

Abstract
Proteins subject to proteolysis or phosphorylation during apoptosis are commonly precipitated by autoantibodies found in the serum of patients with systemic lupus erythematosus (SLE). We screened a panel of murine monoclonal and human monospecific sera reactive with known autoantigens for their ability to selectively precipitate phosphoproteins from apoptotic Jurkat T cell lysates. Sera known to recognize the U1–small nuclear ribonucleoprotein (snRNP) complex (confirmed by their ability to precipitate U1–snRNA) selectively precipitated a phosphoprotein complex (pp54, pp42, pp34, and pp23) from apoptotic lysates. Monoclonal antibodies reactive with U1–snRNP proteins precipitated the same phosphoprotein complex from apoptotic lysates. The phosphorylation and/or recruitment of these proteins to the U1–snRNP complex is induced by multiple apoptotic stimuli (e.g., Fas ligation, gamma irradiation, or UV irradiation), and is blocked by overexpression of bcl-2. The U1–snRNP-associated phosphoprotein complex is immunoprecipitated by monoclonal antibodies reactive with serine/arginine (SR) proteins that comprise a structurally related family of splicing factors. The association of phosphorylated SR proteins with the U1–snRNP complex in cells undergoing apoptosis suggests a mechanism for regulation of alternative splicing of apoptotic effector molecules.
Components of ribonucleoproteins (RNPs)1 such as Ro, La, heterogeneous nuclear (hnRNP), and small nuclear (snRNP) are commonly recognized by autoantibodies found in the serum of patients with autoimmune disease (1–4). The mechanisms by which these and other autoantigens escape tolerance are largely unknown. The observation that keratinocytes subjected to ultraviolet radiation express autoantigens such as Ro, La, and the U1-70 kD snRNP protein at cell surface blebs suggests that apoptotic cells may play an important role in the production of autoantibodies (5–7). This is supported by experiments demonstrating the development of autoantibodies after immunization of mice with apoptotic cells (8). Proteolytic cleavage of at least 13 known protein autoantigens by individual interleukin-1β converting enzyme (ICE) family proteases (now collectively termed cysteine protease with aspartic acid substrate specificity, or “caspases” [9]) during programmed cell death further supports this hypothesis. To date, over half of all caspase targets are autoantigens or are constituents of larger complexes that contain a protein that is cleaved, and include the U1-70 kD snRNP (10), poly A ribose polymerase (PARP; reference 11), DNA-dependent protein kinase (DNA-PK; 12), hnRNP C1 and C2 (13), lamins A, B, and C (14), the nuclear mitotic apparatus protein (NuMA; 15, 16), topoisomerases 1 and 2 (16), the nucleolar protein UBF/NOR-90 (16), and α fodrin (17, 18).
Although proteolysis could expose novel epitopes required for the production of autoantibodies, only a fraction of the known autoantigens are cleaved during apoptosis. Recently, we reported that phosphoproteins are commonly precipitated from apoptotic cell extracts by autoantibodies derived from patients with systemic lupus erythematosus (SLE), suggesting that protein modifications accompanying apoptosis might generally predispose to autoantibody formation (19). We previously identified seven phosphoproteins (termed pp200, pp54, pp46, pp42, pp34, pp23, and pp17) in Jurkat T cells that are specifically precipitated with autoimmune sera in response to apoptotic stimuli (19). We also showed that a serine kinase activity is present in immunoprecipitates prepared from apoptotic Jurkat cell extracts using sera from patients with SLE and SLE overlap syndromes. We proposed that phosphorylation of autoantigens may be a common sequela of apoptotic cell death, and we postulated that these phosphoproteins, like other kinase substrates, such as c-jun, may be involved in the effector arm of the cell death pathway.
Well-characterized, monospecific human sera have been used in several recent studies to identify autoantigens that are cleaved during apoptosis (12, 16). We have used a similar approach to identify autoantigens that are selectively phosphorylated during apoptosis. Although most of the sera did not precipitate phosphoproteins from radiolabeled apoptotic lysates, five sera known to recognize the U1–snRNP complex precipitated phosphoproteins migrating with apparent molecular masses of 54, 42, 34, and 23 kD by SDS-PAGE. A series of human autoimmune sera directed against the U1–snRNP, but not the U2–snRNP, also coprecipitated this same phosphoprotein complex. Identical results were obtained using anti-U1A human variable domain antibody fragments and monoclonal antibodies directed against individual components of U–snRNPs. Because the relative migration of these U1–snRNP-associated phosphoproteins resembled the serine/arginine (SR) complex of splicing factors, we used antibodies reactive with SR proteins to precipitate phosphoproteins from apoptotic lysates. A monoclonal antibody specific for a phosphoepitope common to all SR proteins (mAb104) and a monoclonal antibody specific for the phosphorylated form of the SR protein SC35 precipitated a similar phosphoprotein complex from these lysates. The identification of SR proteins as potential substrates for a serine kinase that is activated during apoptosis has important implications for understanding cell death pathways, RNA splicing, and the immune response in diseases that are characterized by the development of autoantibodies.
Materials and Methods
Cell Culture.
Jurkat cells were grown in 5% CO2 at 37°C using RPMI 1640 (BioWhittaker, Inc., Walkersville, MD) supplemented with 9% heat-inactivated FCS (HI-FCS; Tissue Culture Biologicals, Tulare, CA), penicillin, and streptomycin (Mediatech, Inc., Herndon, VA). Cells were grown and harvested at mid-log phase. Jurkat T cells engineered to stably overexpress bcl-2 (or empty vector), a gift from John Reed (the La Jolla Cancer Research Foundation, La Jolla, CA), were grown in RPMI medium as described above supplemented with G418 (GIBCO BRL, Gaithersburg, MD) at a final concentration of 500 μg/ml. Protein overexpression was confirmed by Western blotting.
Metabolic Labeling.
Labeling was performed as described (19). In brief, Jurkat cells were incubated at a density of 2 × 106 cells/ ml in labeling medium containing the following: 45% RPMI 1640, 45% RPMI 1640 lacking either phosphate (GIBCO BRL) or methionine and cysteine (GIBCO BRL), 2 mM glutamine (Mediatech, Inc.), 5% HI-FCS, and 5% HI-FCS that had been dialyzed to equilibrium against a buffer containing 10 mM Hepes and 140 mM NaCl (Sigma Chemical Co., St. Louis, MO). [32P]orthophosphate or [35S]methionine and cysteine (Dupont-NEN, Boston, MA) was added at a concentration of 0.1 mCi/ml. Cells were incubated at 37°C for 10–16 h to allow the cells to reach steady state before each treatment, unless otherwise indicated. For two-dimensional tryptic phosphopeptide mapping experiments, cells were labeled for 2 h, followed by a 3-h stimulation with anti-Fas antibodies (7C11) in labeling media composed of 90% RPMI 1640 lacking phosphate, 2 mM glutamine, 10% dialyzed HI-FCS, and 0.15 mCi/ml 32P-orthophosphate.
Cell Lysis.
Lysis of cells was performed using NP-40 (Sigma Chemical Co.) lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris, pH 7.8, and 1 mM EDTA). NP-40 lysis buffer was supplemented immediately before use with 1 mM sodium vanadate (Sigma Chemical Co.) and a 100× protease inhibitor cocktail prepared by dissolving 10 mg chymostatin, 1.5 mg leupeptin, 7 mg pepstatin A, 850 mg phenylmethylsulfonyl fluoride, 500 mg benzamidine, and 5 mg aprotonin in 50 ml of ethanol by stirring overnight (20). The solution was sterilized by filtration and stored at room temperature. All chemicals were purchased from Sigma Chemical Co. After addition of 1 ml lysis buffer, the lysate was incubated on ice for 30 min, centrifuged in a refrigerated microfuge (5402; Eppendorf Inc., Hamburg, Germany) at 14,000 rpm for 15 min, and then the supernatant was used immediately for each experiment.
UV Irradiation.
Labeled Jurkat cells were plated on 100 × 15-mm polystyrene petri dishes (Nunc, Thousand Oaks, CA) at a concentration of 2 × 106 cells/ml and irradiated in a Stratalinker 2400 (Stratagene, La Jolla, CA) at a distance of 9 cm for 18 s. After irradiation, cells were incubated at 37°C for the indicated times before harvesting.
Gamma Irradiation.
Labeled cells were placed in a 50-ml conical tube and irradiated at a dose of 3,300 rad from a Cesium 137 source using an irradiator (Gammacell 1000; Nordion International, Kanata, Ontario, Canada). After irradiation, cells were placed in culture dishes at 37°C and incubated for the indicated times before harvesting.
Cellular Activation.
Labeled Jurkat cells were treated with the following antibodies: anti-Fas antibody 7C11 (provided by Michael Robertson, Indiana University, Bloomington, IN) from hybridoma supernatant at a final dilution of 1:500; and anti-CD3 antibody (Coulter Immunology, Hialeah, FL) at a concentration of 5 μg/ml followed by goat anti–mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) at the same concentration. Cells were incubated at 37°C for the indicated times before harvesting.
Immunoprecipitation and Western Blot Analysis.
Lysates were precleared once with 25 μl of a 50% solution of protein A–Sepharose (Pharmacia, Uppsala, Sweden) in PBS and 5 μg rabbit anti–mouse (RAM) IgG (Jackson ImmunoResearch Laboratories) for 1 h, followed by two preclears with protein A–Sepharose overnight. Mouse monoclonal antibodies (5 μg) and 5 μg RAM (IgG or IgM), or 2 μl patient serum alone were used in precipitation experiments. Immunoprecipitation experiments using anti-U1A human antibody fragments were performed as described (21). The following human polyclonal antibodies were stored at −70°C until used: anti-Ro, anti-La, anti–Smith complex (Sm), anti–Jo-1, antinucleolar, anticentromere, anti–Scl-70, anti-DNA, and anti-U1– RNP (Arthritis Foundation/CDC Reference Sera, Atlanta, GA); anti-Th/To, anti–U3-fibrillarin, anti-signal recognition particle (SRP), anti–PL-7, and anti–PL-12 (T. Medsger and N. Fertig, University of Pittsburgh School of Medicine, Pittsburgh, PA); two different anti-U1/U2 monospecific sera (Sera Ya and Ga; reference 22) and anti-SRP (J. Craft, Yale University School of Medicine, New Haven, CT); anti-NuMA (serum AS), and anti-UBF (serum JO; E. Tan and C. Casiano, The Scripps Institute, La Jolla, CA); anti–RNA polymerase I/III and II (serum KA), anti–polymerase I/III (serum IM), anti-Th/To, anti–U3-fibrillarin, anti-Ku, and anti–Scl-70 (M. Kuwana, Keio University Medical School, Tokyo, Japan); anti-ribosomal P and antihistone/U– RNP (Immunovision Inc., Springdale, AZ); anti–U1-70 kD snRNP protein (A. Rosen, the Johns Hopkins University School of Medicine, Baltimore, MD); anti-sp140, and anti-sp100 (D. Bloch, Massachusetts General Hospital, Boston, MA); seven human sera specific for the U1–snRNP complex (B83, B152, B175, H34, H165, K4, and L41) and a control serum specific for both U1 and U2–snRNPs (V26) have been reported previously (23, 91). Serum from patients with SLE and mixed connective tissue disease (MCTD) with high titers of antibodies against Sm and RNP components, respectively, were provided by P.H. Schur (Brigham and Women's Hospital, Boston, MA). Autoimmune serum capable of precipitating pp54, pp42, pp34, and pp23 (corresponding to patients 1, 8, 11, and 12) were described previously (19). Serum from a fifth patient (patient 3) also coprecipitated these proteins but was unavailable in sufficient quantity to complete the studies described below (19). The following mouse monoclonal antisera were stored at −70°C until used: anti-lamin B (E3), and anti–lamin A+B (E6; E.A. Nigg, University of Geneva, Geneva, Switzerland); anti–lamin B (Calbiochem-Novabiochem Corp., San Diego, CA); anti–proliferating cell nuclear antigen (PCNA; Zymed Laboratories, Inc., South San Francisco, CA); anti–DNA-PK (D. Weaver, Dana Farber Cancer Institute, Boston, MA); two monoclonal anti-Ku antibodies (C. Zhang, Dana Farber Cancer Institute, Boston, MA); anti-Ki67 (D. Bloch, Massachusetts General Hospital, Boston, MA); anti–U1-70 kD (reference 24; S. Hoch, the Agouron Institute, La Jolla, CA); anti-Sm Y12 (J. Craft, Yale University School of Medicine, New Haven, CT); anti-U1A/U2B′′ 9A9 (25); anti-U2B′′ 4G3 (25); anti-Ro60 2G10 (26); anti-Ro52 and anti-La SW5 (27) have been described previously; mAb104 monoclonal directed against SR proteins (reference 28; R. Reed, Harvard University School of Medicine, Boston, MA); anti-SC35 (Sigma Chemical Co.); and antifibrillarin monoclonal antibodies (72B9.D31 and 17C12.G9) and two monoclonal antibodies directed against other U3–snRNP components (7G3.B7 and 6G10.D3; K.M. Pollard, Scripps Institute, La Jolla, CA). Serum from healthy control patients was a gift from P. Fraser (Brigham and Women's Hospital, Boston, MA). Immunoprecipitations were performed after addition of 1% BSA (Intergen Company, Purchase, NY) in PBS to a total volume of 500 μl and rotation in a 4°C cold room for 2–24 h. Comparison of precipitates showed no difference between incubation times for periods of up to 72 h. Precipitates were harvested by centrifuging for 15 s at 14,000 rpm in a refrigerated Eppendorf microfuge, washing three times with NP-40 lysis buffer supplemented with protease inhibitor cocktail, resuspending in SDS loading buffer with 9% 2-mercaptoethanol, boiling for 5 min, and separating by PAGE as described (29). Proteins were transferred to nitrocellulose (Schleicher & Schuell, Inc., Keene, NH) for Western blotting or tryptic mapping experiments, or to polyvinylidene difluoride (PVDF; Dupont-NEN, Boston, MA) for phosphoamino acid analysis, and either exposed for autoradiography or subjected to Western blot analysis as indicated (30). The anti–bcl-2 mouse monoclonal antibody 4D7 (PharMingen, San Diego, CA) was used for blotting studies at a dilution of 1:1,000. Nitrocellulose blots were blocked with 5% Blotto (Bio-Rad Laboratories, Hercules, CA) in PBS overnight at 4°C. Bands were visualized using RAM conjugated to horseradish peroxidase (Amersham Corp., Arlington Heights, IL) at a dilution of 1:7,500 in 5% Blotto in PBS, and developed using ECL chemiluminescence performed according to the manufacturer's instructions (Amersham Corp.).
Phosphoamino Acid Analysis.
Immunoprecipitates that had been electrophoresed and transferred to PVDF were rinsed thoroughly with water, exposed for radiography, and then appropriate bands were excised with a razor blade. The radiolabeled bands were then subjected to acid hydrolysis as described (31).
RNA Isolation and Identification.
Immunoprecipitates from 32P-labeled Jurkat cells were prepared as described above. After the third NP-40 lysis buffer wash, the immunoprecipitate was digested in a volume of 300 μl for 1 h at 37°C in a solution containing 50 μg/ml proteinase K (Sigma Chemical Co.), 10 mM Tris, pH 7.8, 10 mM EDTA, and 0.5% SDS. The RNA was isolated after two extractions with a phenol/chloroform/isoamyl alcohol (25:24:1) mixture (GIBCO BRL). The RNA was precipitated overnight at −70°C after the addition of 20 μl 3 M sodium acetate, 400 μl ethanol, and 10 μg transfer RNA (Sigma Chemical Co.) as a carrier. The pellet was obtained after a 15-min centrifugation in an Eppendorf centrifuge maintained at 4°C. The pellet was washed once with 70% ethanol, dried in a fume hood, and subjected to PAGE on 6% sequencing gels. A small amount of whole cell lysate was also processed as above and included as an internal standard on each gel.
Two-Dimensional Phosphopeptide Analysis.
Two-dimensional tryptic phosphopeptide mapping was performed as described (32) using trypsin (Worthington Biochemical Corp., Freehold, NJ) at a concentration of 0.1 mg/ml in 50 mM ammonium bicarbonate. Plates were exposed to film at −70°C with an intensifying screen for 2 d.
Results
Autoimmune Sera Precipitate Phosphoproteins from Lysates Prepared from Jurkat T Cells Undergoing Fas-induced Apoptosis.
Sera reactive with known autoantigens were first tested for their ability to precipitate the expected protein or complex from extracts prepared from 35S-labeled Jurkat cells, as detected by SDS-PAGE followed by autoradiographic exposure. Sera that precipitated ambiguous patterns of proteins were subjected to further analysis by Western blotting of both whole cell Jurkat extracts and immunoprecipitates prepared as above, to confirm that the well-characterized sera precipitated the expected target antigen. Most of the sera were derived from patients with autoimmune disease. In addition, six murine monoclonal antibodies reactive with known autoantigens (Ku, DNA-PK, lamins A and B, Ki67, and PCNA) were included in this screen.
Jurkat cells metabolically labeled with 32P-orthophosphate were cultured for 3 h in the absence or presence of a monoclonal antibody reactive with Fas (anti-7C11), solubilized in NP-40 lysis buffer, and immunoprecipitated using patient serum or monoclonal antibodies as previously described (19). Immunoprecipitates were separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and then subjected to autoradiography. Most sera did not precipitate unique phosphoproteins from apoptotic lysates. For example, sera reactive with nuclear proteins implicated in DNA replication, binding, or repair (i.e., Ku, DNA-PK, PCNA, Scl-70, and histone) failed to reproducibly precipitate new phosphoproteins from apoptotic Jurkat cell extracts (data not shown). Similarly, proteins present in the nuclear matrix or involved in mitosis (i.e., lamins A and B, centromere A and B, Ki-67, NuMA, sp140, and sp100), the nucleolus (i.e., Th/To, UBF/NOR-90, RNA polymerase I, II, and III, and U3–snRNPs), or cytoplasmic components of the translational apparatus (i.e., Jo-1, Pl-7, Pl-12, SRP, and ribosomal P) were unmodified in these experiments (data not shown). In contrast, several sera specific for U–snRNP complexes precipitated phosphoproteins of 54, 42, 34, and 23 kD from apoptotic Jurkat cell extracts (Fig. 1 A). The phosphorylation of these proteins did not result from a nonspecific, general increase in kinase activity after Fas engagement, as 32P-labeled, whole cell extracts prepared from untreated and apoptotic cells were identical when analyzed by SDS PAGE (data not shown). Moreover, this pattern was similar to that observed using four distinct sera described in our previous report, suggesting that these four proteins (termed pp54, pp42, pp34, and pp23) may be previously unrecognized components of U–snRNP complexes (19). As reported previously (19), the constitutive phosphorylation of La (Fig. 1 A, Ro and La) was unaltered in cells undergoing apoptosis.
Figure 1.


Human autoimmune sera specific for U–snRNP complexes precipitate phosphoproteins from apoptotic Jurkat cell lysates. (A) Jurkat cells were labeled with 32P orthophosphate and lysed either before (−) or 3 h after (+) the addition of anti-Fas 7C11. Proteins were then precipitated using the indicated autoimmune serum, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and exposed for autoradiography. Individual sera are described in detail in Materials and Methods. U-serum 1, Immunovision antihistone/RNP; U-serum 2, CDC/AF reference serum 4 (anti-U1–RNP); U-serum 3, serum Ga; U-serum 4, serum Ya; U-serum 5, CDC/AF reference serum 5 (anti-Sm). The relative migration of molecular mass markers in kilodaltons is indicated on the left side of the gel. (B) Immunoprecipitation from 35S-labeled Jurkat cells. Jurkat cells were labeled with 35S-methionine and cysteine and lysed either before (−) or 3 h after (+) the addition of anti-Fas 7C11 before immunoprecipitation using sera derived from the indicated patient. Immunoprecipitates were separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and then subjected to autoradiographic analysis. The relative migration of molecular mass markers in kilodaltons is indicated on the left side of each panel. Bands corresponding to the U–snRNP proteins A, B, B′, and C are indicated on the right side of the panel.
Autoimmune Sera Coprecipitate U–snRNP Proteins.
Immunoprecipitates were prepared from 35S-labeled lysates prepared before (−) or 3 h after (+) Fas ligation (Fig. 1 B). Although immunoprecipitates prepared from apoptotic and nonapoptotic lysates contained 35S-labeled proteins migrating similarly to the phosphoproteins identified in Fig. 1 A, we were unable to establish the relationship between the 32P- and 35S-labeled proteins. Nevertheless, we did not observe the appearance of new 35S-labeled proteins corresponding to components of the phosphoprotein complex. Moreover, all five sera precipitated U–snRNP components (e.g., the A, B, B′, and C proteins, indicated by arrows) from labeled Jurkat cell extracts (Figs. 1 B and 2, also see below). Six other sera from patients with SLE and MCTD possessing high titers of antibodies against Sm or RNP components (as determined by ELISA assay), also precipitated U–snRNP components from 35S-labeled Jurkat cell extracts, as well as all four phosphoproteins from apoptotic cell extracts prepared from 32P-labeled Jurkat cells (data not shown). These results demonstrate that autoimmune sera capable of precipitating U–snRNP proteins coprecipitate a phosphoprotein complex containing pp54, pp42, pp34, and pp23 from apoptotic Jurkat cell lysates.
Figure 2.

Coprecipitation of U1–snRNA using selected autoantisera. Jurkat cells were labeled with 32P-orthophosphate and solubilized in NP-40 lysis buffer. After immunoprecipitation with the indicated serum, RNA was extracted and separated on 6% sequencing gels before drying and autoradiographic exposure. The relative migration of known RNA moieties is depicted on the right side of the figure. The serum specificity is indicated above each sample. Lanes are numbered at the bottom of the panel. Lanes 1–4, patients 1, 8, 11, 12 (19); U-serum 1, Immunovision antihistone/RNP; U-serum 2, CDC/AF reference serum 4 (anti-U1–RNP); U-serum 3, serum Ga; U-serum 4, serum Ya; U-serum 5, CDC/AF reference serum 5 (anti-Sm); U-serum 6, anti–U1-70 kD serum (gift of A. Rosen).
Autoimmune Sera Coprecipitate the U1–snRNA Molecule and pp54, pp42, pp34, and pp23.
To further establish that pp54, pp42, pp34, and pp23 are components of U–snRNP complexes, we used all five sera shown in Fig. 1 B, four sera previously reported to precipitate these four phosphoproteins (patients 1, 8, 11, and 12; reference 19), and control sera to identify the RNA molecules present in individual immunoprecipitates. Jurkat cells metabolically labeled with 32P-orthophosphate were solubilized in NP-40 lysis buffer, RNA was extracted from washed immunoprecipitates, separated on a 6% polyacrylamide gel, and subjected to autoradiography. As shown in Fig. 2, all sera capable of precipitating the phosphoprotein complex also precipitate the U1–snRNA. A similar result was obtained for another well-characterized serum previously shown to recognize the U1-70 kD protein (Fig. 2, lane 6, U-serum 6, a gift of A. Rosen; reference 10). We noted that one sample (U-serum 2, Fig. 2, lane 10) specifically precipitated the U1– but not the U2–snRNA molecule, suggesting that pp54, pp42, pp34, and pp23 may be components of the U1–snRNP complex. To confirm this observation, we screened seven additional human sera previously shown to specifically precipitate the U1–snRNP complex (91), and serum V26 (which precipitates both the U1– and U2–snRNPs; reference 23) for their ability to coprecipitate pp54, pp42, pp34, and pp23 from 32P-labeled, apoptotic Jurkat cell extracts. All 7 sera coprecipitated the U1–snRNA (Fig. 3 A) and all four phosphoproteins (Fig. 3 B). Identical results were obtained using serum V26 (data not shown). Interestingly, individual sera precipitated phosphoproteins of varying intensity (e.g., all four phosphoproteins are precipitated by serum B83, Fig. 3 B, lane 2, but mainly pp42 and pp54 are precipitated by serum H165, Fig. 2, lane 10). This may result from heterogenous populations of antibodies directed against individual U1–snRNP components, or by antibodies that recognize the phosphoprotein directly. None of the control antibodies directed against Ro, La, ribosomal P, or SRP precipitated either the U–snRNAs or any of the four phosphoproteins (data not shown). Several sera also precipitated phosphoproteins between 96 and 200 kD that were no longer detected after Fas stimulation (e.g., B152, H34, and K4, Fig. 3 B, lanes 3, 7, and 11), again suggesting that these sera likely recognize heterogeneous epitopes of the U1– snRNP particle. It is unknown if this represents dephosphorylation, caspase cleavage, or dissociation of these phosphoproteins from the immunoprecipitate after the apoptotic stimulus. These results suggest that the U1–snRNP is a dynamic particle that is altered by caspases (U1-70 kD protein; reference 10), and potentially by kinases (pp54, pp42, pp34, and pp23; reference 19) and phosphatases (the high molecular mass protein complex shown in Fig. 3 B) during apoptosis.
Figure 3.


U1-specific autoantisera coprecipitate the U1–snRNA molecule and pp54, pp42, pp34, and pp23 from apoptotic extracts. (A) Jurkat cells were labeled with 32P-orthophosphate and lysed in NP-40 lysis buffer. After immunoprecipitation with the indicated serum, RNA was extracted and separated on 6% sequencing gels before drying and autoradiographic exposure. Patient sera specific for the U1–snRNP complex were used in lanes 1–7. A patient serum (V26, lane 8) capable of precipitating both the U1– and U2–snRNPs is shown for comparison. The relative migration of the U1– and U2–snRNAs is depicted on the right side of the figure. (B) Jurkat cells were labeled with 32P-orthophosphate and lysed either before (−) or 3 h after (+) the addition of anti-Fas (7C11) before immunoprecipitation using sera derived from the indicated patient. Immunoprecipitates were separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and subjected to autoradiographic analysis. Sera correspond to the seven U1-specific autoantisera shown in Fig. 3 A. The relative migration of molecular size markers in kilodaltons is indicated on the left side of the figure. Bands corresponding to pp54, pp42, pp34, and pp23 are shown on the right side of the panel. A high molecular mass complex is indicated with a large arrowhead. Lanes are numbered at the bottom of the figure.
Monoclonal Antibodies Directed Against U1–snRNP Components Precipitate pp54, pp42, pp34, and pp23 from Apoptotic Jurkat Cell Lysates.
To confirm our results described above using human autoimmune serum, and to determine whether pp54, pp42, pp34, and pp23 exist in both the U1– and U2–snRNP splicing complexes, we used a series of monoclonal antibodies directed against proteins common or unique to each U–snRNP complex. Specifically, we tested antibodies directed against the U1-70 kD component of the U1–snRNP, the U2B′′ protein of the U2–snRNP (4G3), the anti-Sm monoclonal antibody Y12, and a monoclonal antibody (9A9) that recognizes an epitope common to both U1A and U2B′′ for their ability to precipitate pp54, pp42, pp34, and pp23 from apoptotic extracts. Jurkat cells metabolically labeled with 32P-orthophosphate were cultured for 3 h in the absence or presence of a monoclonal antibody reactive with Fas (anti-7C11), solubilized in NP-40 lysis buffer, and individually immunoprecipitated using each of these antibodies; control antibodies directed against other RNA-binding proteins included monoclonal antibodies against Ro60, Ro52, La, and the anti-T cell intracellular antigen-related protein (TIAR) antibody 6E3 (33). As shown in Fig. 4 A, the Smith antibody Y12, and the 9A9 monoclonal antibody specific for the U1A and U2B′′ proteins (both of which recognize components common to both U1– and U2–snRNPs) precipitate all four phosphoproteins from apoptotic Jurkat cell lysates (Fig. 4, A and B, lanes 2 and 8). These bands are absent in the lanes corresponding to the immunoprecipitation using anti-U2B′′ (Fig. 4, A and B, lane 6) and anti–U1-70 kD monoclonal antibodies (A and B, lane 4, see Discussion). Interestingly, increased phosphorylation of a 90-kD protein is observed after Fas stimulation when immunoprecipitates are prepared using anti-U2B′′ (4G3) antibody (Fig. 4, A and B, lane 6), and on a short exposure of the lanes corresponding to the U1A/U2B′′ immunoprecipitate (Fig. 4, A and B, lanes 7 and 8, data not shown), suggesting that a specific phosphoprotein (henceforth called pp90) is associated with the U2–snRNP during apoptosis. Bands corresponding to pp90, pp54, pp42, pp34, and pp23 are absent using monoclonal antibodies directed against TIAR (6E3, Fig. 4, A and B, lane 10), Ro60 (lane 12), Ro52 (lane 14), La (lane 16), or the putative apoptosis effector TIA-1 (data not shown), another autoantigen that is known to be reversibly phosphorylated during Fas-mediated apoptosis, but at an earlier time point (19, 34, 35). The same experiment performed in cells labeled with [35S]methionine and cysteine (Fig. 4 B) demonstrates no difference between immunoprecipitates prepared from apoptotic and nonapoptotic cell extracts, consistent with the results shown in Fig. 1 B. Phosphoamino acid analysis of all four proteins precipitated using anti-U1A/U2B′′ (9A9) demonstrates exclusive phosphorylation of pp54, pp42, pp34, and pp23 on serine residues (Fig. 4 C).
Figure 4.



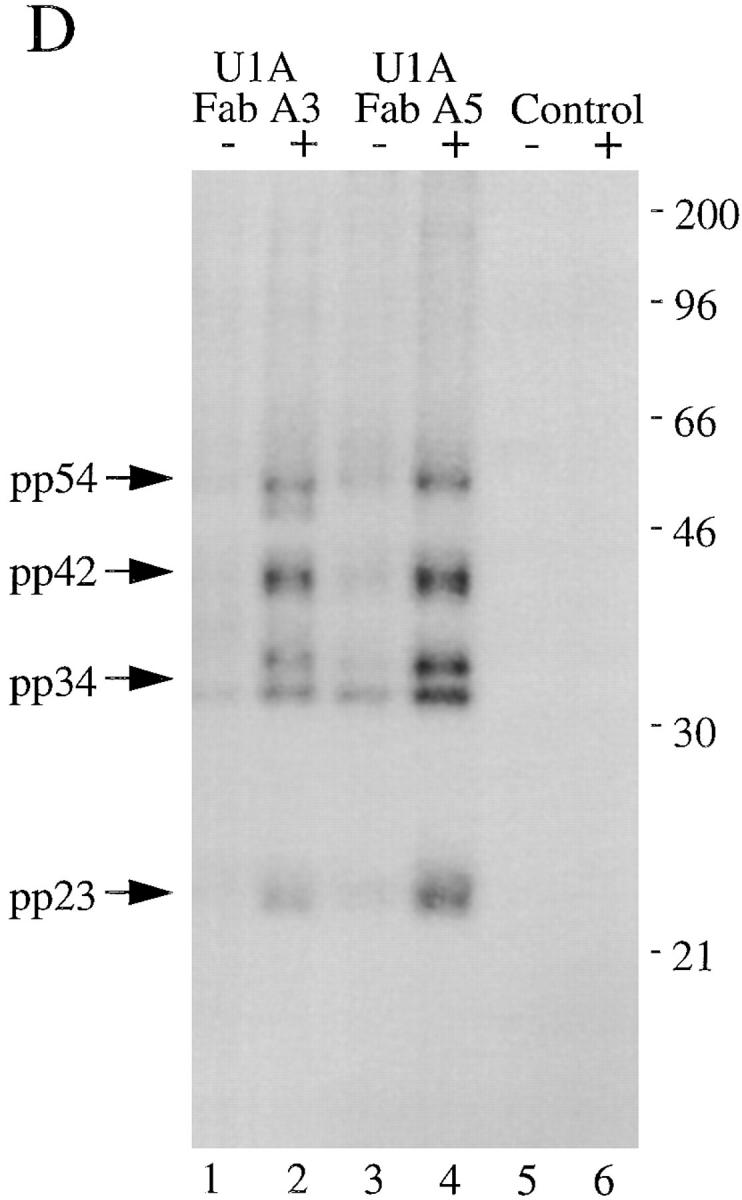
Monoclonal antibodies directed against U1–snRNP components precipitate pp54, pp42, pp34, and pp23 from extracts prepared from apoptotic Jurkat cells. (A) Jurkat cells were labeled with 32P-orthophosphate and lysed either before (−) or 3 h after (+) the addition of anti-Fas 7C11. Proteins were then precipitated using the indicated autoimmune serum, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and then exposed for autoradiography. The relative migration of molecular mass markers in kilodaltons is indicated on the left side of the gel. Bands corresponding to pp90, pp54, pp42, pp34, and pp23 are shown on the right side of the panel. Lanes are numbered at the bottom of the panel. (B) The identical experiment in 35S-labeled Jurkat cells. The relative migration of molecular size markers in kilodaltons is indicated on the left side of the gel. Lanes are numbered at the bottom of the figure. (C) Phosphoamino acid analysis of pp54, pp42, pp34, and pp23. Jurkat cells were labeled with 32P-orthophosphate, treated with the anti-Fas monoclonal antibody 7C11, and solubilized using NP-40 lysis buffer after 3 h. Proteins were then precipitated with the anti-U1A/U2B′′ monoclonal antibody 9A9, separated on a 12% SDS–polyacrylamide gel, transferred to PVDF, and exposed for autoradiography. Individual phosphoproteins were localized on the membrane, excised, and then subjected to acid hydrolysis. Phosphoamino acids were separated by two-dimensional electrophoresis in pH 1.9 buffer in the horizontal dimension, followed by pH 3.5 buffer in the vertical dimension before autoradiographic analysis. Individual proteins are labeled on the side of each panel. Migration of phosphoamino acid standards are labeled with circles as follows: phosphoserine (pS), phosphothreonine (pT), phosphotyrosine (pY). (D) Anti-U1A antibody fragments coprecipitate pp54, pp42, pp34, and pp23 from apoptotic Jurkat cell lysates. Labeled Jurkat cell extracts were prepared as above. Proteins were precipitated using the indicated anti-U1A antibody fragments, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and then exposed for autoradiography. The relative migration of molecular mass markers in kilodaltons is indicated on the right side of the gel. Bands corresponding to pp54, pp42, pp34, and pp23 are shown on the left side of the panel.
The failure of the U1-70 kD monoclonal antibody to precipitate pp54, pp42, pp34, and pp23 from apoptotic lysates appeared to be inconsistent with the hypothesis that these phosphoproteins are specifically associated with the U1–snRNP during apoptosis. To address this apparent paradox (see Discussion), we used two previously described human variable domain antibody fragments directed against a different, unique component of the U1–snRNP (the U1A protein) and repeated the immunoprecipitation experiments (21). Both antibodies (Fig. 4 D, lanes 1–4) coprecipitate a phosphoprotein complex containing pp54, pp42, pp34, and pp23, but not pp90. A control antibody fragment directed against bovine serum albumin precipitates only a faint, nonspecific 60-kD protein (Fig. 4 D, lanes 5 and 6). Taken together, these results demonstrate an association between a phosphoprotein complex (containing pp54, pp42, pp34, and pp23) and the U1–snRNP during apoptosis and suggests that pp90 may be associated specifically with the U2–snRNP during apoptosis.
Association of pp54, pp42, pp34, and pp23 with U–snRNPs Accompanies Apoptosis but Not T Cell Receptor Stimulation.
Previously, we had demonstrated that, in addition to death induced by Fas ligation, phosphorylated autoantigens are also immunoprecipitated during apoptosis triggered by other stimuli including gamma and UV irradiation, but not by T cell receptor stimulation (19). We repeated this experiment using the anti-U1A/U2B′′ (9A9) monoclonal antibody in immunoprecipitation experiments using 32P-labeled Jurkat lysates prepared from cells subjected to apoptotic stimuli or an activation stimulus over a 5-h time course (Fig. 5). This analysis reveals that phosphorylated autoantigens are precipitated beginning at the 3-h time point after Fas cross-linking (Fig. 5, lanes 1–4) or UV irradiation (Fig. 5, lanes 11–14), and much less intense bands are observed 5 h after gamma irradiation (Fig. 5, lanes 5–7), consistent with our initial observations (19). In contrast, ligation of the T cell receptor complex using a monoclonal antibody reactive with CD3, a stimulus that induces IL-2 production and enhances proliferation in these cells (data not shown), induced neither precipitation of phosphoproteins (Fig. 5, lanes 8–10) nor DNA fragmentation (data not shown and reference 19) over the course of this experiment.
Figure 5.

Phosphoprotein components of the U1–snRNP complex are precipitated after multiple apoptotic stimuli but not an activation stimulus. Jurkat cells were labeled with 32P-orthophosphate, treated with the indicated stimulus, and then solubilized using NP-40 lysis buffer at the indicated times. Proteins were then precipitated with anti-U1A/U2B′′, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and then exposed for autoradiography. The apoptotic stimulus is indicated above each panel. The time in hours after each stimulus is indicated above each lane. The relative migration of molecular mass markers in kilodaltons is indicated on the left side of the panel. Bands corresponding to pp54, pp42, pp34, and pp23 are shown on the right side of the panel. Lanes are numbered at the bottom of each panel.
Bcl-2 Overexpression Blocks Gamma Irradiation–induced Apoptosis and Precipitation of pp54, pp42, pp34, and pp23.
Next, we asked whether the precipitation of these phosphorylated U1–snRNP components could be blocked by overexpression of the bcl-2 protein, which has been shown to efficiently block apoptosis induced by multiple apoptotic stimuli, including gamma irradiation and UV irradiation (19, 36–39). As shown in Fig. 6, Jurkat T cells stably transformed with either bcl-2 (lanes 1–4) or empty vector (lanes 5–8) were labeled with 32P-orthophosphate and subjected to gamma irradiation. Cells were solubilized at the indicated times and lysates were precipitated using the anti-U1A/U2B′′ (9A9) monoclonal antibody. Whereas precipitation of phosphorylated pp54, pp42, pp34, and pp23 is rapidly induced in Jurkat (neo) control cells in response to gamma irradiation (Fig. 6, lanes 5–8), the intensity of the bands corresponding to the four phosphoproteins is unchanged in Jurkat (bcl-2) transformants treated with this same stimulus (Fig. 6, lanes 1–4). Short exposure of the gel in Fig. 6 showed that bcl-2 overexpression also inhibited the appearance of pp90 (data not shown). Ectopic expression of bcl-2 effectively inhibited apoptosis in response to gamma irradiation, as judged by the induction of DNA fragmentation (data not shown and reference 19). Taken together, these results demonstrate that the coprecipitation of all five phosphorylated snRNP factors correlates with the induction of apoptosis and is downstream of the inhibitory effects of bcl-2.
Figure 6.

In vivo phosphorylation of U1–snRNP components is inhibited in gamma-irradiated Jurkat cells overexpressing bcl-2. Jurkat (bcl-2) transformants (lanes 1–4) or Jurkat (neo) control transformants (lanes 5–8) were labeled with 32P-orthophosphate, subjected to gamma irradiation, solubilized in NP-40 lysis buffer, precipitated using anti-U1A/U2B′′ antibodies, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and then exposed for autoradiography. The relative migration of molecular size markers in kilodaltons is indicated on the left side of the figure. Bands corresponding to pp54, pp42, pp34, and pp23 are shown on the right side of the figure. The time, in hours, from initial exposure to gamma irradiation is indicated at the top of each lane. Lane numbers appear at the bottom of the figure.
Monoclonal Antibodies Specific for SR Splicing Factors Precipitate a Phosphoprotein Complex Containing pp54, pp42, pp34, and pp23 from Apoptotic Jurkat Cell Lysates.
The identification of pp54, pp42, pp34, and pp23 as components of the U1–snRNP complex prompted us to search for known phosphoprotein components of these splicing particles. Review of the literature identified the SR family of splicing factors as excellent candidates. Five of the most abundant SR proteins are virtually identical in molecular mass to pp54, pp42, pp34, and pp23 (i.e., SRp55, SRp40, SC35, ASF/ SF-2, and SRp20) and are phosphorylated on serine residues in vivo and in vitro (40). To test the possibility that pp54, pp42, pp34, and pp23 might be SR proteins, the experiment shown in Fig. 7 was performed. Jurkat cells metabolically labeled with 32P-orthophosphate were cultured for 3 h in the absence (−) or presence (+) of a monoclonal antibody reactive with Fas (anti-7C11), solubilized in NP-40 lysis buffer, and then immunoprecipitated using 9A9 (anti-U1A/U2B′′), mAb104 (a monoclonal antibody specific for the phospho-SR domain of these factors; reference 28), or anti-SC35 (a monoclonal antibody specific for the phosphorylated form of SC35). Labeled proteins were separated by SDS PAGE and transferred to nitrocellulose. Fig. 7 A demonstrates that both mAb104 and anti-SC35 coprecipitate the same phosphoprotein complex from apoptotic lysates. To confirm that the respective proteins precipitated with each antibody are identical, bands corresponding to pp54, pp42, pp34, and pp23 (from immunoprecipitates prepared independently using anti-SC35 and 9A9, Fig. 7 A, lanes 2 and 4) were localized by autoradiography, excised, and then subjected to two-dimensional phosphotryptic mapping. As shown for one of the bands (pp34), the phosphotryptic maps are identical (Fig. 7 B). Similar results were obtained when comparing phosphopeptide maps corresponding to pp54, pp42, and pp23 (data not shown). Taken together, these results demonstrate that a phosphoprotein complex is selectively associated with the U1–snRNP complex during apoptosis, and identify members of the SR family of splicing factors as likely components of this phosphoprotein complex.
Figure 7.

Monoclonal antibodies specific for SR proteins precipitate pp54, pp42, pp34, and pp23 from apoptotic Jurkat cell extracts. (A) Jurkat cells were labeled with 32P-orthophosphate and lysed either before (−) or 3 h after (+) the addition of anti-Fas 7C11. Proteins were then precipitated using the indicated monoclonal antibody, separated on a 12% SDS–polyacrylamide gel, transferred to nitrocellulose, and exposed for autoradiography. The relative migration of pp34 is indicated on the left side of the gel. Lanes are numbered at the bottom of the panel. The relative migration of molecular size markers in kilodaltons is indicated on the right side of the figure. (B) Two-dimensional phosphotryptic map of pp34. The 34-kD band from the U1A/U2B′′ immunoprecipitate (lane 2) and the anti-SC35 immunoprecipitate (lane 4) were excised and digested with trypsin. Phosphopeptides were separated electrophoretically at pH 1.9 in the first dimension, and by thin-layer chromatography in the second dimension before autoradiographic exposure. Direction of separation is shown by arrows. E, electrophoresis; C, chromatography; O, origin.
Discussion
Autoimmune sera have been used extensively as probes to identify, characterize, and clone proteins and RNA molecules with important cellular functions. Proteins such as La and the U1-70 kD component of the U1–snRNP complex were cloned using serum from SLE patients to screen a phage expression library (41, 42), and antibodies directed against Sm proteins and the Th/To antigen have been used to identify their function in vitro by inhibiting specific steps in mRNA and nucleolar RNA processing, respectively (43, 44). More recently, serum from such patients has proven useful for the identification of proteins that are cleaved by caspases during apoptosis. We have used a similar strategy to identify proteins that may be phosphorylated during stress-induced apoptosis (19). Before our recent report (18), only a few such proteins had been identified, and several of these proteins have been implicated as critical effectors of cell death. The TIA-1 autoantigen (Utz, P.J., and P. Anderson, unpublished data) is phosphorylated by Fas-activated serine/threonine kinase (FAST kinase) during apoptosis (35), and it is postulated that TIA-1 and a related protein, TIAR, differentially regulate RNA metabolism in response to apoptotic stimuli (Anderson, P., and N. Kedersha, unpublished observations; reference 34). Another kinase (termed JNK) phosphorylates and activates the c-jun transcription factor in response to multiple apoptotic stimuli (45–48). Overexpression of an NH2-terminal deletion mutant of the c-jun protein acts as a dominant negative suppressor of apoptosis. Recent reports, however, suggest that kinases such as JNK are activated after both lethal and nonlethal stimuli, and thus may be dispensable for execution of the apoptotic program under some circumstances (49, 50).
The U–snRNPs are a group of related nuclear particles containing a unique, uridine-rich, structural RNA (termed the U–snRNA) and a core of six or more polypeptides (51). The most abundant of these, the U1–, U2–, U5–, and U4/U6–snRNP complexes are known autoantigens (22, 51–54) and play critical roles in the splicing of pre-mRNA molecules. During splicing, U–snRNPs assemble into a macromolecular structure termed a “spliceosome” whose function is to efficiently and precisely process introns from pre-mRNA before export of the mature mRNA from the nucleus. The fidelity of this complex process is facilitated by other splicing factors that transiently associate with the U–snRNP complexes, particularly the U1– and U2–snRNPs (55, 56). Splicing factors belonging to the SR family are highly conserved proteins containing one or more RNA recognition motifs (RRMs) at their NH2 termini and a SR repeat of varying length in their COOH termini (57). Structural analysis of the SR protein ASF/SF2 demonstrates that the SR domains are required for protein phosphorylation and constitutive RNA splicing but are dispensable for alternative splicing. Targeted disruption of the RRM domains blocks RNA binding and constitutive splicing activity (58, 59). At least eight proteins containing SR domains have been identified in humans, including the U1-70 kD protein, SRp75, SRp54, SRp40, ASF/SF2, SC35, U2AF35, and SRp20. Six of these eight proteins (SRp54, SRp40, ASF/SF2, SC35, U2AF35, and SRp20) are similar in size to the proteins described above (pp54, pp42, pp34, and pp23). It has been postulated that SR proteins enhance splicing by binding to the U1–snRNP during the formation of a commitment complex, thus stabilizing the spliceosome assembly (60–62). Individual SR proteins can substitute for the U1–snRNP in in vitro splicing assays (63), and SR proteins have been implicated in regulation of both constitutive and alternative splicing of several mRNAs (57, 64–69).
Reversible protein phosphorylation is thought to regulate both constitutive and alternative mRNA splicing. Experiments using phosphatase inhibitors, nonhydrolyzable ATP analogues, or purified phosphatases in in vitro splicing reactions demonstrates a requirement for reversible protein phosphorylation for mRNA splicing (70–72), and several kinases capable of phosphorylating SR proteins have been identified. The U1-70 kD snRNP protein is an in vivo and in vitro substrate for an unidentified serine kinase that copurifies with the U1–snRNP complex (73). A second kinase, SR protein kinase-1 (SRPK-1), capable of phosphorylating multiple different SR proteins has also been identified (40, 74–76). Interestingly, this kinase is active during mitosis, phosphorylates substrates exclusively on serine residues, copurifies with snRNP complexes, and disrupts both nuclear speckles and in vitro pre-mRNA splicing (40). All five of the known in vitro substrates for SRPK-1 are identical in size to the proteins described in this report and include SRp55 (pp54), SRp40 (pp40), SC35 (pp34), ASF/SF2 (pp34), and SRp20 (pp23; references 19, 40). A related kinase, Clk/Sty, has also been shown to phosphorylate SR proteins in vitro (76, 77). Despite these intriguing reports, to date there have been no studies directly linking a serine kinase to the phosphorylation of splicing factors during stress-induced apoptosis. Experiments designed to identify whether SRPK-1, Clk/Sty, the U1-70 kD kinase or a novel serine kinase is responsible for the apoptosis-specific phosphorylation of SR proteins, and the role that this modification plays in apoptosis and alternative mRNA splicing, are in progress.
The evidence suggesting that pp54, pp42, pp34, and pp23 are components of the U1–snRNP is compelling. First, all 4 autoimmune sera from our initial report (19) and 20 sera described herein simultaneously precipitate all 4 phosphoproteins (Figs. 1 and 3 B) together with the U1 RNA (Figs. 2 and 3 A), from lysates prepared from Fas-treated Jurkat cells. Second, two different monoclonal antibodies (Y12 and 9A9) that recognize core (Sm) components of the U1– snRNP complex also precipitate these same four phosphoproteins from extracts prepared from apoptotic Jurkat cells, whereas monoclonal antibodies directed against six other RNA-binding proteins do not (Fig. 4). Third, two human variable domain antibody fragments directed against overlapping epitopes of the U1A protein coprecipitate pp54, pp42, pp34, and pp23 from apoptotic Jurkat cell extracts (Fig. 4 D). Finally, the anti-U1A/U2B′′ (9A9) monoclonal antibody precipitates all four phosphoproteins from extracts prepared from cells subjected to multiple different apoptotic stimuli but not after engagement of the T cell receptor, and the association of these phosphoproteins with the U–snRNPs is blocked in cells engineered to overexpress bcl-2 (Figs. 5 and 6). Thus, all of the experiments described using SLE sera from our initial report have been replicated using the anti-U1A/U2B′′ (9A9) monoclonal antibody (19).
It remains to be determined whether phosphoproteins are also associated with the U2– and other U–snRNP complexes during apoptosis. Two human sera and four monoclonal antibodies specific for components of the U3–snRNP complex failed to coprecipitate pp54, pp42, pp34, or pp23 (data not shown). A monoclonal antibody directed against the U2B′′ protein that uniquely precipitated the U2–snRNA (data not shown) was incapable of precipitating pp54, pp42, pp34, and pp23 in most (>10) experiments. Rarely, faint bands migrating at 54, 42, 34, and 23 kD were observed on long exposures (data not shown). While this may represent the direct association of the U2–snRNP and the SR proteins, it is equally plausible that these bands represent the association of SR proteins, U1– and U2–snRNPs in an active spliceosome complex.
The identification of pp54, pp42, pp34, and pp23 as SR proteins is suggested by several observations. The respective SR proteins SRp54, SRp42, SC35, ASF/SF2, and SRp20 have similar migration patterns on SDS PAGE and are phosphorylated exclusively on serine residues (74). SR proteins also interact with components of the spliceosome and copurify with the U1–snRNA during gel filtration analysis (78, 79) and sucrose gradient centrifugation (our unpublished data). All four proteins (pp54, pp42, pp34, and pp23) comigrate with their respective SR counterparts during two-dimensional gel electrophoresis, and anti-SC35 is capable of coprecipitating the U1–snRNA (data not shown). Finally, an identical phosphoprotein complex is precipitated by two monoclonal antibodies specific for the phosphorylated forms of SR proteins (Fig. 7; reference 28, 80). A much more difficult question is whether these proteins are stable components of the U1–snRNP that are phosphorylated de novo after an apoptotic stimulus, or rather are recruited to the U1–snRNP complex during apoptosis. SR proteins have few methionine and cysteine residues (two SR proteins have no methionines other than the initiator), perhaps explaining why bands corresponding to these proteins are not consistently observed when immunoprecipitates are prepared from 35S-labeled Jurkat cells. Although the de novo phosphorylation model is favored by the identification of at least three kinases capable of phosphorylating SR domain–containing proteins (40, 73–77), we cannot exclude the possibility that a small fraction of the phosphorylated forms of pp54, pp42, pp34, and pp23 (i.e., an amount below the level of detection obtained by metabolic labeling with [35S]methionine and cysteine) are recruited to the U1–snRNP complex during apoptosis. The answer to this important question awaits the development of other reagents, including anti-SR antibodies that recognize nonphosphorylated SR proteins and epitope-tagged SR proteins for use in transfection experiments.
The inability of the U1-70 kD monoclonal antibody to coprecipitate pp54, pp42, pp34, and pp23 appeared to contradict our argument that these phosphoproteins are associated with the U1–snRNP during apoptosis. This prompted us to test monoclonal antibodies specific for other components of the U1–snRNP for their ability to precipitate this phosphoprotein complex (Fig. 4 D). We hypothesize that only a subfraction of the U1–snRNP complexes present in a cell is associated with SR proteins. In this model, the U1A/U2B′′ (9A9), anti-Sm (Y12), and anti-U1A monoclonal antibodies recognize this population, while the U1-70 kD monoclonal antibody recognizes a different population that is incapable of interacting with SR proteins, perhaps by a steric hindrance. It is also possible that caspase-mediated cleavage of U1-70 kD during apoptosis disrupts the interaction of U1-70 kD with other SR proteins, an event that may explain the observation that overexpression of the COOH terminus of U1-70 kD (which contains tandem SR domains that are separated after caspase cleavage; reference 81) acts as a dominant negative suppressor of RNA splicing and RNA transport (82). Several reports identifying a direct interaction between U1-70 kD and SR proteins support both possibilities (61, 62).
In addition to transcriptional and translational regulation of apoptosis, our results suggest that a third regulatory mechanism for programmed cell death is at the level of messenger RNA splicing. It has been shown that cells expressing the larger splice variant of the bcl-x gene (bcl-xL) are protected against cell death, while cells expressing the short form lacking the highly conserved BH1 and BH2 interaction domains (bcl-xS) have an increased susceptibility to cell death (36, 83–85). Similar regulation has been described for the Caenorhabditus elegans ced-4 gene product (86), for the death domain-containing receptor LARD (87), and for caspase 2 (Nedd2/Ich1) in which the protein product of the larger splice variant (Ich1L) is proapoptotic and the shorter variant (Ich1S) is protective (36, 83–85). It has been shown that reversible phosphorylation of SR proteins (e.g., ASF/SF2) can alter their ability to select alternative mRNA splice sites (65, 88, 89). It is tempting to speculate that SR protein phosphorylation may regulate levels of prosurvival factors such as bcl-xL and Ich1S, or of proapoptotic factors such as bcl-xS and Ich1L, thus altering the susceptibility of a particular cell to an apoptotic trigger. Although this is unlikely to be an important mechanism after engagement of a dedicated death receptor such as Fas or the TNF receptor, both of which rapidly activate irreversible caspase cascades, alternative splicing of bcl-x, caspase 2, and other unidentified mRNAs may be a critical checkpoint when cells are subjected to slowly lethal or sublethal stimuli.
Autoantibodies reactive with core components of the U1–snRNP (anti-Sm) are specifically found in patients with SLE. The observation that snRNP particles reside in plasma membrane blebs formed at the surface of cells undergoing apoptosis suggests that antigens presented in this manner might bypass normal mechanisms of tolerance. In addition to its subcellular localization, the U1–snRNP complex undergoes profound structural alterations in cells undergoing apoptosis. These structural alterations could produce novel peptide epitopes to which T cells have not been rendered tolerant. This may be particularly important for the production of autoantibodies reactive with the U1–snRNP complex, which is subject to the phenomenon of “epitope spreading” whereby an immune response to one component of the particle promotes the formation of antibodies reactive with other components of the particle (90). We propose that a T cell response directed against modified components of the U1–snRNP complex (e.g., caspase-cleaved U1-70 kD and/or phosphorylated SR-derived peptides) may promote the formation of antibodies reactive with other components of the complex. By driving the maturation of potentially self reactive B cells specific for components of the U1–snRNP particle, T cells recognizing these neoepitopes could be essential for autoantibody production. It is currently unknown whether human autoantisera directly recognize SR proteins. With the identification of monoclonal antibodies capable of recognizing pp54, pp42, pp34, and pp23 in apoptotic Jurkat cells, it should now be possible to address these and several other important questions. Are SR proteins stable components of the U1–snRNP complex that are phosphorylated during apoptosis, or are they recruited to the complex during cell death? What is the kinase responsible for SR protein phosphorylation, and what is its role in programmed cell death pathways and RNA splicing? Answers to these important questions and the identification of other posttranslational modifications of autoantigens during apoptosis are certain to yield valuable clues to the pathogenic mechanisms underlying autoimmune diseases such as SLE, scleroderma, MCTD, and Sjögren's Syndrome. Further studies may identify components of this putative kinase pathway as novel molecular targets for pharmacologic therapy of autoimmune disease.
Footnotes
1 Abbreviations used in this paper: DNA-PK, DNA-dependent protein kinase; HI-FCS, heat-inactivated FCS; hnRNP, heterogeneous nuclear RNP; MCTD, mixed connective tissue disease; NuMA, nuclear mitotic apparatus protein; PARP, poly A ribose polymerase; PCNA, proliferating cell nuclear antigen; PVDF, polyvinylidene difluoride; RNP, ribonucleoprotein; snRNP, small nuclear RNP; SLE, systemic lupus erythematosus; Sm, Smith complex; SR, serine/arginine; SRP, signal recognition particle; TIAR, T cell intracellular antigen-related protein.
The authors thank J. Craft and members of the laboratories of P. Anderson, R. Reed, and M. Streuli for insights and helpful comments; Q. Medley and A. Da Silva for assisting with two-dimensional peptide mapping; V. Shifrin and Q. Medley for critical review of the manuscript; the Brigham & Women's Hospital Clinical Immunology Laboratory; P.H. Schur, P. Fraser, J. Craft, M. Kuwana, C. Casiano, E. Tan, E.A. Nigg, C. Zhang, D. Weaver, T. Medsger, N. Fertig, D. Bloch, A. Rosen, S. Hoch, N. Kedersha, R. Reed, K.M. Pollard, and M. Robertson for providing monoclonal and polyclonal antibodies used in this study; R. de Wildt and R.M.A. Hoet for providing the anti-U1A human variable chain antibody fragments; and J. Reed for the gift of the bcl-2– and neo-overexpressing Jurkat cells.
This work was supported in part by the Arthritis Foundation (P.J. Utz and P. Anderson); the National Institutes of Health grants AI33600 and CA67929 (P. Anderson); and the Peabody Foundation (P. Anderson). P. Anderson is a Scholar of the Leukemia Society of America. The work of W.J. van Venrooij was supported in part by the Netherlands Foundation for Chemical Research (SON) with financial aid from the Netherlands Organization for Scientific Research (NWO) and the Netherlands Technology Foundation (STW).
Address correspondence to Paul J. Utz, Department of Medicine, Division of Rheumatology, Immunology, and Allergy, Brigham & Women's Hospital, Smith Bldg., Rm. 608, 75 Francis St., Boston, MA 02115. Phone: 617-525-1216; Fax: 617-525-1310; E-mail: pjutz@rics.bwh.harvard.edu
References
- 1.Astaldi-Ricotti G, Bestagno M, Cerino A, Negri C, Caporali R, Cobianchi F, Longhi M, Montecucco C. Antibodies to hnRNP core protein A1 in connective tissue diseases. J Cell Biochem. 1989;40:43–47. doi: 10.1002/jcb.240400105. [DOI] [PubMed] [Google Scholar]
- 2.Van Venrooij W, Sillekens P. Small-nuclear RNA-associated proteins: autoantigens in connective tissue diseases. Clin Exp Rheum. 1989;7:635–639. [PubMed] [Google Scholar]
- 3.von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin Arthritis Rheum. 1995;24:323–358. doi: 10.1016/s0049-0172(95)80004-2. [DOI] [PubMed] [Google Scholar]
- 4.Montecucco M, Caporali R, Negri C, deGennaro F, Cerino A, Bestagno M, Cobianchi F, Astaldi-Ricotti G. Antibodies from patients with rheumatoid arthritis and systemic lupus erythematosus recognize different epitopes of a single heterogeneous nuclear RNP core protein. Arthritis Rheum. 1990;33:180–186. doi: 10.1002/art.1780330205. [DOI] [PubMed] [Google Scholar]
- 5.LeFeber WP, Norris DA, Ryan SR, Huff JC, Lee LA, Kubo M, Boyce ST, Kotzin BL, Weston WL. Ultraviolet light induces binding of antibodies to selected nuclear antigens on cultured keratinocytes. J Clin Invest. 1984;74:1545–1551. doi: 10.1172/JCI111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Golan TD, Elkon KB, Gharavi AE, Krueger JG. Enhanced membrane binding of autoantibodies to cultured keratinocytes of systemic lupus erythematosus patients after ultraviolet B/ultraviolet A irradiation. J Clin Invest. 1992;90:1067–1076. doi: 10.1172/JCI115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface blebs on cultured keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mevorach D, Zhou J, Elkon K. Immunization of mice with apoptotic cells induces low levels of autoantibodies. Arthritis Rheum. 1996;39:S143. . (Abstr.) [Google Scholar]
- 9.Alnemri E, Livingston D, Nicholson D, Salveson G, Thornberry N, Wong W, Yuan J. Human ICE/ CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 10.Casciola-Rosen LA, Miller DK, Anhalt GJ, Rosen A. Specific cleavage of the 70 kDa protein component of the U1 small nuclear riboprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–30760. [PubMed] [Google Scholar]
- 11.Lazebnik Y, Kaufmann S, Desnoyers S, Poirier G, Earnshaw W. Cleavage of poly (ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 12.Casciola-Rosen LA, Anhalt GJ, Rosen A. DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J Exp Med. 1995;182:1625–1634. doi: 10.1084/jem.182.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterhouse N, Kumar S, Song Q, Strike P, Sparrow L, Dreyfuss G, Alnemari ES, Litwack G, Lavin M, Watters D. Heterogeneous ribonucleoproteins C1 and C2, components of the spliceosome, are specific targets of interleukin 1β-converting enzyme-like proteases in apoptosis. J Biol Chem. 1996;271:29335–29341. doi: 10.1074/jbc.271.46.29335. [DOI] [PubMed] [Google Scholar]
- 14.Lazebnik Y, Takahashi A, Moir R, Goldman R, Poirier G, Kaufmann S, Earnshaw W. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weaver V, Carson C, Walker P, Chaly N, Lach B, Raymond Y, Brown D, Sikorska M. Degradation of nuclear matrix and DNA cleavage in apoptotic thymocytes. J Cell Sci. 1996;109:45–56. doi: 10.1242/jcs.109.1.45. [DOI] [PubMed] [Google Scholar]
- 16.Casiano CA, Martin SJ, Green DR, Tan EM. Selective cleavage of nuclear autoantigens during CD95 (Fas/Apo-1)-mediated T cell apoptosis. J Exp Med. 1996;184:765–770. doi: 10.1084/jem.184.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marin S, O'Brien G, Nishioka W, McGahon A, Mahboubi A, Saido T, Green D. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem. 1995;270:6425–6428. doi: 10.1074/jbc.270.12.6425. [DOI] [PubMed] [Google Scholar]
- 18.Haneji N, Nakamura T, Takio K, Yanagi K, Higashiyama H, Saito I, Noji S, Sugino H, Hayashi Y. Identification of alpha-fodrin as a candidate autoantigen in primary Sjogren's syndrome. Science. 1997;276:604–607. doi: 10.1126/science.276.5312.604. [DOI] [PubMed] [Google Scholar]
- 19.Utz PJ, Hottelet M, Schur P, Anderson P. Proteins phosphorylated during stress-induced apoptosis are common targets for autoantibody production in patients with systemic lupus erythematosus. J Exp Med. 1997;185:843–854. doi: 10.1084/jem.185.5.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karwan R, Bennett JL, Clayton DA. Nuclear RNase MRP processes RNA at multiple discrete sites: interaction with an upstream G box is required for subsequent downstream cleavages. Genes Dev. 1991;5:1264–1276. doi: 10.1101/gad.5.7.1264. [DOI] [PubMed] [Google Scholar]
- 21.de Wildt R, Finnern R, Ouwehand W, Griffiths A, van Venrooij W, Hoet R. Characterization of human variable domain antibody fragments against the U1 RNA-associated A protein, selected from a synthetic and a patient-derived combinatorial V gene library. Eur J Immunol. 1996;26:629–639. doi: 10.1002/eji.1830260319. [DOI] [PubMed] [Google Scholar]
- 22.Craft J, Mimori T, Olsen T, Hardin J. The U2 small nuclear ribonucleoprotein particle as an autoantigen. J Clin Invest. 1988;81:1716–1724. doi: 10.1172/JCI113511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sillekens P, Beijer R, Habets W, van Venrooij W. Molecular cloning of the cDNA for the human U2 snRNA-specific U1A′ protein. Nucleic Acids Res. 1989;17:1893–1906. doi: 10.1093/nar/17.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billings P, Allen R, Jensen F, Hoch S. Anti-RNP monoclonal antibodies derived from a mouse strain with lupus-like autoimmunity. J Immunol. 1982;128:1176–1180. [PubMed] [Google Scholar]
- 25.Habets W, Hoet M, de Jong B, van der Kemp A, van Venrooij W. Mapping of B cell epitopes on small nuclear ribonucleoproteins that react with human autoantibodies as well as with experimentally-induced mouse monoclonal antibodies. J Immunol. 1989;143:2560–2566. [PubMed] [Google Scholar]
- 26.Veldhoven CIA, Pruijn G, Meilof J, Thijssen J, van der Kemp A, van Venrooij W, Smeenk R. Characterization of murine monoclonal antibodies against 60 kDa Ro/SS-A and La/SS-B autoantigens. Clin Exp Immunol. 1995;101:45–54. doi: 10.1111/j.1365-2249.1995.tb02275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pruijn G, Thijssen J, Smith P, Williams D, van Venrooij W. Anti-La monoclonal antibodies recognizing epitopes within the RNA-binding domain of the La protein show different capacities to immunoprecipitate RNA-associated La. Eur J Biochem. 1995;232:611–619. doi: 10.1111/j.1432-1033.1995.611zz.x. [DOI] [PubMed] [Google Scholar]
- 28.Roth M, Murphy C, Gall J. A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosome loops and small granules in the amphibian germinal vesicle. J Cell Biol. 1990;111:2217–2223. doi: 10.1083/jcb.111.6.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laemmli E. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Harlow, E., and D. Lane. 1988. Immunoblotting. In Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Publications, Cold Spring Harbor, NY. 474–510.
- 31.Coligan, J., A. Kruisbeek, D. Margulies, E. Shevach, and W. Strober. 1994. Two dimensional phosphopeptide mapping. In Current Protocols in Immunology. John Wiley & Sons, Inc. 3:11.2.1–11.2.8.
- 32.Medley Q, Kedersha N, O'Brien S, Tian Q, Schlossman S, Streuli M, Anderson P. Characterization of GMP-17, a granule membrane protein that moves to the plasma membrane of natural killer cells following target recognition. Proc Natl Acad Sci USA. 1996;93:685–689. doi: 10.1073/pnas.93.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taupin J, Tian Q, Kedersha N, Robertson M, Anderson P. The RNA-binding protein TIAR is translocated from the nucleus to the cytoplasm during Fas-mediated apoptotic cell death. Proc Natl Acad Sci USA. 1995;92:1629–1633. doi: 10.1073/pnas.92.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian Q, Streuli M, Saito H, Schlossman S, Anderson P. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell. 1991;67:629–639. doi: 10.1016/0092-8674(91)90536-8. [DOI] [PubMed] [Google Scholar]
- 35.Tian Q, Taupin J, Elledge S, Robertson M, Anderson P. Fas-activated serine/threonine kinase (FAST) phosphorylates TIA-1 during Fas-mediated apoptosis. J Exp Med. 1995;182:865–874. doi: 10.1084/jem.182.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boise L, Gottschallk A, Quintans J, Thompson C. Bcl-2 and Bcl-2-related proteins in apoptosis regulation. Curr Top Microbiol Immunol. 1995;200:107–121. doi: 10.1007/978-3-642-79437-7_8. [DOI] [PubMed] [Google Scholar]
- 37.Itoh N, Tsujimoto Y, Nagata S. Effect of bcl-2 on Fas antigen-mediated cell death. J Immunol. 1993;151:621–627. [PubMed] [Google Scholar]
- 38.Reed J. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sentman C, Shutter J, Hockenberry D, Kanagawa O, Korsmeyer S. Bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell. 1991;67:879–888. doi: 10.1016/0092-8674(91)90361-2. [DOI] [PubMed] [Google Scholar]
- 40.Gui J-F, Lane W, Fu X-D. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 41.Chambers J, Keene J. Isolation and analysis of cDNA clones expressing human lupus La antigen. Proc Natl Acad Sci USA. 1985;82:2115–2119. doi: 10.1073/pnas.82.7.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Query C, Bentley R, Keene J. A common RNA recognition motif identified within a defined U1 RNA binding domain of the 70K small nuclear ribonuclearprotein component. Cell. 1989;57:89–101. doi: 10.1016/0092-8674(89)90175-x. [DOI] [PubMed] [Google Scholar]
- 43.Padgett R, Mount S, Steitz J, Sharp P. Splicing of messenger RNA precursors is inhibited by antisera to small nuclear ribonucleoprotein. Cell. 1983;35:101–107. doi: 10.1016/0092-8674(83)90212-x. [DOI] [PubMed] [Google Scholar]
- 44.Gold HA, Topper JN, Clayton D, Craft J. The RNA processing enzyme RNase MRP is identical to the Th RNP and related to RNase P. Science. 1989;245:1377–1380. doi: 10.1126/science.2476849. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y-R, Meyer CF, Tan T-H. Persistent activation of c-Jun N-terminal kinase 1 (JNK1) in gamma radiation-induced apoptosis. J Biol Chem. 1996;271:631–634. doi: 10.1074/jbc.271.2.631. [DOI] [PubMed] [Google Scholar]
- 46.Derijard B, Hibi M, Wu I-H, Barrett T, Bing S, Deng T, Karin M, Davis R. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 47.Kyriakis J, Banerjee P, Nikolakaki E, Dai T, Rubie E, Ahmad M, Avruch J, Woodgett J. The stress-activated protein kinase subfamily of c-jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 48.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 49.Liu Z-G, Hsu H, Goeddel D, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 50.Lenczowski J, Dominguez L, Eder A, King L, Zacharchuk C, Ashwell J. Lack of a role for jun kinase and AP-1 in Fas-induced apoptosis. Mol Cell Biol. 1997;17:170–181. doi: 10.1128/mcb.17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Craft J. Antibodies to snRNPs in systemic lupus erythematosus. Rheum Dis Clin North Amer. 1992;18:311–335. [PubMed] [Google Scholar]
- 52.Lerner M, Steitz J. Antibodies to small nuclear RNAs complexed with proteins are produced by patients with systemic lupus erythematosus. Proc Natl Acad Sci USA. 1979;76:5495–5499. doi: 10.1073/pnas.76.11.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okano Y, Medsger T. Newly identified U4/U6 snRNP-binding proteins by serum autoantibodies from a patient with systemic sclerosis. J Immunol. 1991;146:535–542. [PubMed] [Google Scholar]
- 54.Okano Y, Medsger T. Novel serum autoantibodies reactive with U5 small nuclear ribonucleoprotein particle (snRNP) specific proteins obtained from a patient with systemic sclerosis-polymyositis overlap. Arthritis Rheum. 1991;34:S103. . (Abstr.) [Google Scholar]
- 55.Horowitz D, Krainer A. Mechanisms for selecting 5′ splice sites in mammalian pre-mRNA splicing. Trends Genet. 1994;100:100–106. doi: 10.1016/0168-9525(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 56.Hodges P, Beggs J. U2 fulfills a commitment. Curr Biol. 1994;4:264–267. doi: 10.1016/s0960-9822(00)00061-0. [DOI] [PubMed] [Google Scholar]
- 57.Screaton G, Caceres J, Mayeda A, Bell M, Plebanski M, Jackson D, Bell J, Krainer A. Identification and characterization of three members of the human SR family of pre-mRNA splicing factors. EMBO (Eur Mol Biol Organ) J. 1995;14:4336–4349. doi: 10.1002/j.1460-2075.1995.tb00108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caceres J, Krainer A. Functional analysis of pre-mRNA splicing factor ASF/SF2 structural domains. EMBO (Eur Mol Biol Organ) J. 1993;12:4715–4726. doi: 10.1002/j.1460-2075.1993.tb06160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zuo P, Manley J. Functional domains of the human splicing factor ASF/SF2. EMBO (Eur Mol Biol Organ) J. 1993;12:4727–4737. doi: 10.1002/j.1460-2075.1993.tb06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fu X-D. Specific commitment of pre-mRNAs to splicing by single SR proteins. Nature. 1993;365:82–85. doi: 10.1038/365082a0. [DOI] [PubMed] [Google Scholar]
- 61.Kohtz J, Jamison S, Will C, Zuo P, Luhrmann R, Garcia-Blanco M, Manley J. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- 62.Wu J, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 63.Crispino J, Blencowe B, Sharp P. Complementation by SR proteins of pre-mRNA splicing reactions depleted of U1 snRNP. Science. 1994;265:1866–1869. doi: 10.1126/science.8091213. [DOI] [PubMed] [Google Scholar]
- 64.Caceres J, Stamm F, Helfman D, Krainer A. Regulation of alternative splicing by overexpression of antagonistic splicing factors. Science. 1994;265:1706–1709. doi: 10.1126/science.8085156. [DOI] [PubMed] [Google Scholar]
- 65.Ge H, Manley J. Primary structure of the human splicing factor ASF reveals similarities with Drosophilaregulators. Cell. 1991;66:373–382. doi: 10.1016/0092-8674(91)90626-a. [DOI] [PubMed] [Google Scholar]
- 66.Krainer A, Mayeda A, Kozak D, Binns G. Functional expression of cloned human splicing factor SF2: homology to RNA binding proteins, U1-70K, and Drosophilasplicing regulators. Cell. 1991;66:383–394. doi: 10.1016/0092-8674(91)90627-b. [DOI] [PubMed] [Google Scholar]
- 67.Fu X-D, Mayeda A, Maniatis T, Krainer A. General splicing factors SF2 and SC35 have equivalent activities in vitro, and both affect alternative 5′ and 3′ splice site selection. Proc Natl Acad Sci USA. 1992;89:11224–11228. doi: 10.1073/pnas.89.23.11224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mayeda A, Krainer A. Regulation of alternative pre-mRNA splicing by hnRNPA1 and splicing factor SF2. Cell. 1992;68:365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- 69.Zahler A, Neugebauer K, Lane W, Roth M. Distinct functions of SR proteins in alternative pre-mRNA splicing. Science. 1993;260:219–222. doi: 10.1126/science.8385799. [DOI] [PubMed] [Google Scholar]
- 70.Mermoud J, Cohen P, Lamond A. Ser/Thr-specific phosphatases are required for both catalytic steps of pre-mRNA splicing. Nucleic Acids Res. 1992;20:5263–5269. doi: 10.1093/nar/20.20.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mermoud J, Cohen P, Lamond A. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. EMBO (Eur Mol Biol Organ) J. 1994;13:5679–5688. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tazi J, Daugeron M-C, Cathala G, Brunel C, Jeanteur P. Adenosine phosphorothioates (ATPαS and ATPγS) differentially affect the two steps of mammalian pre-mRNA splicing. J Biol Chem. 1992;267:4322–4326. [PubMed] [Google Scholar]
- 73.Woppmann A, Will C, Kornstadt U, Zuo P, Manley J, Luhrmann R. Identification of an snRNP-associated kinase activity that phosphorylates arginine/serine rich domains typical of splicing factors. Nucleic Acids Res. 1993;21:2815–2822. doi: 10.1093/nar/21.12.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gui J-F, Fu X-D. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature. 1994;369:678–682. doi: 10.1038/369678a0. [DOI] [PubMed] [Google Scholar]
- 75.Gui J-F, Tronchere H, Chandler S, Fu X-D. Purification and characterization of a serine kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc Natl Acad Sci USA. 1994;91:10824–10828. doi: 10.1073/pnas.91.23.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colwill K, Feng L, Yeakley J, Gish G, Caceres J, Pawson T, Fu X-D. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. J Biol Chem. 1996;271:24569–24575. doi: 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- 77.Melcher M, Thorner J. Identification and characterization of the CLK1 gene product, a novel CaM kinase-like protein kinase from the yeast Saccaromyces cerevisiae. J Biol Chem. 1996;271:29958–29968. doi: 10.1074/jbc.271.47.29958. [DOI] [PubMed] [Google Scholar]
- 78.Staknis D, Reed R. SR proteins promote the first specific recognition of pre-mRNA and are present together with the U1 small nuclear ribonucleoprotein particle in a general splicing enhancer complex. Mol Cell Biol. 1994;14:7670–7682. doi: 10.1128/mcb.14.11.7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Staknis D, Reed R. Members of a family of proteins (the RD family) detected by a U1 70K monoclonal antibody are present in spliceosomal complexes. Nucleic Acids Res. 1995;23:4081–4086. doi: 10.1093/nar/23.20.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zahler A, Lane W, Stolk J, Roth M. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 81.Casciola-Rosen L, Nicholson D, Chong T, Rowan K, Thornberry N, Miller D, Rosen A. Apopain/ CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med. 1997;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Romac J-J, Keene J. Overexpression of the arginine-rich carboxy-terminal region of U1 snRNP 70K inhibits both splicing and nucleocytoplasmic transport of mRNA. Genes Dev. 1995;9:1400–1410. doi: 10.1101/gad.9.11.1400. [DOI] [PubMed] [Google Scholar]
- 83.Fang W, Rivard J, Mueller D, Behrens T. Cloning and molecular characterization of mouse bcl-x in B and T lymphocytes. J Immunol. 1994;153:4388–4398. [PubMed] [Google Scholar]
- 84.Boise L, Gonzalez-Garcia M, Postema C, Ding L, Lindsten T, Turka L, Mao X, Nunez G, Thompson C. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 85.Yang E, Korsmeyer S. Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- 86.Shaham S, Horvitz H. An alternatively spliced C. elegans ced-4 RNA encodes a novel cell death inhibitor. Cell. 1996;86:201–208. doi: 10.1016/s0092-8674(00)80092-6. [DOI] [PubMed] [Google Scholar]
- 87.Screaton G, Xu X-N, Olsen A, Cowper A, Tan R, McMichael A, Bell J. LARD: A new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc Natl Acad Sci USA. 1997;94:4615–4619. doi: 10.1073/pnas.94.9.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krainer A, Conway G, Kozak D. Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 1990;4:1158–1171. doi: 10.1101/gad.4.7.1158. [DOI] [PubMed] [Google Scholar]
- 89.Kuo H, Nasim F, Grabowski P. Control of alternative splicing by the differential binding of U1 small nucleoprotein particle. Science. 1991;251:1045–1050. doi: 10.1126/science.1825520. [DOI] [PubMed] [Google Scholar]
- 90.Craft J, Fatenejad S. Self antigens and epitope spreading in systemic autoimmunity. Arthritis Rheum. 1997;40:1374–1382. doi: 10.1002/art.1780400803. [DOI] [PubMed] [Google Scholar]
- 91.Hoet RM, de Weerd P, Klein J, Gunnewiek, Koornneef I, van Venrooij WJ. Epitope regions on U1 small nuclear RNA recognized by anti-U1RNA-specific autoantibodies. J Clin Invest. 1992;90:1753–1762. doi: 10.1172/JCI116049. [DOI] [PMC free article] [PubMed] [Google Scholar]