

Abstract
The crystal structure of the complex between hen egg lysozyme and the Fv fragment of a humanized antilysozyme antibody was determined to 2.7-Å resolution. The structure of the antigen combining site in the complex is nearly identical to that of the complexed form of the parent mouse antibody, D1.3. In contrast, the combining sites of the unliganded mouse and humanized antilysozymes show moderate conformational differences. This disparity suggests that a conformational readjustment process linked to antigen binding reverses adverse conformations in the complementarity determining regions that had been introduced by engineering these segments next to human framework regions in the humanized antibody.
Humanized antibodies were designed to limit the response of the human immune system to rodent monoclonal antibodies used in therapy of human disease (1). They represented an advance over chimeric antibodies (2–4), which were engineered with murine immunoglobulin variable domains and human constant domains, in that the foreign content of humanized variable regions was substantially reduced. This was achieved by combining the short, hypervariable complementarity determining regions (CDRs)1 of a murine antibody, which fold to form an antigen combining site of unique structure, with human variable domain framework regions, which appear to be conserved in sequence throughout all races (5–9). The resulting molecule has the same specificity as the murine antibody, but substitution of human sequences confers a much longer in vivo lifetime and nearly eliminates immunogenic side effects (10).
Framework and CDR segments can be identified from sequence information alone, since they are defined by homology rather than structure (11, 12). At the three-dimensional level, however, the two sets of residues are in intimate contact and mutually influence each other's conformation (13, 14). Human framework residues can alter the conformation of transposed mouse CDRs and thereby disrupt antigen binding. Since human and mouse framework regions differ by upwards of 50 out of 170 residues (15), the potential for this sort of disruption is high. Nevertheless, humanizing by grafting mouse CDRs onto human frameworks usually transfers antigenic specificity, though sometimes additional framework mutations are required to put antigen affinity on a par with the starting mouse antibody (13).
In this report, we describe intrinsic aspects of immunoglobulin structure that may partly account for the ease with which CDRs and framework regions from different animal species and unrelated antigenic specificities may be combined to give a functional molecule. We have determined the crystal structure of the complex between hen egg white lysozyme and the immunoglobulin heavy and light chain variable domains (Fv) of a humanized version of the mouse antilysozyme D1.3 (HuLys). This is the first reported structure of a complex of antigen with a humanized antibody, and completes a family of crystallographically determined component structures. D1.3 was previously determined in the free form and complexed with lysozyme, both at 1.8-Å resolution (16, 17). HuLys was determined in the unliganded form at 2.9-Å resolution (14) . The human immunoglobulins NEW and REI, from which the HuLys heavy and light chain framework regions were taken, respectively, have previously been determined in their unliganded form, both at 2.0-Å resolution (18–20). The availability of mouse, human, and antigen complex structures for reference has now allowed us to identify conformational differences in the humanized Fv that result from protein engineering and ligand binding.
Materials and Methods
Protein Expression.
The HuLys protein used in this study was a high affinity form with variable domain sequences identical to those published previously (21). This molecule was expressed as an Fv fragment in the Escherichia coli strain 25F2, using the vector pAK19 (22). In brief, 15 liters of defined medium (23) supplemented with 0.15% casamino acids, 1% glucose, and 20 μg/ml tetracycline was inoculated with 400 ml of a starter culture (absorbance at 600 nm 1.5–2) and grown in a fermentor (New Brunswick Scientific MPP, Edison, NJ) for 20 h at 37°C. Cells were harvested by centrifugation and subjected to osmotic shock to release periplasmic proteins (24). The periplasmic fraction was clarified by centrifugation (30 min at 14,500 g) and passed through a lysozyme–Sepharose column. After extensive washes with PBS and high salt buffer (500 mM NaCl, 50 mM Tris, pH 8.5), the Fv was eluted with 50 mM diethylamine.
Crystallization.
The purified Fv was complexed with lysozyme and crystallized from phosphate buffer. Lysozyme (three times crystallized) was purchased from Sigma Chemical Co. (St. Louis, MO) and dissolved in PBS. HuLys–lysozyme complex was prepared by mixing the two in equimolar proportions, letting the solution sit for 30 min, and diluting to 10 μg/ml with PBS. There was immediate slight cloudiness upon mixing; the solution was spun in a bench-top centrifuge before setting up crystallizations. Crystallization was by vapor diffusion; both hanging drops and sitting drops in microbridges were used. Protein concentration was 7 or 10 mg/ml. The reservoir was 1.6 or 1.7 M phosphate (made by mixing equal volumes of K2HPO4 and NaH2PO4), and 0.1 M Hepes, pH 6.5. Equal volumes of protein and reservoir solutions were mixed to make the drop.
Data Collection.
Two x-ray diffraction data sets were collected to 2.9-Å resolution and 2.7-Å resolution, each from a single crystal at 4°C, using an r-axis detector. The two data sets were processed (Table 1) with DENZO and SCALEPACK (25). The lower resolution data set, truncated to 3.5 Å, was used initially for determining the molecular replacement solution. Refinement was carried out when the higher resolution data set became available.
Table 1.
Data Collection
| Parameter | Data set 1 value | Data set 2 value | ||
|---|---|---|---|---|
| Resolution | Å | Å | ||
| Entire data set | 50.0–3.5 | 50.0–2.7 | ||
| Last shell | 3.56–3.0 | 2.75–2.70 | ||
| Reflections | ||||
| Total | 73,185 | 120,820 | ||
| Unique | 9,493 | 22,379 | ||
| Completeness | % | % | ||
| Entire data set | 85.1 | 93.2 | ||
| Last shell | 76.3 | 73.5 | ||
| R value* | ||||
| Entire data set | 0.078 | 0.075 | ||
| Last shell | 0.226 | 0.452 | ||
| Average I/σI | 10.3 | 9.6 |
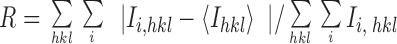
Molecular Replacement Solution.
A search model for molecular replacement was constructed by superposing molecule 2 of uncomplexed HuLys (14), the molecule whose CDRs are less affected by crystal packing, upon the murine complexed structure and combining the HuLys and lysozyme structures. Molecular replacement was carried out using the program AMoRe (26) with the 3.5-Å resolution data set. The space group was determined to be P41212 from the four possibilities P4x212, x = 0, 1, 2, 3, by solution of the translation function. (There were no 00l reflections in the data set.) The unit cell dimensions were a = b = 97.7 Å, and c = 174.9 Å. After refinement of the translation functions for the two molecules in the asymmetric unit, the correlation coefficient was 0.63 and the R value was 0.38 (15–3.5 Å).
Crystallographic Refinement.
The 2.7-Å resolution data set was partitioned by X-PLOR (27) into two sets, one for refinement and calculation of the working R value, and the other for calculation of the free R value (28, 29). A rigid-body refinement was performed at 3.5-Å resolution using X-PLOR, first with each complex as a rigid body, and then with each chain as a rigid body. The refinement resulted in a drop of both working R and free R from 0.46 to 0.32 (10–3.5 Å). Alternating rounds of positional and individual B value refinement, using both X-PLOR and TNT (30), and model building, using QUANTA (Molecular Simulations, Inc., Burlington, MA), were performed (Table 2). No solvent molecules were included in the model. The values for working R and free R dropped from 0.35 to 0.21 and from 0.34 to 0.30, respectively (10–2.7 Å). A PROCHECK analysis (31) of the structure showed no residues in disallowed regions of a Ramachandran plot except for residue L51 of both molecules. This is also seen in the uncomplexed HuLys and REI and many other Fab structures. The residue is in a γ-turn conformation (32). Residue numbering follows the Kabat system (15). In this paper we precede each residue number with a chain designator, e.g., L51 for light chain residue 51.
Table 2.
Refinement
| Parameter | Value | |
|---|---|---|
| Resolution | 10.0–2.7 Å | |
| Reflections | ||
| Total (F >20) | 18,822 | |
| Working set | 16,927 | |
| Test set | 1,895 | |
| Atoms | 5,490 | |
| R value | ||
| Working | 0.208 | |
| Free | 0.297 | |
| rms deviation from ideality | ||
| Bond lengths | 0.014 Å | |
| Bond angles | 1.8° | |
| PROCHECK analysis, percent residues in: | ||
| Most favored regions | 79.3 | |
| Additional allowed regions | 19.2 | |
| Generously allowed regions | 1.2 | |
| Disallowed regions | 0.3 |
Results
Antigen–Antibody Interaction.
The most striking feature of the structure is apparent when the antigen-contacting residues in the mouse and humanized Fv–lysozyme complexes are compared to the same residues seen in the free Fv structures. The unliganded structures are shown superposed in Fig. 1 A. A multitude of conformational differences are observed between the unliganded mouse and humanized combining sites. In contrast, the antigen-contacting residues in the HuLys and D1.3 complexes have virtually identical conformations (Fig. 1 B). Although these superpositions were calculated using only alpha carbon (Cα) atoms of the polypeptide chains, it is clear that the side chain conformations are also nearly identical in the complexed structures.
Figure 1.
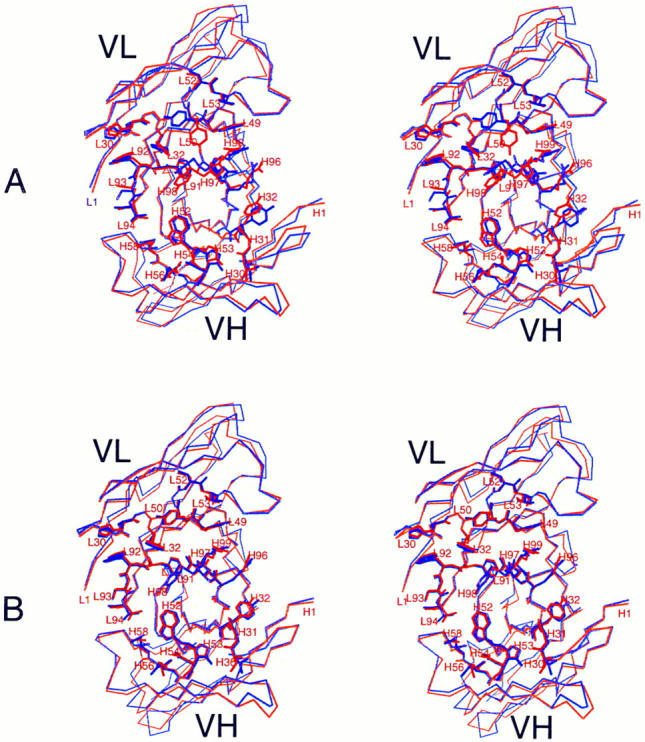
Antigen combining site of free and complexed HuLys and D1.3, wall-eyed stereo views. (A), unliganded forms. Molecule 2 of free HuLys (14) is shown superposed on free D1.3 Fv. (B), Complexed forms. Molecule 1 of the HuLys–lysozyme complex crystal form is shown superposed on D1.3 Fv from the D1.3–hen lysozyme complex. Lysozyme has been removed from the illustration. Superpositions were performed with QUANTA, using the Cα atoms of antigen-contacting residues. A list of lysozyme-contacting residues in D1.3, which match those seen in the HuLys– lysozyme complex, is given in reference 35. The structures are shown from analogous “antigen eye views” looking directly at the antigen combining site of each antibody. For simplicity, residues that do not directly contact antigen are drawn as a Cα trace, whereas antigen-contacting residues are fully drawn. HuLys, red; D1.3, purple. The illustration was drawn using the program MOLSCRIPT (46).
Several of the differences between complexed and uncomplexed forms in Fig. 1 occur not because of antigen binding, but because the antigen-binding residues in the uncomplexed Fv are involved in crystal packing interactions. The largest shift amongst antigen-contacting residues of the murine Fv (0.69–1.06 Å) occurs for heavy chain residues H96-H98 (16), and probably arises from contacts with symmetry-related molecules in the uncomplexed Fv. A comparison of the uncomplexed and complexed forms of HuLys Fv shows that the largest shift amongst antigencontacting residues occurs for residue L93; the Cα–Cα distances after CDR superposition are 1.20 and 1.31 Å for molecules 1 and 2, respectively, versus molecule 2 of the uncomplexed Fv structure. This large shift is not seen upon examination of the D1.3 structures, possibly because in uncomplexed D1.3, this residue may be held in a complexed-like conformation due to crystal packing interactions. All other shifts in antigen-contacting residues are less than twice the root mean square (rms) distance for the superposition of all six CDRs.
The rearrangements accompanying lysozyme binding represent a broader effect than symmetry-related intermolecular contacts alone can account for. Quantitative evidence that the CDRs of the liganded forms of HuLys and D1.3 are more structurally similar than in the free Fv molecules is presented in Table 3. The rms differences in position of Cα atoms in the mouse and humanized CDRs, which comprise 56 residues in each molecule, is 0.37 Å when the Fv–lysozyme complexes are compared, versus 0.63 Å comparing the CDRs in the free Fvs (33). The liganded CDRs are closer in structure than any other residue subset in Table 1. In addition, the binding of lysozyme by HuLys causes the CDRs to more closely resemble the unbound D1.3 CDRs. Even superposition of the lysozyme molecule in the HuLys complex onto the lysozyme molecule in the D1.3 complex gives a rms distance one fourth larger than for the corresponding CDRs. Lysozyme itself forms crystal packing interactions that differ in the mouse and HuLys complexes, which give rise to some of the differences.
Table 3.
rms Distances between Superimposed HuLys and Cα Atoms
| HuLys | D1.3 | Framework | CDR | Lysozyme | ||||
|---|---|---|---|---|---|---|---|---|
| Å | Å | Å | ||||||
| Liganded | Liganded | 0.67 | 0.37 | 0.43 | ||||
| Free | Free | 0.68 | 0.63 | – | ||||
| Liganded | Free | 0.62 | 0.44 | – | ||||
Distances were calculated between Cα atoms of each segment after superposition of those atoms. Each heavy chain and each light chain of the two HuLys molecules was superposed separately on the corresponding D1.3 chain. Each set of distances was separated into two lists, one containing framework residue distances and one containing CDR residue distances. Each entry in the Table was then calculated from the four (liganded HuLys) or two (free HuLys) corresponding lists of distances. Both liganded HuLys molecules in the crystallographic asymmetric unit were used in the calculation, but only one of the two free HuLys molecules was included because the CDRs of the heavy chain of the other are somewhat distorted by symmetry-related contacts. For lysozyme superpositions, the two lysozyme molecules of the HuLys complex were superposed on the D1.3 lysozyme. D1.3 structures used for comparison were obtained from the Brookhaven Protein Data Bank, and correspond to the accession numbers 1VFA (free Fv) and 1 VFB (Fv-lysozyme complex).
Contacts between HuLys and lysozyme were identified using the program PAIRS (34). An analysis of these contacts is given in Table 4. Only direct protein–protein hydrogen bonds are considered, as we have not included water molecules in the HuLys structure due to its lower resolution. Solvent molecules do in fact contribute greatly to the stabilization of the complex, as was seen in the higher resolution D1.3 complex structure (35). The hydrogen bond distances in the D1.3 and HuLys complexes are similar, with those in HuLys marginally longer. Hydrogen bonds between HuLys and lysozyme seen in both HuLys complexes are seen in all cases as well in the D1.3-lysozyme complex. This comparison of antibody–antigen contacts in the HuLys and D1.3 complexes was extended to include van der Waals interactions. The comparison showed three interactions in D1.3 closer than 4 Å, between Trp H52 and lysozyme Gly 117, which are not seen in HuLys. No interactions were found in both HuLys molecules that were not also seen in D1.3.
Table 4.
Comparison of Hydrogen-bonded Antibody–Antigen Contact Distances in HuLys and D1.3
| Fv atom | Lysozme atom | D1.3 distance | HuLys distance (molecules 1,2) | |||
|---|---|---|---|---|---|---|
| Å | Å | |||||
| Ly30 His ND1 | 129 Leu OT2 | 3.4 | 3.3, 4.6 | |||
| L50 Tyr OH | 18 Asp OD1 | 3.2 | 3.2, 3.7 | |||
| L50 Tyr OH | 18 Asp OD2 | 2.7 | 2.7, 2.9 | |||
| L53 Thr OG1 | 19 Asn ND2 | 2.8 | 3.2, 3.1 | |||
| L91 Phe O | 121 Gln NE2 | 2.8 | 3.0, 3.0 | |||
| L93 Ser N | 121 Gln OE1 | 2.9 | 2.9, 2.8 | |||
| H53 Gly N | 117 Gly O | 2.8 | 3.1, 3.3 | |||
| H96 Arg NH2 | 102 Gly O | 2.7 | 2.8, 4.0 | |||
| H97 Asp OD1 | 24 Ser OG | 2.8 | 2.8, 3.2 | |||
| H97 Asp OD1 | 27 Asn ND2 | 3.1 | 3.5, 3.1 | |||
| H97 Asp OD2 | 22 Gly O | 3.0 | 3.0, 3.0 | |||
| H97 Asp OD2 | 24 Ser N | 2.9 | 2.9, 3.0 | |||
| H98 Tyr OH | 119 Asp OD1 | 2.7 | 2.9, 2.7 | |||
| H98 Tyr OH | 120 Val N | 3.2 | 3.3, 3.2 | |||
| H98 Tyr OH | 121 Gln N | 3.0 | 3.2, 2.9 | |||
| H99 Arg NH1 | 22 Gly O | 2.8 | 3.0, 3.0 | |||
| H99 Arg NH2 | 22 Gly O | 3.4 | 2.7, 3.2 | |||
All direct D1.3-lysozyme hydrogen bonds shorter than 3.5 Å are listed. These residues are a subset of a larger list, which includes residues that hydrogen bond via water molecules.
We interpret the shift of the HuLys antigen contacting residues from rough congruence to exact congruence with the cognate residues in D1.3 as evidence for a conformational correction mechanism that allows the antibody to attain precise stereochemical complementarity with antigen. The unliganded conformation of HuLys observed crystallographically (14) is unlikely to interact with the antigen as strongly as the conformation both HuLys and D1.3 adopt in the complex. During the antigen recognition process, the HuLys combining site must undergo a correction to the more favorable lysozyme binding conformation. Exactly such a mechanism was hypothesized in the initial report of humanizing the D1.3 heavy chain (36). X-ray data do not allow us to distinguish whether this involves an “induced fit” rearrangement of combining site residues after initial complex formation (37, 38) or isomerization of HuLys to the high affinity form before antigen encounter (39–41). D1.3 itself undergoes a small conformational change accompanying lysozyme complex formation (16), but this differs somewhat from the change in HuLys, as evidenced by the moderate disparity in D1.3 and HuLys unliganded conformations.
Conformational Correction in Framework.
A second conformational correction mechanism appears to involve a subtle rearrangement of HuLys framework residues proximal to the CDRs. Evidence for this rearrangement is shown in Fig. 2. In 2 A, the framework of the Fv structure has been divided into layers according to CDR proximity. Residues within 4 Å of a CDR form the first layer, residues approaching between 4 and 8 Å the next layer, and so on. Fig. 2 C shows, for each layer, the degree of sequence identity between HuLys and cognate residues in the two parent antibody molecules. Here, HuLys and D1.3 have 100% sequence identity in the CDRs, whereas in all framework layers HuLys and the human molecules are more similar in primary structure. Fig. 2 B depicts a parameter more relevant to homology at the three-dimensional level. Here, the distance from each HuLys Cα to the cognate Cα in superposed D1.3, REI, or NEW molecules was determined. The difference between the HuLys to human and HuLys to mouse distance was then calculated, and the median for each residue layer was compiled. This difference is a measure of whether a particular layer of HuLys is generally more similar in conformation to D1.3 or to REI and NEW. We found that the CDRs of the mouse and humanized molecules are much more structurally similar than the HuLys CDRs are to the CDRs of NEW and REI. Likewise, in the distal layers of the Fv, the humanized framework is very similar to the human molecules at the three-dimensional level. Although the sequence–structure correlations at opposite poles of the Fv are intuitive and unremarkable, the sequence–structure correlation in layers of framework residues near the framework–CDR interface is counterintuitive. The humanized sequence remains very close to the human sequences, but the framework of the humanized molecule is conformationally more similar to the mouse structure. The HuLys CDRs, which have virtually no homology to NEW and REI, thus appear to induce a D1.3-like conformation in adjacent framework layers.
Figure 2.


CDR-induced changes in framework conformation. (A) Cα trace of the HuLys Fv from molecule 1 of the HuLys–lysozyme complex crystal form, stereo view. Framework layers are colored according to CDR proximity. CDRs are colored red. Framework residues that have at least one nonhydrogen atom within 4 Å of any nonhydrogen CDR atom are colored maroon. Residues whose nearest atom to a CDR atom is within 4–8 Å are colored purple, within 8–12 Å colored green, and 12–27 Å colored black. (B) Conformational similarity between HuLys and D1.3 or HuLys and REI/NEW, by framework layer. Heavy and light chains of the mouse and human structures were independently superposed on HuLys molecules 1 and 2 using PDBFIT (33). The distance between cognate Cα atoms was calculated, and for each cognate set the value of the HuLys to D1.3 distance was subtracted from the HuLys to REI/NEW distance. The median difference for each framework layer is shown in the bar graph. A positive value indicates that the HuLys to human distance is greater than the HuLys to mouse distance for a majority of residues in that layer, and therefore that the HuLys structure is less similar to the human and more similar to the mouse structure for that layer. For these calculations we used a total of 112 CDR Cα atoms in the two crystallographically independent HuLys Fv molecules, 92 in the 0–4 Å layer, 88 in the 4–8 Å layer, 74 in the 8–12 Å layer, and 80 in the 12–27 Å layer. (C) Sequence identity between HuLys, D1.3, and REI/NEW, by framework layer. Open bars, HuLys compared to REI or NEW; filled bars, HuLys compared to D1.3. The humanized, mouse, and human protein sequences were aligned by Kabat residue number. The number of positions with identical residues in HuLys and D1.3 or HuLys and REI/NEW were then tabulated for each framework layer, and this value is presented in the figure as a percentage. The illustration was drawn using the program MOLSCRIPT (27).
Discussion
In HuLys, and possibly many other humanized antibodies, a heirarchy of immunoglobulin structural properties allows humanizing to “work”. As a first approximation, mouse CDRs retain function on human frameworks because backbone conformations of hypervariable loops usually follow one of a small number of canonical structures regardless of chemical environment (42). Human frameworks can usually be found that will support a set of mouse CDRs identically to the mouse frameworks with which the CDRs were first isolated. In HuLys, we have identified a fine-tuning mechanism in which the mouse CDRs force nearby humanized framework residues toward the backbone conformation found in the corresponding D1.3 frameworks. The implication for humanizing is that although point mutations may have to be introduced into the frameworks to improve antigen binding by a humanized antibody, generally only a few are required, and these are necessary to recreate local structural motifs, such as the CDR-separating side chain at H71 (43). Finer-scale tailoring of framework structures appears unnecessary. At the combining site, the conformation of a humanized antibody free in solution may differ somewhat from that of the parent mouse antibody, as was found by comparing HuLys and D1.3 (14). However, a second conformational correction mechanism compensates for imprecisions in lock and key complementarity that would otherwise result from apposing rigid bodies. Interaction with antigen erases these differences by selecting or inducing a conformation of HuLys nearly identical to that in the D1.3–lysozyme complex. A concomitant shift towards the uncomplexed mouse antibody conformation occurs as well. Lastly, 25 tightly ordered water molecules form a layer at the D1.3–lysozyme interface (35, 44, 45). These water molecules bridge many antibody–antigen contacts, are thermodynamically significant in the antigen-binding reaction, and many are conserved in free, complexed, and mutant D1.3 structures, and hence must be considered an intrinsic part of the combining site. Though we cannot identify ordered solvent in HuLys at 2.7-Å resolution, it would seem essential for antigen recognition that CDRs in a humanized antibody recreate a water network similar to that in the combining site of the parent mouse antibody.
Footnotes
We thank Steve Sheriff, Thomas B. Lavoie, and Greg Winter for suggestions on the manuscript.
We gratefully acknowledge financial support from the Arnold and Mabel Beckman Foundation and the Department of the Army Breast Cancer Research Program (grant DAMD 17-97-1-7124).
Address correspondence to Jefferson Foote, Division of Molecular Medicine, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. North, C3-168, PO Box 19024, Seattle, WA 98109-1024. Phone: 1-206-667-6720; Fax: 1-206-667-6524; E-mail: jfoote@fhcrc.org
Abbreviations used in this paper: Cα, alpha carbon; CDR, complementarity determining region; Fv, dimer of immunoglobulin heavy and light chain variable domains; rms, root mean square.
References
- 1.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321:522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 2.Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci USA. 1984;81:6851–6855. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulianne GL, Hozumi N, Shulman MJ. Production of functional chimaeric mouse/human antibody. Nature. 1984;312:643–646. doi: 10.1038/312643a0. [DOI] [PubMed] [Google Scholar]
- 4.Neuberger MS, Williams GT, Mitchell EB, Jouhal SS, Flanagan JG, Rabbitts TH. A hapten-specific chimaeric IgE antibody with human physiological effector function. Nature. 1985;314:268–270. doi: 10.1038/314268a0. [DOI] [PubMed] [Google Scholar]
- 5.Schaible G, Rappold GA, Pargent W, Zachau HG. The immunoglobulin kappa locus: polymorphism and haplotypes of Caucasoid and non-Caucasoid individuals. Hum Genet. 1993;91:261–267. doi: 10.1007/BF00218268. [DOI] [PubMed] [Google Scholar]
- 6.Matsuda F, Shin EK, Nagaoka H, Matsumura R, Haino M, Fukita Y, Taka-Ishi S, Imai T, Riley JH, Anand R, Soeda E, Honjo T. Structure and physical map of 64 variable segments in the 3′ 0.8-megabase region of the human immunoglobulin heavy-chain locus. Nat Genet. 1993;3:88–94. doi: 10.1038/ng0193-88. [DOI] [PubMed] [Google Scholar]
- 7.Cook GP, Tomlinson IM, Walter G, Riethman H, Carter NP, Buluwela L, Winter G, Rabbitts TH. A map of the human immunoglobulin Vhlocus completed by analysis of the telomeric region of chromosome 14q. Nat Genet. 1994;7:162–168. doi: 10.1038/ng0694-162. [DOI] [PubMed] [Google Scholar]
- 8.Sasso EH, Buckner JH, Suzuki LA. Ethnic differences in polymorphism of an immunoglobulin Vh3 gene. J Clin Invest. 1995;96:1591–1600. doi: 10.1172/JCI118198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milner, E.C.B. 1996. Organization of the human VH locus and rearrangement patterns of the VH3 gene family. In Human B Cell Superantigens. M. Zouali, editor. Springer, New York. 1–10.
- 10.Hale G, Dyer MJS, Clark MR, Phillips JM, Marcus R, Riechmann L, Winter G, Waldmann H. Remission induction in non-Hodgkin lymphoma with reshaped human monoclonal antibody CAMPATH-1H. Lancet. 1988;2:1394–1399. doi: 10.1016/s0140-6736(88)90588-0. [DOI] [PubMed] [Google Scholar]
- 11.Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 1970;132:211–250. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabat EA, Wu TT. Attempts to locate complementarity-determining residues in the variable positions of light and heavy chains. Ann NY Acad Sci. 1971;190:382–393. doi: 10.1111/j.1749-6632.1971.tb13550.x. [DOI] [PubMed] [Google Scholar]
- 13.Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988;332:323–327. doi: 10.1038/332323a0. [DOI] [PubMed] [Google Scholar]
- 14.Holmes MA, Foote J. Structural consequences of humanizing an antibody. J Immunol. 1997;158:2192–2201. [PubMed] [Google Scholar]
- 15.Kabat, E.A., T.T. Wu, H.M. Perry, K.S. Gottesman, and K. Coeller. 1991. Sequences of Proteins of Immunological Interest. 5th ed. US Department of Health and Human Services, Public Health Service, National Institutes of Health, Bethesda.
- 16.Bhat TN, Bentley GA, Fischmann TO, Boulot G, Poljak RJ. Small rearrangements in structures of Fv and Fab fragments of antibody D1.3 on antigen binding. Nature. 1990;347:483–485. doi: 10.1038/347483a0. [DOI] [PubMed] [Google Scholar]
- 17.Amit AG, Mariuzza RA, Phillips SEV, Poljak RJ. Three-dimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science. 1986;233:747–753. doi: 10.1126/science.2426778. [DOI] [PubMed] [Google Scholar]
- 18.Poljak RJ, Amzel LM, Avey HP, Chen BL, Phizackerly RP, Saul F. Three-dimensional structure of the Fab′ fragment of a human immunoglobulin at 2.8 Å resolution. Proc Natl Acad Sci USA. 1973;70:3305–3310. doi: 10.1073/pnas.70.12.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saul FA, Poljak RJ. Crystal structure of human immunoglobulin fragment Fab New refined at 2.0 Å resolution. Proteins. 1992;14:363–371. doi: 10.1002/prot.340140305. [DOI] [PubMed] [Google Scholar]
- 20.Epp O, Lattman EE, Schiffer M, Huber R, Palm W. The molecular structure of a dimer composed of the variable portions of the Bence-Jones protein REI refined at 2.0 Å resolution. Biochemistry. 1975;14:4943–4952. doi: 10.1021/bi00693a025. [DOI] [PubMed] [Google Scholar]
- 21.Foote J, Winter G. Antibody residues affecting conformation of the hypervariable loops. J Mol Biol. 1992;224:487–499. doi: 10.1016/0022-2836(92)91010-m. [DOI] [PubMed] [Google Scholar]
- 22.Carter P, Kelley RF, Rodrigues ML, Snedecor B, Covarrubias M, Velligan MD, Wong WLT, Rowland AM, Kotts CE, Carver ME, et al. High level Escherichia coli expression and production of a bivalent humanized antibody fragment. Bio/ Technology. 1992;10:163–167. doi: 10.1038/nbt0292-163. [DOI] [PubMed] [Google Scholar]
- 23.Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skerra A, Plückthun A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. . Science. 1988;240:1038–1041. doi: 10.1126/science.3285470. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr Sect A Found Crystallogr. 1994;50:157–163. [Google Scholar]
- 27.Brünger, A.T. 1992. X-PLOR Manual, Version 3.1. Yale University Press, New Haven.
- 28.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 29.Kleywegt GJ, Jones TA. When freedom is given, liberties are taken. Structure. 1995;3:535–540. doi: 10.1016/s0969-2126(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 30.Tronrud DE, TenEyck LF, Matthews BW. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr Sect A Found Crystallogr. 1987;42:489–501. [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemistry of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Milner-White EJ, Ross BM, Ismail R, Belhadj-Mostefa K, Poet R. One type of gamma-turn, rather than the other gives rise to chain-reversal in proteins. J Mol Biol. 1988;204:777–782. doi: 10.1016/0022-2836(88)90368-3. [DOI] [PubMed] [Google Scholar]
- 33.McRee DE. A visual protein crystallographic software system for X11/Xview. J Mol Graphics. 1992;10:44–46. [Google Scholar]
- 34.Padlan EA. Anatomy of the antibody molecule. Mol Immunol. 1994;31:169–217. doi: 10.1016/0161-5890(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 35.Bhat TN, Bentley GA, Boulot G, Green MI, Tello D, Dall'Acqua W, Souchon H, Schwarz FP, Mariuzza RA, Poljak RJ. Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc Natl Acad Sci USA. 1994;91:1089–1093. doi: 10.1073/pnas.91.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verhoeyen ME, Milstein C, Winter G. Reshaping human antibodies: grafting an antilysozyme activity. Science. 1988;239:1534–1536. doi: 10.1126/science.2451287. [DOI] [PubMed] [Google Scholar]
- 37.Herron JN, He XM, Ballard DW, Blier PR, Pace PE, Bothwell ALM, Voss EW, Jr, Edmundson AB. An autoantibody to single-stranded DNA: comparison of the three-dimensional structures of the unliganded Fab and a deoxynucleotide-Fab complex. Proteins. 1991;11:159–175. doi: 10.1002/prot.340110302. [DOI] [PubMed] [Google Scholar]
- 38.Rini JM, Schulze-Gahmen U, Wilson IA. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science. 1992;255:959–965. doi: 10.1126/science.1546293. [DOI] [PubMed] [Google Scholar]
- 39.Lancet D, Pecht I. Kinetic evidence for hapten-induced conformational transition in immunoglobulin MOPC 460. Proc Natl Acad Sci USA. 1976;73:3548–3553. doi: 10.1073/pnas.73.10.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens FJ, Chang C-H, Schiffer M. Dual conformations of an immunoglobulin light-chain dimer: heterogeneity of antigen specificity and idiotope profile may result from multiple variable-domain interaction mechanisms. Proc Natl Acad Sci USA. 1988;85:6895–6899. doi: 10.1073/pnas.85.18.6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci USA. 1994;91:10370–10374. doi: 10.1073/pnas.91.22.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;96:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 43.Tramontano A, Chothia C, Lesk AM. Framework residue 71 is a major determinant of the position and conformation of the second hypervariable region in the Vhdomains of immunoglobulins. J Mol Biol. 1990;215:175–182. doi: 10.1016/S0022-2836(05)80102-0. [DOI] [PubMed] [Google Scholar]
- 44.Braden BC, Fields BA, Poljak RJ. Conservation of water molecules in an antibody-antigen interaction. J Mol Recognit. 1995;8:317–325. doi: 10.1002/jmr.300080505. [DOI] [PubMed] [Google Scholar]
- 45.Goldbaum FA, Schwarz FP, Eisenstein E, Cauerhff A, Mariuzza RA, Poljak RJ. The effect of water activity on the association constant and the enthalpy of reaction between lysozyme and the specific antibodies D1.3 and D44.1. J Mol Recognit. 1996;9:6–12. doi: 10.1002/(SICI)1099-1352(199601)9:1%3C6::AID-JMR240%3E3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 46.Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]