet adhesion. Similarly, platelet β1 integrins were the primary receptors mediating the binding of activated platelets to HUVEC in suspension (data not shown). However, platelet adhesion to adherent HUVEC monolayers did not involve platelet β1 integrins (see below), which further indicates that exposed matrix proteins on the HUVEC monolayer did not serve as ligands for activated platelet receptors.
Figure 4.

Platelet adhesion to HUVEC matrix after blockade of platelet receptors. Thrombin-activated platelets treated with antagonists to different platelet receptors were allowed to adhere to HUVEC matrix for 30 min. Platelet binding was then determined by a fluorescence plate reader as described above. The following concentrations were used: 40 μg/ml for mAb and a dilution of 1:250 for polyclonal antibodies. Results are expressed as the mean ± SD of the percentage of adherent platelets of at least three experiments. *P <0.01 by Student's t test, compared with adhesion of activated platelets treated with the isotype-specific control mAb.
Activated Platelets Bind to HUVEC by GPIIbIIIa.
As shown in Fig. 5 A, thrombin-activated platelet adhesion to HUVEC was found to be mediated by the platelet receptor GPIIbIIIa. Blockade of phosphatidlyserine by annexin V and enzymatic degradation of sialylated glycoproteins by neuraminidase did not affect adhesion. As well, incubation of platelets with mAbs to β1 integrins (clone P4C10), GPIbα (clones SZ2 and 2E4), GPIX (clone BL-H6), GPIV (clone FA6.152), and P-selectin (clones G1 and WAPS12.2) did not reduce platelet binding to HUVEC. Similar to HUVEC, treatment of platelets with the anti–PECAM-1 mAb M89D3 led to a slight decrease of platelet binding, which, however, was not significant. Because in separate experiments we found that platelets did not bind to recombinant PECAM-1 (our unpublished observation), involvement of PECAM-1 in the interaction with HUVEC seemed very unlikely. On the other hand, when platelets were treated with either RGD peptides or the anti-GPIIbIIIa mAb P2, platelet binding was reduced by >50%. Furthermore, EDTA (which dissociates GPIIbIIIa) decreased platelet binding almost to the level of resting platelets. Because it was not possible to remove the mAbs from the platelet suspension after activation with thrombin (centrifugation would cause an irreversible clot), it was necessary to consider the possibility that unbound mAbs could also block HUVEC receptors. However, when using mAbs and peptides at concentrations corresponding to the concentration in the platelet suspension after dilution in the adhesion buffer, platelet binding was not affected (data not shown).
Figure 5.

Platelet adhesion to HUVEC monolayers after blockade of platelet receptors. Thrombin-activated platelets treated with either antagonists to different platelet receptors (A) or antibodies to adhesive proteins (B) were allowed to adhere to a HUVEC monolayer for 30 min. Platelet binding was determined by flow cytometry as described above. The following concentrations were used: 5 μM annexin V, 0.5 U/ml neuraminidase, 50 μg/ml GRGES and GRGDS peptides, 1 mM EDTA, 40 μg/ml mAb, and a dilution of 1:250 for polyclonal antibodies. Results are expressed as the mean ± SD of the median fluorescence (FL-1-H) of at least four experiments. *P <0.01 and P <0.05 by Student's t test, compared with adhesion of activated platelets treated with the isotype-specific control mAb.
Platelet-associated Adhesive Proteins Mediate Binding to HUVEC Monolayers.
Upon activation, platelets express different adhesive proteins, such as fibrinogen, fibronectin, and vWF (28, 29). Because all three of these proteins are known to mediate platelet aggregation, their involvement in the binding to HUVEC seemed very likely. Indeed, as shown in Fig. 5 B, blockade of vWF, fibrinogen, and fibronectin on platelets decreased adhesion to HUVEC monolayers by 56, 46, and 39%, respectively. This result indicates that, similar to platelet aggregation, platelet-exposed adhesive proteins participate in activated platelet binding to HUVEC monolayers.
Exogenous Adhesive Proteins Enhance Platelet Binding to HUVEC.
To investigate further the role of adhesive proteins in the binding mechanism, platelet adhesion was examined in the presence of purified fibrinogen, fibronectin, vWF, and albumin (Fig. 6). All proteins but albumin were found to induce a significant increase in platelet binding. The minimal concentrations required for a maximal increase were 75 μg/ml for fibrinogen, 15 μg/ml for fibronectin, and 0.25 μg/ml for vWF, which correspond to plasma concentrations of 2.5, 5, and 2.5%, respectively. Addition of vWF increased platelet adhesion only in the presence of ristocetin, but had no effect when added alone. This indicates an involvement of endothelial and/or platelet GPIbα. Participation of endothelial GPIbα seemed more likely, because blockade of GPIbα on HUVEC, but not platelets, was found to affect platelet adhesion (Figs. 4 A and 5 A). Albumin added at concentrations up to 4 mg/ml did not alter platelet binding, confirming that the effect of adhesive proteins was specific.
Figure 6.

Platelet adhesion to HUVEC monolayers after addition of adhesive proteins. Thrombin-activated platelets were allowed to adhere to HUVEC monolayers for 30 min in the presence of different adhesive proteins. Platelet binding was then determined by flow cytometry as described above. The following concentrations were used: 4 mg/ml albumin, 75 μg/ ml fibrinogen, 15 μg/ml fibronectin, and 0.25 μg/ml vWF, which corresponded to a plasma concentration of 10, 2.5, 5, and 2.5%, respectively. Ristocetin was added together with vWF at a final concentration of 1 mg/ml. Results are expressed as the mean ± SD of the median fluorescence (FL-1-H) of at least four experiments. **P <0.05 by Student's t test, compared with adhesion without proteins.
Activated Platelet Adhesion to HUVEC Requires Bridging Molecules.
Results to this point suggested that adhesive proteins may play an important role in mediating the binding of activated platelets to HUVEC monolayers. This hypothesis was further supported by experiments using zinc or manganese, rather than thrombin, to activate platelets (Fig. 7). Both cations are known to activate GPIIbIIIa without inducing secretion and aggregation (30, 31). In the presence of 0.5 mM zinc or 2 mM manganese only, platelets did not adhere to HUVEC. However, when exogenous fibrinogen (375 μg/ml) was added, both zinc- and manganese-activated platelets bound to HUVEC to the same degree as thrombin-activated platelets. Again, this binding could be partly inhibited by the anti-GPIIbIIIa mAb P2 and the antifibrinogen mAb D1G10VL2. Hence, the ability of platelets to bind to HUVEC monolayers depended not only on an activated GPIIbIIIa receptor, but also on the presence of a bridging adhesive protein such as fibrinogen.
Figure 7.

Adhesion of resting platelets to HUVEC monolayers in the presence of zinc or manganese. Resting platelets were allowed to adhere to HUVEC monolayers for 30 min in the presence of 0.5 mM zinc (open bars) or 2 mM manganese (filled bars) with or without the addition of 375 μg/ml fibrinogen. Both zinc and manganese activate platelets without inducing secretion. Platelet binding was then determined by flow cytometry as described above. Adhesion was also measured in the presence of 40 μg/ml anti-GPIIbIIIa mAb P2 and antifibrinogen mAb D1G10VL2. Results are expressed as the mean ± SD of the median fluorescence (FL-1-H) of at least three experiments. *P <0.01 by Student's t test, compared with adhesion with fibrinogen.
vWF and Ristocetin Enhance Platelet Binding to HUVEC.
Fig. 8 further demonstrates a GPIbα-dependent component of platelet adhesion to HUVEC. In the presence of 1 μg/ml vWF, ristocetin dose dependently increased binding of resting and activated platelets. However, when HUVEC were treated with the anti-GPIbα mAb SZ2, binding of resting and activated platelets decreased by ∼30 and 23%, respectively. Treatment of resting platelets with the mAb SZ2 reduced binding to HUVEC by ∼80%, whereas treatment of activated platelets did not affect adhesion significantly. This result indicates that the vWF/ristocetin-induced increase in the binding of resting platelets to HUVEC depended on both platelet and endothelial GPIbα, whereas increased binding of activated platelets required only endothelial GPIbα.
Figure 8.

Platelet adhesion to HUVEC monolayers after addition of vWF and ristocetin. Resting (filled bars) and thrombin-activated platelets (open bars) were allowed to adhere to HUVEC monolayers for 30 min in the presence of 1 μg/ml vWF and increasing concentrations of ristocetin. Platelet binding was then determined by flow cytometry as described above. To test involvement of endothelial and platelet GPIbα, HUVEC and platelets were pretreated separately with 40 μg/ml anti–human GPIbα mAb, clone SZ2. Results are expressed as the mean ± SD of the median fluorescence (FL-1-H) of at least four experiments. *P <0.01 and **P <0.05 by Student's t test, compared with the corresponding controls without vWF and ristocetin.
Discussion
In this report we show that adherence of thrombin-activated platelets to HUVEC monolayers is mediated by platelet GPIIbIIIa and distinct endothelial cell adhesive molecules (Fig. 9). As has previously been demonstrated (32), flow cytometry is a very sensitive and accurate method to measure heterotypic adhesion of platelets and its use allowed us to exclude the involvement of matrix proteins in the adhesive interaction.
Figure 9.

Proposed model of adhesion of activated platelets to HUVEC. Upon activation, platelet GPIIbIIIa receptors bind different adhesive proteins, such as fibrinogen, vWF, and fibronectin. These proteins are either present in plasma or secreted by the platelets themselves. Dependent upon the bound adhesive protein, adhesion of platelets to endothelial cells is mediated by different endothelial counter receptors. Whereas fibronectin binds only to αvβ3-integrin, fibrinogen binds to ICAM-1 or αvβ3-integrin and vWF binds primarily to αvβ3-integrin or possibly also to GPIbα.
Besides other mechanisms, activated platelet adhesion to leukocytes and tumor cells has been found, similar to platelet aggregation, to involve fibrinogen bound to GPIIbIIIa (33–35). Thus, it seemed very likely that the same mechanism mediated platelet binding to endothelial cells. Indeed, blockade of GPIIbIIIa by the mAb P2 or RGD peptides decreased platelet binding to HUVEC monolayers by ∼50%, which is comparable to a recent report (7). Although both RGD peptides and the mAb P2 have been shown to block completely platelet aggregation (19), platelet binding to HUVEC was not inhibited 100% by these antagonists. Because platelets were activated before treatment with antagonists, adhesion of small preformed platelet aggregates to HUVEC had to be considered. However, when platelets were treated with the mAb P2 before thrombin activation to prevent aggregate formation, platelet binding to HUVEC did not further decrease (data not shown). Thus, activated platelets that are unable to aggregate may still adhere to HUVEC, perhaps by an additional binding mechanism. However, blockade of β1 integrins, GPIbα, GPIX, GPIV, P-selectin, or PECAM-1 did not reveal an inhibitory effect.
We found that adhesive proteins play an important role in activated platelet adhesion to HUVEC. Blockade of platelet-exposed fibrinogen resulted in a significant decrease of bound platelets. Moreover, addition of fibrinogen further increased platelet adhesion, indicating that platelets may be able to use both endogenous and exogenous fibrinogen to bind to endothelial cells. Importantly, blockade of fibronectin and vWF, which are both also exposed on GPIIbIIIa on activated platelets (29), decreased adhesion as well. This is in accordance with previous studies showing that platelet-released fibronectin and vWF are capable of cross-linking platelets or platelets and tumor cells (35, 36). It is conceivable that thrombospondin (TSP)-1, another platelet-released adhesive protein, may also participate in the binding to HUVEC. Although blockade of the major TSP-1 receptor on platelets, GPIV (CD36), did not affect platelet adhesion to HUVEC, treatment of platelets with the anti–TSP-1 mAb 5G11 revealed a minimal inhibitory effect (data not shown). This indicates that TSP-1 may be bound by a different receptor, most likely GPIIbIIIa. However, because this inhibition was very modest and this mAb does not block platelet aggregation (37), involvement of TSP-1 remains uncertain.
The significant role of adhesive proteins in platelet adhesion to HUVEC monolayers is further demonstrated by the fact that binding was not observed when platelets were activated without secretion by using zinc or manganese. Although these cations activate the GPIIbIIIa receptor, only the addition of exogenous fibrinogen allowed platelets to bind to HUVEC or to form aggregates (30, 31), indicating that platelet–platelet and platelet–HUVEC interactions are mediated by the same bridging mechanism. Thus, it is possible that the reduced fluorescence of HUVEC after treatment of platelets with anti-GPIIbIIIa antagonists may be due to the reduction of platelet aggregates rather than of platelet-HUVEC interactions. However, extensive platelet aggregate formation was not observed on TEM, suggesting that the inhibition was primarily due to blockade of platelet adhesion to HUVEC.
Interestingly, evaluation of the HUVEC receptors involved in activated platelet adhesion revealed participation of three different receptors. Treatment of HUVEC with RGD peptides resulted in the greatest inhibition of platelet adhesion, suggesting that RGD-dependent adhesive protein receptors were predominantly involved. However, blockade of β1 integrins on HUVEC did not affect platelet adhesion. This is in accordance with a previous study showing that α5β1 integrins serve as RGD-dependent fibrinogen receptors only when HUVEC are suspended, but not attached (38). Under our experimental conditions, αvβ3 integrin was the only RGD-binding receptor found to be involved in platelet binding, although its blockade by the mAb LM609 did not reduce platelet adhesion to the level observed with RGD peptides. Similarly, two previous reports have shown that platelet adhesion to activated endothelial cells could be partly inhibited by the same anti-αvβ3 mAb (11, 12). Because αvβ3 integrin binds different adhesive proteins (39), it is likely that platelets bind to endothelial αvβ3 integrin by forming bridges with fibrinogen, fibronectin, and vWF.
Besides the binding site for β2 integrins, endothelial ICAM-1 has recently been shown to contain an epitope for the binding of fibrinogen that mediates the binding of leukocytes (22, 40). We now present evidence that a component of platelet binding to HUVEC uses this mechanism. Treatment of HUVEC with the mAb 2D5, which blocks the fibrinogen-binding site of ICAM-1, decreased binding of activated platelets by ∼50%. The fact that the other anti–ICAM-1 mAbs, that recognize either the epitope for β2 integrins (R6.5 and 6E6) or rhinoviruses (VF27) also decreased platelet binding suggests some cross-reactivity with the fibrinogen-binding region. This region has recently been localized within the first Ig domain of ICAM-1 (41), which is also involved in the αLβ2-dependent binding of leukocytes (42) and rhinoviruses (23).
Besides αvβ3 integrin and ICAM-1, we additionally found evidence for involvement of endothelial GPIbα in the binding of activated platelets to HUVEC monolayers. Platelet adhesion could be blocked significantly by treating HUVEC with the anti-GPIbα mAb SZ2. Furthermore, addition of exogenous vWF increased platelet binding only in the presence of ristocetin, which is known to allow soluble vWF to interact with GPIbα. Also, this vWF/ristocetin-dependent binding of activated platelets to HUVEC could be partially inhibited with the mAb SZ2. However, the mAb 2E4, another blocking anti-GPIbα mAb, did not exhibit a significant inhibitory effect, raising some question as to the significance of endothelial GPIbα in platelet binding. Indeed, it has been debated whether HUVEC express GPIbα. Whereas one study has recently questioned the synthesis of GPIbα in HUVEC (43), other investigators have found that GPIbα on HUVEC participated in different functions, including binding to vWF, binding of sickle erythrocytes, and homotypic binding of HUVEC in the presence of vWF and ristocetin (44–47). We found that unstimulated HUVEC expressed GPIbα at a modest level when measured by flow cytometry with the mAb SZ2 and 2E4 (data not shown). Thus, based on our data, we conclude that platelet-exposed vWF may bind not only to αvβ3 integrin but, in part, also to GPIbα. The potential relevance of this observation is enhanced by a recent study showing that human endothelial cells also express GPIbα in vivo (48).
Although activated platelet adhesion to endothelial cells was demonstrated some 20 years ago (2), the involvement of either surface-bound or exposed intercellular matrix proteins has not been conclusively evaluated. We believe that our studies measuring platelet binding to HUVEC monolayers by flow cytometry convincingly demonstrate that activated platelets can bind directly to endothelial cells rather than to exposed intercellular matrix proteins. Moreover, we present evidence that endothelial cell surface–associated matrix proteins are likewise not involved in the binding of activated platelets. Although suspended HUVEC were found to express vWF, collagen IV, and fibronectin, platelet adhesion to HUVEC monolayers was not inhibited by blocking mAbs to these proteins. HUVEC-associated vWF and fibronectin participated in platelet binding only when HUVEC were detached and suspended before the addition of platelets. Furthermore, we found that activated platelet adhesion to HUVEC matrix (or to HUVEC in suspension) was mediated by platelet β1 integrins, whereas binding to confluent HUVEC monolayers was dependent upon GPIIbIIIa. This participation of different platelet receptors in binding to matrix versus cells further confirms that HUVEC-associated matrix proteins were not significantly involved in activated platelet binding to HUVEC monolayers.
It is notable that platelet adhesion was not affected by blockade of platelet P-selectin, a major pathway for platelet binding to leukocytes and tumor cells (49, 50). Both anti– P-selectin mAbs G1 and WAPS12.2 have previously been shown to block platelet adhesion and rolling, respectively (6, 24). Similarly, neuraminidase-induced degradation of endothelial sialylated glycoproteins that may be potential endothelial ligands for platelet P-selectin also did not alter platelet binding. Although in our assays HUVEC were not stimulated by any exogenous agonists, platelet-induced stimulation of HUVEC could not be excluded. Thus, we additionally tested the contribution of activation-dependent adhesion molecules, including P-selectin, E-selectin, and VCAM-1. However, none of these proteins participated in the binding mechanism. This may be due, in part, to the brief incubation with platelets (30 min), which may have been too short to induce significant upregulation of E-selectin and VCAM-1. Finally, PECAM-1 has recently been proposed to participate in platelet binding to endothelium (13). However, treatment of platelets and HUVEC with two different anti–PECAM-1 mAbs produced only minimal inhibition that was not significant.
As summarized in Fig. 9, our data provide evidence that, similar to platelet aggregation, activated platelet adhesion to HUVEC monolayers is mediated by a GPIIbIIIa-dependent bridging mechanism, involving platelet-bound fibrinogen, fibronectin, and vWF. Consequently, administration of GPIIbIIIa-blocking drugs may inhibit not only the formation of platelet aggregates, but also their adhesion to the intact endothelium. This effect might be important, particularly in the microvasculature, as it has recently been shown in an in vivo thrombosis model using the anti-GPIIbIIIa mAb 7E3 (51). Furthermore, the involvement of the endothelial cell receptors ICAM-1, αvβ3 integrin, and GPIbα in the binding of activated platelets suggests that blockade of these receptors may have antithrombotic effects. Finally, because these endothelial adhesion molecules are known to become upregulated by various exogenous stimuli, endothelial cell activation may further increase binding of activated platelets and thus contribute to a prothrombotic state.
Acknowledgments
This work was supported by the United States Public Health Service Grants HL 18645 and 30541. T. Bombeli was supported by the Swiss Foundation for Medical and Biological Grants and the Anniversary Foundation of Swiss Life for Public Health and Medicinal Research, Switzerland.
Footnotes
1 Abbreviations used in this paper: HUVEC, human umbilical vein endothelial cells; ICAM, intercellular adhesion molecule; PECAM, platelet endothelial cell adhesion molecule; RGD, Arg-Gly-Asp; TEM, transmission electron microscopy; TSP, thrombospondin; VCAM, vascular cell adhesion molecule; vWF, von Willebrand factor.
The authors gratefully acknowledge Dr. R. Rothlein, Dr. D.C. Altieri, Dr. M.R. Zocchi, and Dr. G.J. Roth for providing antibodies. We also thank Dr. J.F. Tait for the generous gift of recombinant annexin V and Dr. S. Lana for performing electron microscopy.
References
- 1.Abrams C, Shattil SJ. Immunological detection of activated platelets in clinical disorders. Thromb Haemostasis. 1991;65:467–473. [PubMed] [Google Scholar]
- 2.Czervionke RL, Hoak JC, Fry GL. Effect of aspirin on thrombin-induced adherence of platelets to cultured cells from the blood vessel wall. J Clin Invest. 1978;62:847–856. doi: 10.1172/JCI109197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fry GL, Czervionke RL, Hoak JC, Smith JB, Haycraft DL. Platelet adherence to cultured vascular cells: influence of prostacyclin (PGI2) Blood. 1980;55:271–275. [PubMed] [Google Scholar]
- 4.Kaplan JE, Moon DG, Weston LK, Minnear FL, Del Vecchio PJ, Shepard JM, Fenton JW. Platelets adhere to thrombin-treated endothelial cells in vitro. Am J Physiol. 1989;257:H423–H433. doi: 10.1152/ajpheart.1989.257.2.H423. [DOI] [PubMed] [Google Scholar]
- 5.Tloti MA, Moon DG, Weston LK, Kaplan JE. Effect of 13-hydroxy octadeca-9,11-dienoic acid (13-HODE) on thrombin induced platelet adherence to endothelial cells in vitro. Thromb Res. 1991;62:305–317. doi: 10.1016/0049-3848(91)90151-l. [DOI] [PubMed] [Google Scholar]
- 6.Diacovo TG, Puri KD, Warnock RA, Springer TA, von Andrian UH. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 1996;273:252–255. doi: 10.1126/science.273.5272.252. [DOI] [PubMed] [Google Scholar]
- 7.Li JM, Podolsky RS, Rohrer MJ, Cutler BS, Massie MT, Barnard MR, Michelson AD. Adhesion of activated platelets to venous endothelial cells is mediated via GPIIb/IIIa. J Surg Res. 1996;61:543–548. doi: 10.1006/jsre.1996.0161. [DOI] [PubMed] [Google Scholar]
- 8.Manduteanu I, Calb M, Lupu C, Simionescu N, Simionescu M. Increased adhesion of human diabetic platelets to cultured valvular endothelial cells. J Submicrosc Cytol Pathol. 1992;24:539–547. [PubMed] [Google Scholar]
- 9.Frenette PS, Johnson RC, Hynes RO, Wagner DD. Platelets roll on stimulated endothelium in vivo: an interaction mediated by endothelial P-selectin. Proc Natl Acad Sci USA. 1995;92:7450–7454. doi: 10.1073/pnas.92.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etingin OR, Silverstein RL, Hajjar DP. Von Willebrand factor mediates platelet adhesion to virally infected endothelial cells. Proc Natl Acad Sci USA. 1993;90:5153–5156. doi: 10.1073/pnas.90.11.5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchanan MR, Bertomeu MC, Haas TA, Orr FW, Eltringham-Smith LL. Localization of 13-hydroxyoctadecadienoic acid and the vitronectin receptor in human endothelial cells and endothelial cell/platelet interactions in vitro. Blood. 1993;81:3303–3312. [PubMed] [Google Scholar]
- 12.Bertomeu MC, Gallo S, Lauri D, Levine MN, Orr FW, Buchanan MR. Chemotherapy enhances endothelial cell reactivity to platelets. Clin Expl Metastasis. 1990;8:511–518. doi: 10.1007/BF00135874. [DOI] [PubMed] [Google Scholar]
- 13.Rosenblum WI, Nelson GH, Wormley B, Werner P, Wang J, Shih CCY. Role of platelet-endothelial cell adhesion molecule (PECAM) in platelet adhesion/aggregation over injured but not denuded endothelium in vivo and ex vivo. Stroke. 1996;27:709–711. doi: 10.1161/01.str.27.4.709. [DOI] [PubMed] [Google Scholar]
- 14.Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maciag T, Cerundolo J, Ilsley S, Kelley PR, Forand R. An endothelial cell growth factor from bovine hypothalamus: identification and partial characterization. Proc Natl Acad Sci USA. 1979;76:5674–5678. doi: 10.1073/pnas.76.11.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baenziger NL, Majerus PW. Isolation of human platelets and platelet surface membranes. Methods Enzymol. 1974;31:149–155. doi: 10.1016/0076-6879(74)31015-4. [DOI] [PubMed] [Google Scholar]
- 17.Carter WC, Wayner EA, Bouchard TS, Kaur P. The role of integrins α2β1 and α3β1in cell-cell and cell-substrate adhesion of human epidermal cells. J Cell Biol. 1990;110:1387–1404. doi: 10.1083/jcb.110.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheresh DA. Human endothelial cells synthesize and express an Arg-Gly-Asp-directed adhesion receptor involved in attachment to fibrinogen and von Willebrand factor. Proc Natl Acad Sci USA. 1987;84:6471–6475. doi: 10.1073/pnas.84.18.6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGregor JL, Brochier J, Wild F, Follea G, Trzeciak MC, James E, Dechavanne M, McGregor L, Clemetson K. Monoclonal antibodies against platelet membrane glycoproteins. Eur J Biochem. 1983;131:427–436. doi: 10.1111/j.1432-1033.1983.tb07281.x. [DOI] [PubMed] [Google Scholar]
- 20.Ruan C, Du X, Xi X, Castaldi PA, Berndt MC. A murine antiglycoprotein Ib complex monoclonal antibody SZ 2 inhibits platelet aggregation induced by both ristocetin and collagen. Blood. 1987;74:570–577. [PubMed] [Google Scholar]
- 21.Smith CW, Rothlein R, Hughes BJ, Mariscalco MM, Rudloff HE, Schmalstieg FC, Anderson DC. Recognition of an endothelial determinant for CD18-dependent human neutrophil adherence and transendothelial migration. J Clin Invest. 1988;82:1746–1756. doi: 10.1172/JCI113788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Languino LR, Duperray A, Joganic KJ, Fornaro M, Thornton GB, Altieri DC. Regulation of leukocyte-endothelium interaction and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc Natl Acad Sci USA. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–853. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 24.Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood. 1990;75:550–554. [PubMed] [Google Scholar]
- 25.Kyan-Aung U, Haskard DO, Poston RN, Thornhill MH, Lee TH. Endothelial leukocyte adhesion molecule-1 and intercellular adhesion molecule-1 mediate the adhesion of eosinophils to endothelial cells in vitro and are expressed by endothelium in allergic cutaneous inflammation in vivo. J Immunol. 1991;146:521–528. [PubMed] [Google Scholar]
- 26.Bevilacqua MP, Stengelin S, Gimbrone MA, Jr, Seed B. Endothelial leukocyte adhesion molecule-1: an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–1165. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- 27.Thornhill MH, Wellicome SM, Mahiouz DL, Lanchbury JS, Kyan-Aung U, Haskard DO. Tumor necrosis factor combines with IL-4 or IFN-γ to selectively enhance endothelial cell adhesiveness for T-cells. The contribution of vascular cell adhesion molecule-1–dependent and –independent binding mechanisms. J Immunol. 1991;146:592–598. [PubMed] [Google Scholar]
- 28.Thurlow PJ, Kenneally DA, Connellan JM. The role of fibronectin in platelet aggregation. Br J Haematol. 1990;75:549–556. doi: 10.1111/j.1365-2141.1990.tb07797.x. [DOI] [PubMed] [Google Scholar]
- 29.Peerschke EI. Adhesive protein expression on thrombin-stimulated platelets: time-dependent modulation of anti-fibrinogen, -fibronectin, and -von Willebrand factor antibody binding. Blood. 1992;79:948–953. [PubMed] [Google Scholar]
- 30.Heyns AP, Eldor A, Yarom R, Marx G. Zinc-induced platelet aggregation is mediated by the fibrinogen receptor and is not accompanied by release or by thromboxane synthesis. Blood. 1985;66:213–219. [PubMed] [Google Scholar]
- 31.Azuma H, Shigekiyo T, Miura S, Uno Y, Saito S. Mechanism of potentiation by manganese ion of aggregation of porcine pancreatic elastase-treated human platelets. Thromb Haemostasis. 1989;62:984–988. [PubMed] [Google Scholar]
- 32.Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Activated and unactivated platelet adhesion to monocytes and neutrophils. Blood. 1991;78:1760–1769. [PubMed] [Google Scholar]
- 33.Spangenberg P, Redlich H, Bergmann I, Losche W, Gotzrath M, Kehrel B. The platelet glycoprotein IIb/IIIa complex is involved in the adhesion of activated platelets to leukocytes. Thromb Haemostasis. 1993;70:514–521. [PubMed] [Google Scholar]
- 34.Spangenberg P. Adhesion of activated platelets to polymorphonuclear leukocytes. Thromb Res. 1994;74:S35–S44. doi: 10.1016/s0049-3848(10)80005-2. [DOI] [PubMed] [Google Scholar]
- 35.Nierodzik ML, Plotkin A, Kajumo F, Karpatkin S. Thrombin stimulates tumor-platelet adhesion in vitro and metastasis in vivo. J Clin Invest. 1991;87:229–236. doi: 10.1172/JCI114976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeMarco L, Girolami A, Zimmerman TS, Ruggeri ZM. von Willebrand factor interaction with the glycoprotein IIb/IIIa complex: its role in platelet function as demonstrated in patients with congenital afibrinogenemia. J Clin Invest. 1986;77:1272–1277. doi: 10.1172/JCI112430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kieffer N, Nurden AT, Hasitz M, Titeux M, Breton-Gorius J. Identification of platelet membrane thrombospondin binding molecules using an anti-thrombospondin antibody. Biochim Biophys Acta. 1988;967:408–415. doi: 10.1016/0304-4165(88)90104-3. [DOI] [PubMed] [Google Scholar]
- 38.Suehiro K, Gailit J, Plow EF. Fibrinogen is a ligand for integrin α5β1on endothelial cells. J Biol Chem. 1997;272:5360–5366. doi: 10.1074/jbc.272.8.5360. [DOI] [PubMed] [Google Scholar]
- 39.Cox D, Aoki T, Seki J, Motoyama Y, Yoshida K. The pharmacology of the integrins. Med Res Reviews. 1994;14:195–228. doi: 10.1002/med.2610140203. [DOI] [PubMed] [Google Scholar]
- 40.Languino LR, Plescia J, Duperray A, Brian AA, Plow EF, Geltosky JE, Altieri DC. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell. 1993;73:1423–1434. doi: 10.1016/0092-8674(93)90367-y. [DOI] [PubMed] [Google Scholar]
- 41.D'Souza SE, Byers-Ward VJ, Gardiner EE, Wang H, Sung SS. Identification of an active sequence within the first immunoglobulin domain of intercellular cell adhesion molecule-1 (ICAM-1) that interacts with fibrinogen. J Biol Chem. 1996;271:24270–24277. doi: 10.1074/jbc.271.39.24270. [DOI] [PubMed] [Google Scholar]
- 42.Staunton DE, Dustin ML, Erickson HP, Springer TA. The arrangement of immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinoviruses. Cell. 1990;61:243–254. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- 43.Perrault C, Lankhof H, Pidard D, Kerbiriou-Nabias D, Sixma JJ, Meyer D, Baruch D. Relative importance of the glycoprotein Ib-binding domain and the RGD sequence of von Willebrand factor for its interaction with endothelial cells. Blood. 1997;90:2335–2344. [PubMed] [Google Scholar]
- 44.Beacham DA, Cruz MA, Handin RI. Glycoprotein Ib can mediate endothelial cell attachment to a von Willebrand factor substratum. Thromb Haemostasis. 1995;73:309–317. [PubMed] [Google Scholar]
- 45.Beacham DA, Tran LP, Shapiro SS. Cytokine treatment of endothelial cells increases glycoprotein Ibα-dependent adhesion to von Willebrand factor. Blood. 1997;89:4071–4077. [PubMed] [Google Scholar]
- 46.Wick TM, Moake JL, Udden MM, McIntire LV. Unusually large von Willebrand factor multimers preferentially promote young sickle and nonsickle erythrocyte adhesion to endothelial cells. Am J Hematol. 1993;42:284–292. doi: 10.1002/ajh.2830420308. [DOI] [PubMed] [Google Scholar]
- 47.Asch AS, Adelman B, Fujimoto M, Nachman RL. Identification and isolation of a platelet GPIb-like protein in human umbilical vein endothelial cells and bovine aortic smooth muscle cells. J Clin Invest. 1988;81:1600–1607. doi: 10.1172/JCI113494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu G, Essex DW, Meloni FJ, Takafuta T, Fujimura K, Konkle BA, Shapiro SS. Human endothelial cells in culture and in vivo express on their surface all four components of the glycoprotein Ib/IX/V complex. Blood. 1997;90:2660–2669. [PubMed] [Google Scholar]
- 49.Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie B. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59:305–312. doi: 10.1016/0092-8674(89)90292-4. [DOI] [PubMed] [Google Scholar]
- 50.Stone JP, Wagner DD. P-selectin mediates adhesion of platelets to neuroblastoma and small cell lung cancer. J Clin Invest. 1993;92:804–813. doi: 10.1172/JCI116654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor FB, Coller BS, Chang AC, Peer G, Jordan R, Engellener W, Esmon CT. 7E3 F(ab′)2, a monoclonal antibody to the platelet GPIIb/IIIa receptor, protects against microangiopathic hemolytic anemia and microvascular thrombotic renal failure in baboons treated with C4b-binding protein and a sublethal infusion of Escherichia coli. . Blood. 1997;89:4078–4084. [PubMed] [Google Scholar]