

Abstract
The effector functions of CD4+ T lymphocytes are generally thought to be controlled by distinct populations of regulatory T cells and their soluble products. The role of B cells in the regulation of CD4-dependent host responses is less well understood. Hepatic egg granuloma formation and fibrosis in murine schistosomiasis are dependent on CD4+ lymphocytes, and previous studies have implicated CD8+ T cells or cross-regulatory cytokines produced by T helper (Th) lymphocytes as controlling elements of this pathologic process. In this report, we demonstrate that B cell–deficient (μMT) mice exposed to Schistosoma mansoni develop augmented tissue pathology and, more importantly, fail to undergo the spontaneous downmodulation in disease normally observed during late stages of infection. Unexpectedly, B cell deficiency did not significantly alter T cell proliferative response or cause a shift in the Th1/Th2 balance. Since schistosome-infected Fc receptor–deficient (FcR γ chain knockout) mice display the same exacerbated egg pathology as that observed in infected μMT mice, the B cell– dependent regulatory mechanism revealed by these experiments appears to require receptor-mediated cell triggering. Together, the data demonstrate that humoral immune response/FcR interactions can play a major role in negatively controlling inflammatory disease induced by CD4+ T cells.
Pathology induced by infection with the trematode Schistosoma mansoni is a product of the immune response to parasite eggs in host tissues. The resulting granulomatous lesions lead to fibrosis, which in turn can produce severe circulatory impairment of the affected organs. The process of egg granuloma formation is dependent on sensitized CD4+ T lymphocytes, with both Th1 and Th2 subsets participating at different times during the development of the lesions.
The granulomas forming around newly deposited eggs are maximal in size during the early, acute phase of infection and become smaller during the later chronic period of the disease. This regression in pathology has been attributed to immunoregulatory events affecting the T cell response to egg Ag, which also declines during the same period. A variety of different mechanisms have been implicated in the regulation of schistosome egg pathology. These include “suppressor” CD8+ T cells (1–4), cross-regulation by cytokines produced by Th1 or Th2 cells (5–8), and the development of antiidiotypic Ab or T cells (9, 10). All of the above regulatory pathways involve the participation of T lymphocytes, acting either directly or indirectly through their helper function for Ab synthesis, on the host effector response to eggs.
Although it is commonly assumed that the regulation of egg pathology is T cell mediated, the possible function of B lymphocytes in this process has until recently been difficult to directly assess. Early studies using mice depleted of B cells as a result of anti-μ chain Ab treatment suggested B lymphocyte involvement but were inconclusive because of the potential side effects of the injection of large quantities of xenogenic Ab (11). A more direct approach for assessing the regulatory role of B cells in schistosome pathology is now possible with the advent of mice genetically B lymphocyte–deficient as a consequence of disruption of the μ-chain locus (12). These animals have no detectable B cells or circulating Ab, yet display normal development of the T lymphocyte compartment. Moreover, in initial studies, we and others have shown that B cell–deficient mice (μMT strain)1 have normal antigen presenting function for priming of CD4+ T cells to most soluble protein Ag as well as unimpaired CD8+ T lymphocyte responses (13–17). Indeed, these knockout (KO) animals develop pulmonary granulomas indistinguishable from those in wild-type (WT) mice after intravenous injection of schistosome eggs and display comparable lymphokine responses to egg Ag (13).
In this report, we have studied the pathogenesis of schistosomiasis in μMT mice during natural infection. The goal of this work was to formally assess the role of B lymphocytes in both hepatic egg pathology and more importantly in the downmodulation of disease and T cell responsiveness occurring in late infection. Unexpectedly, these animals showed markedly exacerbated hepatic granuloma formation and fibrosis during acute infection and failed to downmodulate pathology during the chronic stage of the disease. Importantly, S. mansoni–infected FcR-deficient mice (18) displayed alterations in immunopathology identical to the μMT animals. In both cases the observed differences in liver pathology occurred independently of significant changes in T lymphocyte reactivity and cytokine production patterns. Taken together these findings reveal a previously unrecognized function for Ig/FcR signaling in the regulation of CD4+ T cell–mediated inflammatory responses.
Materials and Methods
Experimental Animals, Infection Procedure, and Ag Preparation.
B cell– deficient μMT mice originally derived on a 129 × C57BL/6 background (12), were backcrossed to C57BL/6 for seven generations and then intercrossed to generate homozygous B cell KO animals. The mice were both bred and maintained at the American Association for Accreditation Laboratory animal care facility of National Institute of Allergy and Infectious Diseases. To rule out possible effects of 129 background genes, most experiments were repeated using μMT mice backcrossed to the C57BL/10 background for 12 generations. The latter B cell–deficient and C57BL/10 control mice were shipped from a specific pathogen– free animal facility at Taconic Farms (Germantown, NY). The μMT mice on the C57BL/6 and C57BL/10 backgrounds gave indistinguishable results in all the assays performed. The experiments shown are those performed with C57BL/6 μMT animals. Mice genetically deleted for the FcR γ subunit were derived as previously described (18) and backcrossed for 12 generations to C57BL/6J. WT C57BL/6 mice purchased from Charles River Laboratories (Wilmington, MA) and The Jackson Laboratory (Bar Harbor, ME) were used as B cell– and FcR-sufficient controls, respectively. Age- (8–12 wk) and sex-matched mice were used in each experiment.
S. mansoni cercariae, a Puerto Rican strain (Naval Medical Research Institute), were obtained from infected Biomphalaria glabrata snails (Biomedical Research Institute, Rockville, MD). Mice were infected by percutaneous exposure of the tail to 35 cercariae unless otherwise indicated. A soluble extract of schistosome eggs (SEA) was prepared as previously described (19).
Parasitological Measurements.
Adult worms were enumerated by hepatic perfusion of mice at 6 or 8 wk after infection (20). Tissue egg counts were performed on liver and intestines digested in 4% potassium hydroxide. Excreted eggs were counted in stool samples collected from individual mice for 24 h immediately before perfusion.
Assessment of Egg Pathology.
Granuloma diameters were measured in histological sections stained with Litt's modification of the Dominici procedure using an ocular micrometer and the volume of each lesion was calculated assuming a spherical shape. Mean granuloma sizes were then computed for each mouse from measurements performed on ∼30 granulomas per animal. The percentage of eosinophils in the same granulomas was estimated by microscopic examination. Liver collagen levels were determined by chemical measurement of hydroxyproline as previously described (21). The statistical significance of differences in granuloma volumes and eosinophil levels between animal groups was evaluated using Student's two-tailed t test and differences in fibrosis by analysis of covariance.
Cell Proliferation and Cytokine Assays.
Single-cell suspensions of spleens from individual mice were prepared and RBC lysed by osmotic treatment. Mesenteric lymph node cell suspensions were pooled from four to six animals per group. All in vitro assays involved culturing of cells in RPMI 1640 medium supplemented with 10% FCS, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ ml streptomycin, 25 mM Hepes, 1 mM sodium pyruvate, nonessential amino acids, and 50 μM 2-ME at 37°C in 5% CO2.
Proliferative responses to SEA were assayed at 8 and 16 wk after infection by exposing splenocytes (3 × 106/ml) from individual B cell KO and WT animals to graded concentrations of the Ag preparation in 200 μl in 96-well flat-bottomed microtiter plates. After 48 h of incubation, [3H]TdR (0.5 μCi/well, New England Nuclear Corp., Boston, MA; sp act: 2 Ci/mmol) was added and incorporation of the isotope was measured 18 h later. The stimulation index was calculated as the ratio between the [3H]TdR incorporated in the presence and absence of SEA.
To measure cytokine secretion, mesenteric lymph node cells (2 × 106/ml) were cultured in 24-well plates in 1 ml volumes and exposed to medium alone, 2.5 μg of plate-bound anti-CD3 mAb (145-2C11; PharMingen, San Diego, CA), or SEA (20 μg/ml) for 72 h. IFN-γ, IL-5, and IL-10 were measured in culture supernatants by specific sandwich ELISA (22, 23). Cytokine levels were calculated by reference to standard curves prepared with known amounts of recombinant cytokine. IL-4 was assayed in culture supernatants using the IL-4–dependent cell line CT.4S as previously described (24, 25). Proliferation of CT.4S cells was quantified by measuring [3H]TdR incorporation, and the amount of cytokine in supernatant was determined by comparison to proliferation induced by known amounts of rIL-4 (Genzyme Corp., Boston, MA).
Reverse Transcriptase PCR Detection of Cytokine mRNAs.
Equivalent specimens of liver tissue from uninfected and infected animals were homogenized in 1 ml RNA STAT-60 using a tissue polytron (Omni International, Waterbury, CT) and total RNA was isolated as recommended by the manufacturer. The RNA was resuspended in diethylpyrocarbonate-treated water and quantitated spectrophotometrically. A reverse transcriptase PCR procedure was performed as previously described (26) to determine relative quantities of mRNA for IL-4, IL-5, IL-10, and IL-13. The amplified DNA was analyzed by electrophoresis, Southern blotting, and hybridization with cytokine-specific probes. The primers and probes have been previously published (26, 27). The chemiluminescent signals were quantified using a 600 ZC scanner (Microtek International, Torrance, CA). The amount of PCR product was determined by comparison of signal density to that of standard curves generated from simultaneously amplified dilutions of cDNA obtained from samples with a high amount of specific cytokine mRNA. Fold increases for individual samples were calculated as the reciprocal of the equivalent dilution of control cDNA from uninfected mouse liver. Amplification of hypoxanthine-guanine phosphoribosyl transferase served as an internal control for the amount of RNA and cDNA from each sample.
Results
B Cell KO Mice Show Normal Susceptibility to Schistosome Infection but Decreased Survival.
In initial experiments, μMT and C57BL/6 WT control animals were percutaneously exposed to different infecting doses of cercariae, and the recovery of adult parasites as well as tissue egg burdens were measured 6–8 wk later. As shown in Fig. 1, A and B, the KO and WT mice were quantitatively indistinguishable in terms of both worm and tissue egg recovery, indicating that neither B cells nor Ab influence the development of S. mansoni during primary infection. Nevertheless, when compared to WT animals, the infected μMT mice showed significantly decreased fecal egg excretion (Fig. 1 C), a parameter that measures the transit of parasite ova into the gut. The latter observation is consistent with previous studies, suggesting a role for Ab in facilitating the transport of eggs through intestinal tissue to the gut lumen (28).
Figure 1.

Worm and tissue egg recoveries in S. mansoni–infected WT and B cell–deficient μMT mice. (A) The absence of B cells does not affect worm burden during primary infection. Adult worms were recovered after hepatic perfusion of infected WT (black bars) and B cell KO (gray bars) animals killed at 6 (challenge with 100 cercariae) and 8 (challenge with 40 and 25 cercariae) wk after infection. Data shown are mean parasite counts (± SEM) of six to nine animals per group. (B) Worm fecundity is unaltered in infected μMT mice. Egg output per worm pair was calculated for individual infected WT or B cell KO mice at 8 wk after infection from the total number of eggs recovered from the liver and intestine. The results shown are the means (± SEM) of data pooled from three experiments in which mice (n = 14/group) were infected with 35 cercariae. (C) Diminished egg excretion in infected B cell KO mice. The number of eggs excreted in the stools collected from individual 8-wk infected WT and B cell KO mice during the 24 h before perfusion was determined. The data shown are the mean (± SEM) egg count values pooled from the same three experiments presented in B.
Despite their unaltered parasite burdens, the μMT mice showed decreased survival during the chronic stage of infection with approximately half the animals dying before week 16 (Fig. 2). Upon autopsy, the μMT mice that succumbed showed extensive hemorrhaging into the gut lumen, a sequela which is only rarely observed in WT animals.
Figure 2.

Enhanced mortality in S. mansoni–infected μMT mice. Groups (n = 10) of WT and KO mice were infected percutaneously with 30 cercariae and animal survival was monitored. The hatched line indicates the 50% survival time point for B cell KO animals. As indicated, no mortality was observed in infected WT animals during the same period. The two experiments shown are representative of four performed.
Exacerbated Egg Pathology in μMT Mice during Acute Infection.
When hepatic granuloma volumes were compared in B cell KO versus WT mice at 8 wk after infection, a significant difference in the tissue response to schistosome eggs was noted (Fig. 3, top left panel). The B cell KO animals developed lesions that were on average 42% larger than those in the infected control group. Although clearly bigger in size, the granulomas in the μMT mice showed no obvious differences in their cellular composition when compared to those occurring in the WT animals. In particular, the granulomas in both types of animals displayed comparable levels of eosinophils (Fig. 3, bottom left panel). The latter parameter is usually dependent on the synthesis of IL-5 (29), a major product of the Th2 response.
Figure 3.

Augmented egg granuloma formation in livers of S. mansoni– infected μMT mice. Upper panels: mean liver granuloma volumes of individual infected animals at 8 (left panel) and 16 wk of infection (right panel). Granuloma diameters were measured (∼30 per mouse) and mean lesion volumes were calculated for each animal and presented as a single point on the graph. The data shown are pooled from the results of 6 individual experiments in which granulomas were measured at 8 wk and from 3 experiments involving measurements at 16 wk. Within each individual experiment performed, granuloma volumes were significantly larger in the B cell KO as compared with the WT mice. Lower panels: mean (± SEM) eosinophil composition of liver granulomas determined by microscopic examination. Percentage of eosinophils was calculated from microscopic analysis of the same granulomas analyzed for lesion size. No statistically significant difference was observed between the values obtained from WT and B cell KO mice.
Downmodulation of Egg Pathology Is Absent in Chronically Infected μMT Mice.
As infection proceeds to the chronic stage, newly formed granulomas become smaller in size (30). As shown in Fig. 3 (top panels) and Fig. 4, this process of downmodulation of immunopathology was found to be defective in B cell KO animals when examined at 16 wk after infection. Granuloma volumes in μMT mice at this stage remained at the same increased size as was detected at 8 wk of infection but were now twice the magnitude of the granulomas in chronically infected WT mice. In agreement with previous observations, the percentage of eosinophils in granulomas from WT animals was significantly diminished at 16 versus 8 wk after infection. A similarly significant, although smaller decrease in eosinophil content was observed as infected KO mice proceeded from the acute to the chronic phase (Fig. 3, bottom panels).
Figure 4.
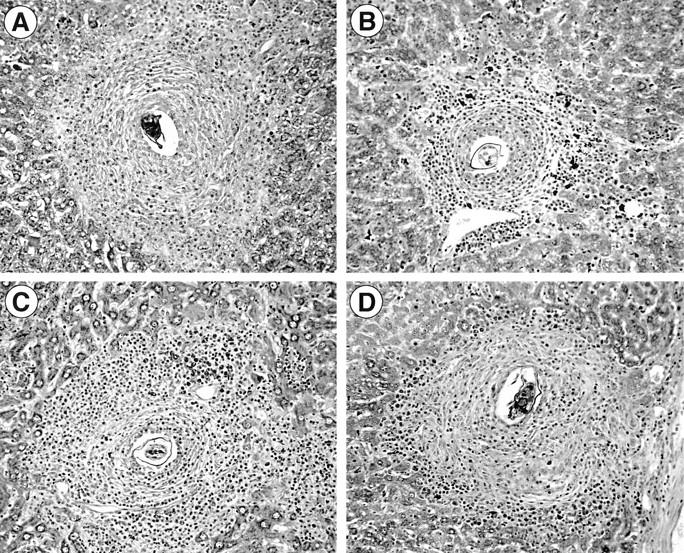
Photomicrographs of representative hepatic granulomas from WT control versus KO animals. The granulomas shown are from 8-wk (A) and 16-wk (B) infected C57BL/6J mice, and from 16-wk infected μMT animals (C) and 16-wk infected FcRγ KO mice (D). Original magnification of Giemsa stain, ×200.
Increased Hepatic Fibrosis in Infected μMT Mice.
Tissue fibrosis is the major pathologic sequela of schistosome egg granuloma formation. At 8 wk after infection, the livers from B cell KO mice showed a small but significant increase in hydroxyproline levels, a chemical measure of collagen deposition (Fig. 5, left panel). This difference in tissue fibrosis was further accentuated at the chronic stage, when the livers from the infected μMT mice displayed almost twice the hydroxyproline levels as those observed in the same tissues from WT animals (Fig. 5, right panel). Thus, in the absence of B cells, both granuloma formation and fibrosis are markedly increased at both the acute and chronic phases of schistosome infection.
Figure 5.

Increased liver fibrosis in S. mansoni–infected μMT mice. Tissue hydroxyproline levels were measured in individual animals at 8 (left panel) and 16 (right panel) wk after infection. Data shown are mean values (± SEM) for four to six mice per group from one representative experiment out of six performed at the acute phase and three assayed at the chronic phase. Differences shown to be statistically significant by analysis of covariance are indicated.
Despite their Augmented Egg Pathology, Infected μMT Animals Exhibit Unaltered T Cell Responses.
S. mansoni egg granuloma formation is known to be dependent on CD4+ T cells and the lymphokines they produce. Therefore, we examined whether the exacerbated pathology observed in infected μMT mice is reflected in enhanced T cell reactivity. When examined during the acute phase (8 wk after infection), spleen cells from μMT mice mounted proliferative responses to SEA that were slightly but insignificantly lower than those measured in WT animals over a wide dose range (Fig. 6, left panel). As expected, proliferative responses in WT mice declined when the infection reached the chronic phase (Fig. 6, right panel). Nevertheless, a similar and even greater suppression of lymphocyte blastogenesis occurred in the chronically infected μMT mice despite their augmented granuloma volumes and fibrosis.
Figure 6.

Proliferative responses of μMT and WT splenocytes to SEA. Spleen cells (3 × 106/ml) from individual 8-wk infected WT or B cell KO mice were cultured with the indicated concentrations of SEA and 3 d later [3H]TdR incorporation was measured after an overnight pulse (left panel). In the case of chronically (16-wk) infected mice, proliferation was assayed in response to a predetermined optimal dose (20 μg/ml) of SEA (right panel). Data shown are mean values (± SEM) of stimulation indices for each group of animals (n = 6–10).
The CD4+ T cell response to schistosome eggs is associated with a cytokine production profile dominated by Th2 lymphokines. This response peaks at 8 wk after infection and diminishes thereafter. To investigate whether the exacerbated pathology in μMT mice is associated with aberrant cytokine production, mesenteric LN cells from infected KO and WT animals were stimulated in vitro with SEA or anti-CD3 mAb and IL-4, IL-5, IL-10, or IFN-γ levels measured in 72-h culture supernatants. At 8 wk after infection, LN from μMT mice produced high levels of all four cytokines when triggered with anti-CD3, and in comparison with WT animals showed no obvious defects in responsiveness (Fig. 7). When stimulated with SEA, the same cells produced a Th2-dominated cytokine profile with only trace levels of IFN-γ. These Ag-specific responses were comparable in the infected μMT and WT mice except in the case of IL-4 and IL-10, where lower cytokine production was observed in the KO animals. In the case of IL-4 secretion, the difference between the KO and WT animals was statistically significant. By the chronic stage of infection, anti-CD3–induced IL-4, IL-5, and IL-10 responses were decreased in both WT and μMT mice, whereas IFN-γ production was unaltered. Similarly, upon SEA stimulation of the same cells, diminished production of IL-4, IL-5, and IL-10 was observed with IFN-γ remaining at trace levels. Although greatly reduced in both groups of animals in comparison with 8-wk infected mice, SEA-induced IL-4 secretion was again significantly lower in the B cell KO versus WT animals (Fig. 7).
Figure 7.

Comparison of cytokine secretion by mesenteric LN cells from infected μMT versus WT mice. Culture supernatants of mesenteric LN cells (2 × 106/ml) pooled from four to six individual mice per group were assayed for the presence of IL-4, IL-5, IL-10, and IFN-γ after 72 h in vitro stimulation with medium, SEA, or plate-bound anti-CD3 Ab. The data shown are from WT (black bars) and B cell KO (gray bars) mice killed at 8 (acute) and 14 (chronic) wk after infection. Supernatants from both time points were analyzed in the same assay and compared to the same standard curve. Bars represent the mean ± SD of ELISA values for IL-5, IL-10, and IFN-γ and values from a CT.4S assay for IL-4. In the case of SEA-induced IL-4 responses at both 8 and 14 wk, the differences between the μMT and WT values were statistically significant (P <0.001). The percentage of CD4+CD44+ cells (previously shown to represent the major population of SEA- responsive T lymphocytes; reference 31) was determined to be comparable in the WT and μMT cell preparations (data not shown).
In Situ Cytokine Expression in Infected μMT versus WT Animals.
Because the results of the LN cell cytokine secretion assays suggested that egg-induced IL-4 and IL-10 responses are partially impaired in μMT mice, we carried out additional experiments to test whether the same differences in the expression of these cytokines are evident in the liver, the major site of granuloma formation. The latter analysis was performed by reverse transcriptase PCR measurement of the cytokine mRNAs in liver tissue in uninfected and acutely and chronically infected KO and WT mice. As shown in Fig. 8, at the acute stage of infection mRNA levels for the Th2 cytokines IL-4, IL-5, IL-10, and IL-13 were comparable in the two groups of animals. Moreover, as expected, the expression of these cytokine mRNAs was decreased at the chronic stage with the exception of IL-10, which increased in the KO but not in the WT animals. The above data argue that the exacerbated egg pathology in μMT mice is unlikely to be the result of an aberrant Th2 cytokine production, and taken together with the in vitro culture findings suggest that the failure of these animals to downmodulate granuloma formation is not due to impaired downregulation of T cell responses.
Figure 8.

Comparison of hepatic cytokine mRNA expression in infected μMT and WT animals. At 8 and 16 wk after infection, liver tissue from individual mice (n = 3–4 per group) were assayed for IL-4, IL-5, IL-10, and IL-13 mRNA expression by reverse transcriptase PCR. Values represent the mean fold increase (as a multiple of control value) ± SE over uninfected WT control liver mRNA expression.
FcRγ-deficient Mice Display Defects in the Regulation of Schistosome Egg Pathology Similar to those Observed in B Cell– deficient Mice.
The above observations on S. mansoni infection in μMT mice pointed to an important role for humoral immune response in the regulation of egg pathology. To both confirm the involvement of Ab and determine whether Fc receptor interactions are required for Ab regulatory activity, we analyzed schistosome immunopathology in FcR γ chain KO mice. These animals possess a normal B cell compartment, but due to the deletion of the gene encoding the common FcR γ chain fail to express FcγRI, FcγRIII, and FcεRI, the major cell surface receptors involved in positive Fc-mediated triggering (18). As shown in Figs. 9 and 4, infected FcRγ KO mice, in common with B cell KO animals, displayed enhanced hepatic granuloma formation during the acute phase of infection, and importantly failed to downmodulate this response during the chronic stage of disease. Moreover, tissue fibrosis was also augmented in chronically infected FcRγ KO animals (Fig. 9, lower right panel). As observed in B cell KO mice, FcRγ KO and WT mice developed comparable levels of tissue eosinophilia at 8 and 16 wk after infection (Fig. 9, lower left panel, and data not shown). Moreover, no significant differences were observed between the two mouse strains when IL-4, IL-5, and IL-10 responses of spleen or mesenteric LN cells to SEA and anti-CD3 were measured in vitro (data not shown). Together these findings indicate that in terms of tissue pathology induced by schistosome infection, B cell– and FcRγ-deficient mice share a common phenotype. Nevertheless, in contrast to the findings with μMT mice, FcRγ KO animals showed no significant decrease in fecal egg excretion (eggs/worm pair/day) at 8 wk after infection (112 ± 16 for WT [n = 15] versus 91 ± 12 for FcRγ KO [n = 13]), nor did they display enhanced mortality throughout the period of the experiments (data not shown). The latter observations suggest that the decreased egg excretion and increased mortality observed in μMT mice are due directly to the absence of B cells or their products but operate via mechanisms that are independent of FcR signaling events.
Figure 9.

Augmented egg granuloma formation in livers of S. mansoni– infected FcR γ chain KO mice. Upper panels: mean (± SEM) liver granuloma volumes of WT and FcRγ KO mice at 8 (left panel) and 16 (right panel) wk after infection. The data shown are pooled from three individual experiments in which both acute and chronic animals were analyzed, and each point represents the value for an individual mouse. Lower left panel: mean (± SEM) eosinophil composition of liver granulomas at 8 wk after infection. Lower right panel: tissue fibrosis (hydroxyproline) measured in two experiments at 16 wk. No significant differences between the WT and FcRγ KO mice were detected in lymphocyte proliferation, Th1/Th2 cytokine expression, or anti-SEA Ab titers in these experiments (data not shown).
Discussion
In this study we demonstrate that B cell–deficient mice as well as animals lacking the common FcR γ chain develop exacerbated egg pathology during both acute and chronic infection with S. mansoni and fail to display the downregulation in granuloma formation typically observed in WT animals. Surprisingly, this augmented pathology occurs in the absence of any appreciable change in T cell proliferative response or shift in Th1/Th2 balance even though egg-induced inflammation is known to be a CD4+ T cell– dependent host reaction. Thus, it appears that Ig/FcR interactions play a major regulatory role in the pathogenesis of experimental schistosomiasis mansoni independent of the level and quality of T cell responsiveness.
When quantitatively infected with S. mansoni, μMT mice gave the same adult worm and tissue egg recoveries as WT animals (Fig. 1). These findings confirm previously published results involving anti–μ treated mice, indicating that worm development and egg deposition during primary infection are not significantly influenced by the host Ab response (11, 32). Nevertheless, our data do not agree with two prior and themselves conflicting reports indicating that in the absence of IgE worm recoveries are either decreased (33) or increased (34) versus control animals. At present the explanation for the observed discrepancy amongst these and our results is unclear, although in the case of our experiments we can definitively exclude the possibility of a residual Ab response of IgE or any isotype in the B cell– deficient animals. Interestingly, however, infected μMT, but not FcRγ KO, mice differed from control animals in displaying a marked reduction in the number of eggs excreted into the feces. The latter finding is consistent with previous reports indicating that Ab facilitate the transport of eggs across the intestinal wall into the lumen (28) and suggest that FcR interactions involving FcγRI, FcγRIII, and FcεRI are not required for this process. Together the data support the hypothesis that the Ab response, rather than limiting worm development, plays an important physiological role in the maintenance of the parasite life cycle.
Another aspect in which infected μMT differed from both WT and FcRγ KO animals was in the increased mortality of the former strain. This pathologic consequence of B cell deficiency, which is associated with hemorrhaging into the gut, is at present poorly understood. It is unlikely to be the cause of the observed exacerbation of hepatic pathology, since that phenomenon was demonstrated in survivors that did not display obvious intestinal pathology as well as in the infected FcRγ KO mice that failed to exhibit significant mortality. One possibility is that in WT (as well as FcRγ KO) animals, Ab produced against endotoxins protect the host against toxic inflammatory responses triggered by gut bacteria that, in the setting of the intestinal alterations induced by schistosomiasis, lead to increased mortality in individual mice (35).
The most striking effects of B cell and FcRγ deficiencies in S. mansoni infection occurred at the level of the downmodulation of hepatic pathology. A large volume of previous work has indicated that the intensity of the host response to schistosome eggs diminishes with the length of the infection. In mice this immunomodulation is evidenced by the decreased size of newly formed granulomas in chronic disease, a phenomenon that has been closely linked to a concomitant decline in T cell responsiveness to egg Ag. A variety of different mechanisms have been proposed to explain the downmodulation of granulomatous hypersensitivity in schistosomiasis, all of which involve active suppression of T cell function. The most extensively investigated mechanisms have been those involving downregulation by CD8+ suppressor cells (3, 4, 9) or inhibitory cytokines produced by cross-regulatory CD4+ Th cells (4, 6–9). However, recent experiments studying the modulation of schistosome pathology in gene KO mice argue against a requirement for either of these pathways. Thus, mice lacking functional CD8 activity (36) or the major cross-regulatory cytokines IFN-γ (36) or IL-10 (36a) display normal downmodulation of both pathology and T cell responsiveness. The latter findings support the involvement of a previously unappreciated mechanism in the immunoregulation of the granulomatous response.
The present study reveals that B cells may provide a crucial activity necessary for the downmodulation of schistosome egg pathology. The observed failure of these mice to downregulate granuloma formation is in fact consistent with several previous observations. First, it is clear that in both mice and humans Ab titers to parasite Ag increase substantially as the infection moves from the acute to the chronic stage (37, 38) and that in the murine model this change is reflected in an expansion in the number of B cells present in the granulomatous lesions themselves (39). Secondly, we have previously observed that mice rendered B cell deficient by treatment with anti-μ sera also develop larger granulomas which are not downmodulated (11). However, these experiments were uninterpretable because of the known peripheral effects of anti-μ treatment on T cell responses (40).
Surprisingly, although downmodulation of egg pathology was absent in μMT mice, normal downregulation of T cell proliferative and lymphokine responses was observed in the same animals. This finding provides striking evidence for a dissociation of the two phenomena and argues against a direct causal relationship. Interestingly, the absence of B cells also resulted in elevated granuloma formation during the acute stage of infection as well as enhanced fibrosis (collagen deposition) at both of the time periods analyzed. Again, the in vitro responses of mesenteric LN cells measured during the acute phase in B cell–deficient mice closely resembled those observed in WT animals with the exception of a decrease in the levels of IL-4 and IL-10 produced. This finding is reminiscent of previously reported results on S. mansoni infection in B cell–deficient (JHD) mice (41), in which decreased IL-4 and IL-10 production in vitro was also described at 7.5–8 wk after infection (42). No analysis of the chronic stage of disease or tissue fibrosis was performed in that study. Nevertheless, the results reported do diverge from those presented here in that no difference in granuloma size was detected between KO and WT animals in acute infection, a discrepancy that could relate to different genetic backgrounds or types of B cell– deficient mouse strains used (13, 43). Furthermore, in the case of our experiments, the observed differences in cytokine production between acutely infected WT and μMT did not reflect a wholesale switch to a Th1 cytokine pattern, since the amounts of both IFN-γ and IL-5 protein secreted in vitro were indistinguishable in the two strains, as were the levels of IL-4, IL-5, IL-10, and IL-13 mRNAs expressed in liver tissue. IL-12, which has been previously demonstrated to suppress hepatic pathology when administered exogenously (44), was not measured in these experiments because it is only minimally expressed endogenously in schistosome infected mice (44, 42).
Because our findings clearly indicated a role for B cells in the downmodulation in liver pathology occurring in chronic infection, we went on to address possible mechanisms. The major immunoregulatory products of B cells are likely to be either immunoglobulins or cytokines. In the case of cytokines, B lymphocyte–produced IL-10 has been proposed as a mediator of granuloma downmodulation by Vellupillai and Harn (45), who demonstrated the induction of such a response by schistosome carbohydrates in vitro. This mechanism is unlikely to explain the effects of the lack of B cells on egg pathology, since IL-10 mRNA expression in liver was found to be increased rather than diminished in chronically infected μMT mice. Moreover, recent studies (36a) indicate that IL-10 KO mice undergo normal downmodulation of granuloma formation in late S. mansoni infection. Thus, although it is impossible at present to exclude the role of other cytokines produced by B cells, it is highly likely that Ab themselves are the B cell products responsible for immunoregulation. This conclusion is supported by the results of preliminary experiments in which sera from chronically infected WT mice transferred a reduction in granuloma size to acutely infected μMT animals (data not shown).
There are two obvious mechanisms by which Ab could control the granulomatous response to schistosome eggs: by neutralizing egg derived inflammatory molecules, or by triggering the production of antiinflammatory mediators from FcR+ cells. The involvement of the latter pathway is strongly suggested by our observation that FcRγ-deficient mice display a nearly identical phenotype to B cell–deficient animals in terms of augmented tissue pathology and the failure to downmodulate granuloma formation during chronic infection. Additionally, infected FcRγ-deficient mice showed no evidence of aberrant T cell function (data not shown). FcRγ-deficient mice do not express the major cell surface receptors involved in Ig binding and cell signaling (FcγRI, which preferentially binds IgG2a; FcγRIII, which binds the immune complexes formed with the Ab of most IgG isotypes; and FcεRI, the high-affinity receptor for IgE). At present, it is unclear which of these three FcRs participate in the immunoregulatory pathway responsible for the downregulation of granulomatous pathology. Because IgG2a Abs are only marginally induced in mice chronically infected with S. mansoni, the involvement of FcγRI is unlikely. Instead, as the dominant isotypes observed in such animals are IgG1 and IgE, FcγRIII and FcεRI are more likely to play a role. Interestingly, we have recently demonstrated that during the acute stage of S. mansoni infection, FcεRI-deficient mice (46) also show increased egg pathology (47), suggesting that FcεRI is one of the receptors involved. Because chronic granuloma formation was not evaluated in that study, it is unclear whether FcεRI is also important for downmodulation. Experiments are in progress in which we are systematically comparing egg pathology in S. mansoni–infected FcεRI-, FcγRII- (48) and FcγRIII-deficient mice (49) in order to evaluate the contribution of each receptor.
Regardless of the particular FcR that mediates the regulatory response, it is clear from data reported here that the humoral Ab production can influence granulomatous pathology independent of detectable effects on CD4+ T cell responsiveness. Possible mediators of this antiinflammatory effect include prostaglandins, phospholipase A, and TGF-β. Interestingly, histamine, which is normally associated with the induction of inflammation, has been shown in previous studies to play a downregulatory role in acute schistosome granuloma formation (50) and is therefore an important candidate for an FcR induced pharmacological mediator of this process. Inherent in these earlier findings as well as in those reported here is the seemingly contradictory concept that FcR-dependent responses that in most settings mediate inflammation (51) can in other contexts be antiinflammatory. In the case of the schistosome infection model studied here, the context is that of a CD4+ T cell–dependent granulomatous tissue reaction. It remains to be seen whether other cell-mediated tissue responses may be negatively regulated through the same humoral mechanism. Interestingly, B cell–deficient mice have been reported to display exacerbated inflammatory responses to myelin basic protein (52), suggesting that this pathway could also contribute to the regulation of autoimmune tissue pathology.
Footnotes
We thank Drs. S.L. James and T. Nutman for reading the manuscript, and R. Dryfuss and S. Everett for help with the photomicrographs.
M.C. Kullberg was supported in part by a grant from the Swedish Medical Research Council.
Address correspondence to Dragana Jankovic, Immunobiology Section, Laboratory of Parasitic Diseases, NIAID, NIH, Building 4, Room 126, 9000 Rockville Pike, MD 20892-0425. Phone: 301-496-8218; Fax: 301-402-0890; E-mail: djankovic@atlas.niaid.nih.gov
Abbreviations used in this paper: μMT, B cell-deficient mice; KO, knockout; SEA, soluble schistosome egg Ag; WT, wild-type.
References
- 1.Colley DG. Adoptive suppression of granuloma formation. J Exp Med. 1976;143:696–700. doi: 10.1084/jem.143.3.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colley DG, Lewis FA, Todd CW. Adoptive suppression of granuloma formation by T lymphocytes and by cells sensitive to cyclophosphamide. Cell Immunol. 1979;46:192–200. doi: 10.1016/0008-8749(79)90258-2. [DOI] [PubMed] [Google Scholar]
- 3.Chensue SW, Wellhausen SR, Boros DL. Modulation of granulomatous hypersensitivity. II. Participation of Lyt1+ and Lyt2+ T lymphocytes in the suppression of granuloma formation and lymphokine production in Schistosoma mansoni-infected mice. J Immunol. 1981;127:363–367. [PubMed] [Google Scholar]
- 4.Chensue SW, Warmington KS, Hershey SD, Terebuh PD, Othman M, Kunkel SL. Evolving T cell responses in murine schistosomiasis. Th2 cells mediate secondary granulomatous hypersensitivity and are regulated by CD8+T cells in vivo. J Immunol. 1993;151:1391–1400. [PubMed] [Google Scholar]
- 5.Pearce EJ, Caspar P, Grzych JM, Lewis FA, Sher A. Downregulation of Th1 cytokine production accompanies induction of Th2 responses by a parasitic helminth, Schistosoma mansoni. . J Exp Med. 1991;173:159–166. doi: 10.1084/jem.173.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flores Villanueva, P.O., H. Reiser, M.J. Stadecker. Regulation of T helper cell responses in experimental murine schistosomiasis by IL-10: effect on expression of B7 and B7-2 costimulatory molecules by macrophages. J Immunol. 1994;153:5190–5199. [PubMed] [Google Scholar]
- 7.Chensue SW, Warmington KS, Ruth J, Lincoln PM, Kunkel SL. Cross-regulatory role of interferon-gamma (IFN-gamma), IL-4 and IL-10 in schistosome egg granuloma formation: in vivo regulation of Th activity and inflammation. Clin Exp Immunol. 1994;98:395–400. doi: 10.1111/j.1365-2249.1994.tb05503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flores Villanueva, P.O., X.X. Zheng, T.B. Storm, M.J. Stadecker. Recombinant IL-10 and IL-10/Fc treatment down-regulate egg antigen-specific delayed hypersensitivity reactions and egg granuloma formation in schistosomiasis. J Immunol. 1996;156:3315–3120. [PubMed] [Google Scholar]
- 9.Abe T, Colley DG. Modulation of Schistosoma mansoniegg–induced granuloma formation. III. Evidence for an anti-idiotypic, I-J–positive, I-J–restricted, soluble T suppressor factor. J Immunol. 1984;132:2084–2088. [PubMed] [Google Scholar]
- 10.Montesano MA, Freeman GL, Secor WE, Colley DC. Immunoregulatory idiotypes stimulate T helper 1 cytokine responses in experimental Schistosoma mansoniinfections. J Immunol. 1997;158:3800–3804. [PubMed] [Google Scholar]
- 11.Cheever AW, Byram JE, Hieny S, von Lichtenberg F, Lunde MN, Sher A. Immunopathology of Schistosoma japonicum and S. mansoniinfection in B cell depleted mice. Parasite Immunol. 1985;7:399–413. doi: 10.1111/j.1365-3024.1985.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 12.Kitamura D, Roes J, Kühn R, Rajewsky K. A B cell–deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 13.Epstein MM, Di Rosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell–deficient mice. J Exp Med. 1995;182:915–922. doi: 10.1084/jem.182.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+T cells to protein antigens in vivo. J Immunol. 1995;155:3734–3741. [PubMed] [Google Scholar]
- 15.Baird A, Parker DC. Analysis of low zone tolerance induction in normal and B cell–deficient mice. J Immunol. 1996;157:1833–1839. [PubMed] [Google Scholar]
- 16.Philips JA, Romball CG, Hobbs MV, Ernst DN, Schultz L, Weigle WO. CD4+T cell activation and tolerance induction in B cell knockout mice. J Exp Med. 1996;183:1339–1344. doi: 10.1084/jem.183.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oxenius A, Campbell KA, Maliszewski CR, Kishimoto T, Kikutani H, Hengartner H, Zinkernagel R, Bachmann MF. CD40–CD40 ligand interactions are critical in T–B cooperation but not for other anti-viral CD4+T cell functions. J Exp Med. 1996;183:2209–2218. doi: 10.1084/jem.183.5.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotropic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 19.Boros DL, Warren KS. Delayed hypersensitivity-type granuloma formation and dermal reaction induced and elicited by soluble factor isolated from Schistosoma mansonieggs. J Exp Med. 1970;132:488–507. doi: 10.1084/jem.132.3.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smithers SR, Terry RJ. The infection of laboratory hosts with cercariae of Schistosoma mansoniand the recovery of adult worms. Parasitology. 1965;55:659–700. doi: 10.1017/s0031182000086248. [DOI] [PubMed] [Google Scholar]
- 21.Bergman I, Loxley R. Two improved and simplified methods for the spectrophotometric determination of hydroxyproline. Anal Biochem. 1963;35:1961–1965. [Google Scholar]
- 22.Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J Exp Med. 1988;168:1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosmann TR, Schumacher JH, Fiorentino DF, Leverah H, Moore KW, Bond MW. Isolation of monoclonal antibodies specific for IL-4, IL-5, IL-6, and a new Th2-specific cytokine (IL-10), cytokine synthesis inhibitory factor, by using a solid phase radioimmunosorbent assay. J Immunol. 1990;145:2938–2945. [PubMed] [Google Scholar]
- 24.Hu-Li J, Ohara J, Watson C, Tsang W, Paul WE. Derivation of a T cell line that is highly responsive to IL-4 and IL-2 (CT.4R) and of an IL-2 hyporesponsive mutant of that line (CT.4S) J Immunol. 1989;142:800–807. [PubMed] [Google Scholar]
- 25.Kullberg MC, Berzofsky JA, Jankovic DLj, Barbieri S, Williams ME, Perlmann P, Sher A, Troye-Blomberg M. T-cell–derived IL-3 induces the production of IL-4 by non-B, non-T cells to amplify the Th2-cytokine response to a non-parasite antigen in Schistosoma mansoni– infected mice. J Immunol. 1996;156:1482–1489. [PubMed] [Google Scholar]
- 26.Wynn TA, Eltoum I, Oswald IP, Cheever AW, Sher A. Endogenous interleukin 12 (IL-12) regulates granuloma formation induced by eggs of Schistosoma mansoniand exogenous IL-12 both inhibits and prophylactically immunizes against egg pathology. J Exp Med. 1994;179:1551–1561. doi: 10.1084/jem.179.5.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svetic A, Finkelman FD, Jian YC, Dieffenbach CW, Scott DE, McCarthy KF, Steinberg AD, Gause WC. Cytokine gene expression after in vivo primary immunization with goat antibody to mouse IgD antibody. J Immunol. 1991;147:2391–2397. [PubMed] [Google Scholar]
- 28.Doenhoff MJ, Musallam R, Bain J, McGregor A. Studies on the host-parasite relationship in Schistosoma mansoni–infected mice: the immunological dependence of parasite egg excretion. Immunology. 1979;35:771–778. [PMC free article] [PubMed] [Google Scholar]
- 29.Sher A, Coffman RL, Hieny S, Scott P, Cheever AW. Interleukin 5 is required for the blood and tissue eosinophilia but not granuloma formation induced by infection with Schistosoma mansoni. . Proc Natl Acad Sci USA. 1990;87:61–65. doi: 10.1073/pnas.87.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boros DL, Pelley RP, Warren KS. Spontaneous down-modulation of granulomatous hypersensitivity in Schistosoma mansoni. . JImmunol. 1975;114:1437–1441. [PubMed] [Google Scholar]
- 31.Vella AT, Pearce EJ. CD4+ Th2 response induced by Schistosoma mansoniegg develops rapidly, through an early, transient, Th0-like stage. J Immunol. 1992;148:2283–2290. [PubMed] [Google Scholar]
- 32.Sher A, Hieny S, James SL, Asofsky R. Mechanisms of protective immunity against Schistosoma mansoniinfection in mice vaccinated with irradiated cercariae. II. Analysis of immunity in hosts deficient in T lymphocytes, B lymphocytes, or complement. J Immunol. 1982;128:1880–1884. [PubMed] [Google Scholar]
- 33.Amiri P, Haak-Frendscho M, Robbins K, McKerrow JH, Stewart T, Jardieu P. Anti–immunoglobulin E treatment decreases worm burden and egg production in Schistosoma mansoni–infected normal and interferon γ knockout mice. J Exp Med. 1994;180:43–51. doi: 10.1084/jem.180.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King CL, Xianli J, Malhotra I, Liu S, Mahmoud AAF, Oettgen HC. Mice with targeted deletion of the IgE gene have increased worm burdens and reduced granulomatous inflammation following primary infection with Schistosoma mansoni. . J Immunol. 1997;158:294–300. [PubMed] [Google Scholar]
- 35.Brunet LR, Finkelman FD, Cheever AW, Kopf MA, Pearce EJ. IL-4 protects against TNF-α–mediated cachexia and death during acute schistosomiasis. J Immunol. 1997;159:777–785. [PubMed] [Google Scholar]
- 36.Yap G, Cheever AW, Caspar P, Jankovic D, Sher A. Unimpaired down-modulations in CD8 T cell– and gamma interferon–deficient mice chronically infected with Schistosoma mansoni. . Infect Immun. 1997;65:2583–2586. doi: 10.1128/iai.65.7.2583-2586.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36a.Wynn, T.A., A.W. Cheever, M.E. Williams, S. Hieny, P. Caspar, R. Kühn, W. Müller, and A. Sher. Interleukin-10 regulates liver pathology in acute murine schistosomiasis mansoni but is not required for immune down-modulation of chronic disease. J. Immunol. In press. [PubMed]
- 37.Sher A, McIntyre S, von Lichtenberg F. Schistosoma mansoni: kinetics and class specificity of hypergammaglobulinemia induced during murine infection. Exp Parasitol. 1977;41:415–422. doi: 10.1016/0014-4894(77)90114-x. [DOI] [PubMed] [Google Scholar]
- 38.Rocklin RE, Brown AP, Warren KS, Pelley RP, Houba V, Siongok TKA, Ouma J, Sturrock RF, Butterworth AE. Factors that modify the cellular-immune response in patients infected by Schistosoma mansoni. . J Immunol. 1980;125:1916–1923. [PubMed] [Google Scholar]
- 39.Boros DL, Amsden AF, Hood AT. Modulation of granulomatous hypersensitivity. IV. Immunoglobulin and Ab production by vigorous and immunomodulated liver granulomas of Schistosoma mansoni–infected mice. J Immunol. 1982;128:1050–1053. [PubMed] [Google Scholar]
- 40.Scott P, Natovitz P, Sher A. B lymphocytes are required for the generation of T cells that mediate healing of cutaneous leishmaniasis. J Immunol. 1986;137:1017–1021. [PubMed] [Google Scholar]
- 41.Chen J, Trounstein M, Alt FW, Kurahara C, Loring JF, Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JHlocus. Int Immunol. 1993;6:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 42.Hernandez HJ, Wong Y, Stadecker MJ. In infection with Schistosoma mansoni, B cells are required for T helper 2 cell responses but not for granuloma formation. J Immunol. 1997;158:4832–4837. [PubMed] [Google Scholar]
- 43.Liu YL, Wu Y, Ramarathinam L, Guo Y, Huszar D, Trounstein M, Zhao M. Gene-targeted B-deficient mice reveal a critical role for B cells in the CD4 T cell response. Int Immunol. 1995;8:1353–1362. doi: 10.1093/intimm/7.8.1353. [DOI] [PubMed] [Google Scholar]
- 44.Wynn TA, Cheever AW, Jankovic D, Poindexter RW, Caspar P, Lewis FA, Sher A. An IL-12-based vaccination method for preventing fibrosis induced by schistosome infection. Nature. 1995;376:594–596. doi: 10.1038/376594a0. [DOI] [PubMed] [Google Scholar]
- 45.Vellupillai P, Harn DA. Oligosaccharide-specific induction of IL-10 production by B220+ cells from schistosome infected mice: a mechanism for regulation of CD4+T-cell subsets. Proc Natl Acad Sci USA. 1994;91:18–23. doi: 10.1073/pnas.91.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dombrowicz D, Flamand V, Brigman KK, Koller BH, Kinet J-P. Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor α chain gene. Cell. 1993;75:969–976. doi: 10.1016/0092-8674(93)90540-7. [DOI] [PubMed] [Google Scholar]
- 47.Jankovic D, Kullberg MC, Dombrowicz D, Barbieri S, Caspar P, Wynn TA, Paul WE, Cheever AW, Kinet J-P, Sher A. FcεRI-deficient mice infected with Schistosoma mansonimount normal Th2-type responses while displaying enhanced liver pathology. J Immunol. 1997;159:1868–1875. [PubMed] [Google Scholar]
- 48.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 49.Hazenbos WLW, Gressner JE, Hofhuis FMA, Kuipers H, Meyer D, Heijnen IAFM, Schmidt RE, Sandor M, Capel PJA, Daeron M, et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in FcγRIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 50.Weinstock JV, Chensue SW, Boros DL. Modulation of granulomatous hypersensitivity. V. Participation of histamine receptor positive and negative lymphocytes in the granulomatous response of Schistosoma mansoni–infected mice. J Immunol. 1983;130:423–427. [PubMed] [Google Scholar]
- 51.Sylvester DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 52.Wolf SD, Dittel BN, Hardardottir F, Janeway CA., Jr Experimental autoimmune encephalomyelitis induction in genetically B cell–deficient mice. J Exp Med. 1996;184:2271–2278. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]