

Abstract
Interleukin-6 (IL-6) is overproduced in the joints of patients with rheumatoid arthritis (RA) and, based on its multiple stimulatory effects on cells of the immune system and on vascular endothelia, osteoclasts, and synovial fibroblasts, is believed to participate in the development and clinical manifestations of this disease. In this study we have analysed the effect of ablating cytokine production in two mouse models of arthritis: collagen-induced arthritis (CIA) in DBA/1J mice and the inflammatory polyarthritis of tumor necrosis factor α (TNF-α) transgenic mice. IL-6 was ablated by intercrossing an IL-6 null mutation into both arthritis-susceptible genetic backgrounds and disease development was monitored by measuring clinical, histological, and biochemical parameters. Two opposite responses were observed; while arthritis in TNF-α transgenic mice was not affected by inactivation of the IL-6 gene, DBA/1J, IL-6−/− mice were completely protected from CIA, accompanied by a reduced antibody response to type II collagen and the absence of inflammatory cells and tissue damage in knee joints. These results are discussed in the light of the present knowledge of cytokine networks in chronic inflammatory disorders and suggest that IL-6 receptor antagonists might be beneficial for the treatment of RA.
Rheumatoid arthritis (RA)1 is a common human autoimmune disease characterized by chronic inflammation of the sinovial joints and by subsequent progressive destruction of articular tissue. Although the etiology and pathogenesis of RA are not yet fully understood, it has become increasingly clear that a series of locally produced cytokines play a central role in disease progression. Indeed, cytokines are responsible both for the mobilization and continuous activation of the inflammatory cell infiltrate and for inducing production of the enzymes that destroy bone and cartilage (for review see reference 1).
The current view of the cytokine network in rheumatoid joints supports the notion that TNF-α activates a cytokine cascade characterized by the simultaneous production of proinflammatory cytokines such as IL-1, IL-6, several chemokines, GM-CSF, and of antiinflammatory factors such as IL-10, IL-1RA, and soluble TNF receptor (for review see reference 2). Disease progression/reactivation or, on the contrary, its silencing, are likely to be due to a dynamic and unstable equilibrium in the production of pro- and antiinflammatory cytokines.
From among these cytokines, IL-6 has been proposed to contribute to the development of arthritis. IL-6 is present at very high levels in serum and synovial fluids of RA and of juvenile RA patients (3–6). Soluble forms of the specific IL-6 receptor subunit α (sIL-6Rα) are elevated (7, 8) and these are known to potentiate IL-6 activity by forming IL-6–sIL-6Rα complexes that bind and homodimerize the signaling-competent transmembrane receptor glycoprotein (gp)130 (9).
Increased IL-6 bioactivity during RA is believed to be responsible for local and systemic effects. IL-6 acts as a stimulator of both B and T cell functions because it promotes proliferation of plasmablastic precursors in the bone marrow and their final stage of maturation into immunoglobulin-producing plasma cells and participates in the activation and proliferation of T cells (for review see reference 10). Moreover, IL-6, in conjunction with sIL-6Rα, has been recently shown to: (a) activate endothelial cell production of a subset of chemokines and adhesion molecules, thus contributing indirectly to recruitment of leukocytes at inflammatory sites (11); and (b) induce synovial fibroblast proliferation (12) and osteoclast formation and activation (13), therefore suggesting a role in synovial fibrosis and in bone resorption in inflamed joints. IL-6 is also known to be an endogenous pirogen (14) and the major cytokine-inducing activation of liver acute phase response genes (15). However, despite all this indirect and circumstantial evidence, a comprehensive investigation of the role of IL-6 in the development of arthritis in animal models of RA has not yet been reported.
The generation of knockout mice for different cytokine genes has led to the conclusion that in most circumstances, ablation of a single cytokine is compatible with normal life (16). Knockout mice can, therefore, be used to evaluate the role played by a specific cytokine in disease development. Using appropriate disease models, IL-6–deficient mice (IL-6−/−) have been shown to be resistant to bone loss after ovariectomy (17), and when bred into the Balb/c genetic background, to be protected from the development of pristane oil-induced plasmacytomas (18). We have hence adopted the same genetic approach to analyze the effects of ablating IL-6 activity in two available mouse arthritis models: collagen-induced arthritis (CIA) in DBA/1J mice and the inflammatory polyarthritis of TNF-α transgenic mice (19). Although the disease's primary cause differs in the two models, they both share numerous RA histological features. Here we provide evidence that IL-6 plays a crucial role in the development of autoimmune CIA, but not in the TNF-α–dependent inflammatory arthritis.
Materials and Methods
Mice.
IL-6–deficient mice (17) were backcrossed into the DBA/1J genetic background (H-2q, Jackson Laboratory, Bar Harbor, ME) for five generations, obtaining DBA/1J, IL-6 mice that were named DIL-6 mice. In all experiments, only DBA/1J, IL-6−/− (DIL-6−/−) and DBA/1J, IL-6+/+ (DIL-6+/+) littermate mice were used.
TNF, IL-6 mice were obtained by intercrossing TNF-α transgenic mice (19) with IL-6−/− mice. Mice carrying the TNF-α transgene and heterozygous for the IL-6 null mutation were intercrossed to obtain TNF, IL-6−/− (TIL-6−/−) and TNF, IL-6+/+ (TIL-6+/+) mice.
DIL-6 and TIL-6 mice were bred at IRBM (Rome, Italy) in a specific pathogen-free animal facility, whereas the parental DBA/1J strain was obtained from Jackson Laboratory. Mice were maintained in standard conditions under a 12-h light–dark cycle, provided irradiated food (4RF21; Mucedola; Settimo Milanese, Milan, Italy) and chlorinated water ad libitum. Procedures involving animals and their care were conducted in conformity with national and international laws and policies.
Induction of CIA, Antibody Treatment, and Clinical Assessment of Arthritis.
Male 8-wk-old DBA/1J and DIL-6 mice were immunized intradermally at the base of the tail with 100 μg of bovine type II collagen (CII; M.M. Griffiths, University of Utah, Salt Lake City, UT) in 0.05 M acetic acid, emulsified with an equal volume of complete Freund's adjuvant, containing 100 μg of H37RA Mycobacterium tuberculosis (Difco, Detroit, MI). On day 21, mice were boosted by intradermal injection with 100 μg of bovine CII in 0.05 M acetic acid emulsified with an equal volume of incomplete Freund's adjuvant (Difco).
Starting from time of the CII booster injection, DBA/1J mice were treated subcutaneously once a week for 6 wk with 0.5 or 1 mg/mouse of the following antibodies: (a) the rat mAb 15A7 (20), which neutralizes the murine IL-6 receptor alpha chain, (b) an isotype-matched IgG2b rat mAb against dinitrophenyl hapten (LO-DNP-57; provided by H. Bazin, University of Louvain, Brussels, Belgium), and (c) total rat IgGs (Sigma Chemical Co., St. Louis, MO).
CIA development was inspected three times per week and inflammation of the four paws was graded from 0 to 4: grade 0, paws with no swelling and focal redness; grade 1, paws with swelling of finger joints; grade 2, paws with mild swelling of ankle or wrist joints; grade 3, paws with severe inflammation of the entire paw; and grade 4, paws with deformity or ankylosis. Each paw was graded and the four scores were totaled so that the possible maximum score per mouse was 16.
The arthritis index of TIL-6 mice was determined by observation of the hindpaw deformation twice a week and was scored as follows: 1, phalanx deformity; 2–6, different grades of limbs deformity; 7, ankylosis detected on flexion and severely impaired movement.
Serum Measurements.
Serum samples of DIL-6 mice were obtained before and 10 wk after the first CII immunization. Analysis of total IgG anti-CII antibodies was performed by ELISA. Microtiter wells were coated with bovine CII dissolved in potassium phosphate buffer, pH 7.6, (0.5 μg/well) overnight at 4°C. Wells were blocked with Superblock Blocking Solution in PBS (Pierce, Rockford, IL) 9 h at 4°C. Sera were diluted in 1% BSA in PNT (1× PBS containing 0.05% Tween 20, and 0.2 M NaCl) and incubated overnight at 4°C. Antibodies anti–bovine CII were revealed by an alkaline-phosphatase–conjugated goat anti–mouse IgG (Promega, Madison, WI). The concentration of antibodies was calculated by comparison with a standard positive control mAb (M.M. Griffiths, University of Utah, Salt Lake City, UT).
ELISA to detect antigen-specific IgG isotypes was performed with a modified protocol of the assay described above. In brief, after incubation wells were washed and different rabbit anti– mouse IgG isotype antibodies (Dako A/S, Glostrup, Denmark) were used. Wells were washed and incubated with an alkaline-phosphatase–conjugated goat anti–rabbit IgG (Promega).
IL-6 serum levels were determined by the hybridoma growth factor assay as previously described (21). Human TNF-α serum levels were measured by ELISA kits (Medgenix EASIA) according to the manufacturer's specifications.
Histology.
DIL-6 mice were killed 7 wk after the first CII immunization and hind limbs were fixed with 4% paraformaldehyde in 1× PBS overnight at 4°C. The joints were decalcified for 12 h (De Castro's buffer: 0.3 M chloral hydrate, 0.72 M HNO3, and 30% ethanol) and then embedded in paraffin blocks. Joint sections were collected and processed with Villanueva staining.
Statistical Analysis.
Results were analyzed using ANOVA, chi-square analysis, and the Spearman correlation coefficient, as appropriate (Statview program, Abacus Concept 1992). A P value of <0.05 was considered significant.
Results
Serum IL-6 Levels Increase in Parallel with the Development of CIA in DBA/1J Mice.
CIA is an animal model for rheumatoid arthritis in which the disease is elicited by immunization of genetically susceptible DBA/1J mice with type II collagen, and it bears many of the histological features and both cellular and humoral immune responses characteristically found in RA (22). To establish possible correlations between IL-6 levels and the severity of arthritis, serum IL-6 levels were evaluated in parallel with disease severity expressed as arthritis index of the affected joints. Mice with macroscopic joint involvement (arthritis index of >1) had serum IL-6 levels (52.2 ± 45.8 U/ml) significantly higher than those of mice without macroscopic involvement (12.5 ± 6.3 U/ml; P = 0.0033) and those of nonimmunized animals (6.3 ± 0.7 U/ml; P = 0.00l). In addition, in mice with macroscopic joint involvement (arthritis index of >1) a significant correlation (regression correlation coefficient of Spearman [Rs] = 0.694; P = 0.008) between serum IL-6 levels and the arthritis index was found (Fig. 1), suggesting a direct correlation between IL-6 production and disease severity.
Figure 1.

Serum levels of IL-6 in DBA/1J mice with CIA correlated with the arthritis index. Type II collagen immunized mice were bled 6 wk after CII immunization. IL-6 activity was measured by hybridoma growth assay and the arthritis index evaluated as described in Materials and Methods. Results were analyzed using the Spearman correlation coefficient. Rs = 0.694; P = 0.008.
Treatment of CIA with an mAb Neutralizing IL-6 Activity.
To investigate the pathogenic role of IL-6 in CIA, we first attempted neutralization of IL-6 in vivo using the mAb 15A7, directed against the murine IL-6 receptor alpha chain (IL-6Rα; reference 20, 23–25). Both 15A7 and control antibodies were administered subcutaneously at weekly intervals starting from the time of the boosting CII injection. In a first experiment the isotype-matched rat mAb LO-DNP-57 (20, 23) was used as a control. Surprisingly, both the 15A7 mAb and the LO-DNP-57 control mAb were able to significantly decrease disease severity, as shown in Fig. 2 A, when used at the dose of 1 mg/mouse but not at the dose of 0.5 mg/mouse. As this result may be due to some unexpected interference with CIA development of the particular antibody used as control, we repeated the treatment using total rat IgGs as control (Fig. 2 B). Again, the 15A7 mAb significantly decreased arthritis development (P <0.02 in weeks 9, 10, and 11 after the first CII immunization), although the total IgG treatment also considerably decreased CIA symptoms, albeit with a statistically significant decrease (P <0.04) only at week 9. These results do not allow any conclusions to be drawn about the role of IL-6 in CIA.
Figure 2.
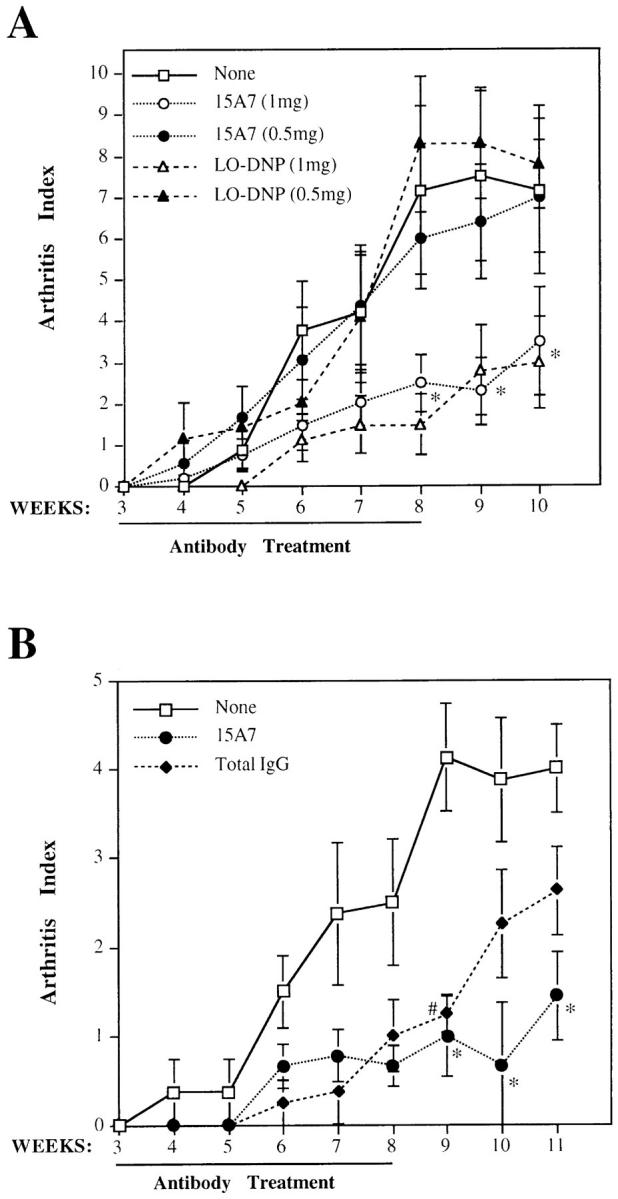
Effects of antibody treatment on arthritis index of CII-immunized DBA/1J mice. (A) Mice (n = 9 for each group) were treated once per week with 1 mg/mouse (white symbols) or 0.5 mg/mouse (black symbols) of either anti–IL-6Rα antibody 15A7 (circles), anti–dinitrophenyl hapten antibody LO-DNP (triangles), or left untreated (squares). *P <0.05 either 15A7 or LO-DNP versus none. (B) Mice were treated with 1 mg/ wk/mouse of either anti–IL6-Rα antibody 15A7 (circles; n = 9), rat total IgG antibodies (diamonds; n = 8), or left untreated (squares; n = 8). *P < 0.02 15A7 versus none; # P <0.04 total IgG versus none. Results were reported as mean ± SD. The weeks after the first immunization with CII and the duration of antibody treatment are indicated on the abscissa.
IL-6 Production Is Absolutely Necessary for CIA.
An alternative strategy to test the role of IL-6 in CIA is ablation of the IL-6 gene in mice genetically susceptible to CIA. We have therefore introduced an IL-6 null mutation (17) into the CIA-susceptible DBA/1J genetic background. After five consecutive backcrosses both DIL-6−/− and DIL-6+/+ mice were generated and used for CIA studies. As the MHC H-2 locus is known to play a major role in the genetic susceptibility to CIA (22), we first confirmed that both DIL-6−/− and DIL-6+/+ mice carried the same H-2 allele (H-2q) as the parental DBA/1J strain (data not shown).
In two independent experiments, a total number of 16 DIL-6−/− and 17 DIL-6+/+ mice were immunized with CII and joint swelling was monitored in the weeks after the injection of the antigen. The results, reported in Fig. 3, show that while DIL-6+/+ mice displayed the expected rate of arthritis development (12/17 animals; 70.6%), DIL-6−/− mice were completely protected (0/16; P <0.0001 by chi-square analysis), suggesting that IL-6 activity is essential for CIA to develop. This conclusion was further supported by histological analysis of the joints. Fig. 4 shows representative sections of six knee joints analyzed for each genotype in which evident inflammatory alterations with proliferating pannus, bone erosion, and mononuclear cell infiltrate were evident only in DIL-6+/+, whereas in DIL-6−/− no pathological alteration was detectable; the joint and the articular discs were normal and no signs of pannus formation and/or inflammatory infiltrate were present.
Figure 3.

IL-6 is absolutely required for the development of CIA. DIL-6+/+ (squares; n = 17) and DIL-6−/− (diamonds; n = 16) mice were observed for arthritis lesions and the percentage of mice that developed CIA is reported. The figure represents two independent experiments. Weeks after the first immunization with CII are indicated.
Figure 4.
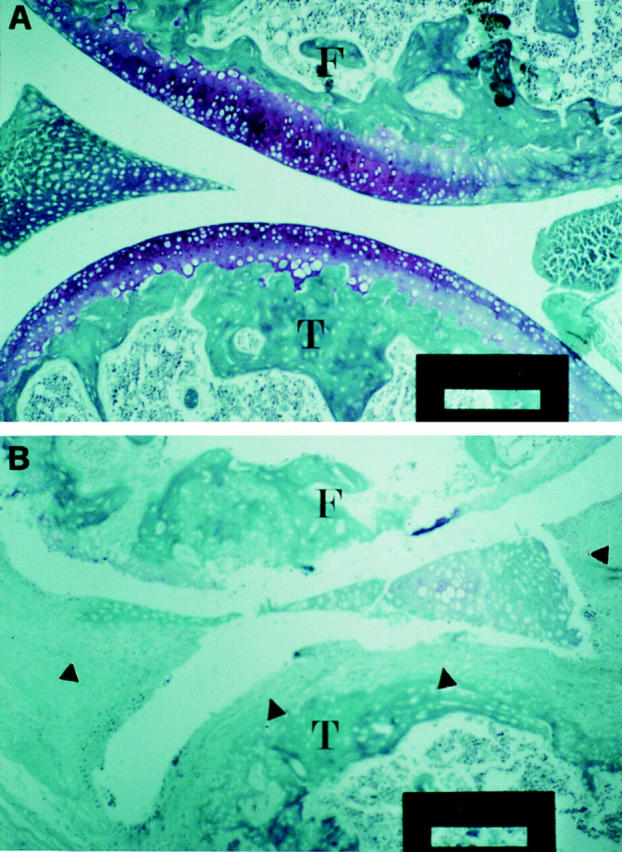
DIL-6−/− mice do not show any histological lesion of the joints. Representative examples of six knee joints for each genotype are shown: DIL-6−/− (A) and DIL-6+/+ (B). In A no signs of inflammatory lesions are detectable. In B severe signs of inflammatory process are depicted; the pannus completely infiltrated the articular cartilage (arrowheads); severe signs of cartilage and bone destruction and mononuclear cells infiltration are shown. Mice were killed 7 wk after first immunization with CII. Paraffin sections were stained with Villanueva staining. T, Tibia; F, femur. Bars: 50 μm.
CIA development has been shown to be dependent on both cellular and humoral immune responses to CII (22). Since IL-6 is a major factor in the growth and differentiation of B cells into antibody-producing cells (10), we measured the level of the total anti-CII IgG. Although DIL-6−/− mice did develop appreciable levels of anticollagen IgGs, they were significantly lower than those found in both DIL-6+/+ mice (Fig. 5 A) and in the parental DBA/1J (not shown). Previous studies have also suggested that antibodies of the IgG2a isotype play a major pathogenic role in CIA development (26, 27). We therefore evaluated the anticollagen IgG isotype distribution in both DIL-6+/+ and DIL-6−/− (Fig. 5 B). Interestingly, although in DIL-6+/+ mice IgG1, IgG2a, and IgG2b were equally present, in DIL-6−/− mice a predominant IgG2a production was observed. Therefore, although the response to CII is quantitatively reduced in the absence of IL-6, it appears qualitatively compatible with disease development.
Figure 5.
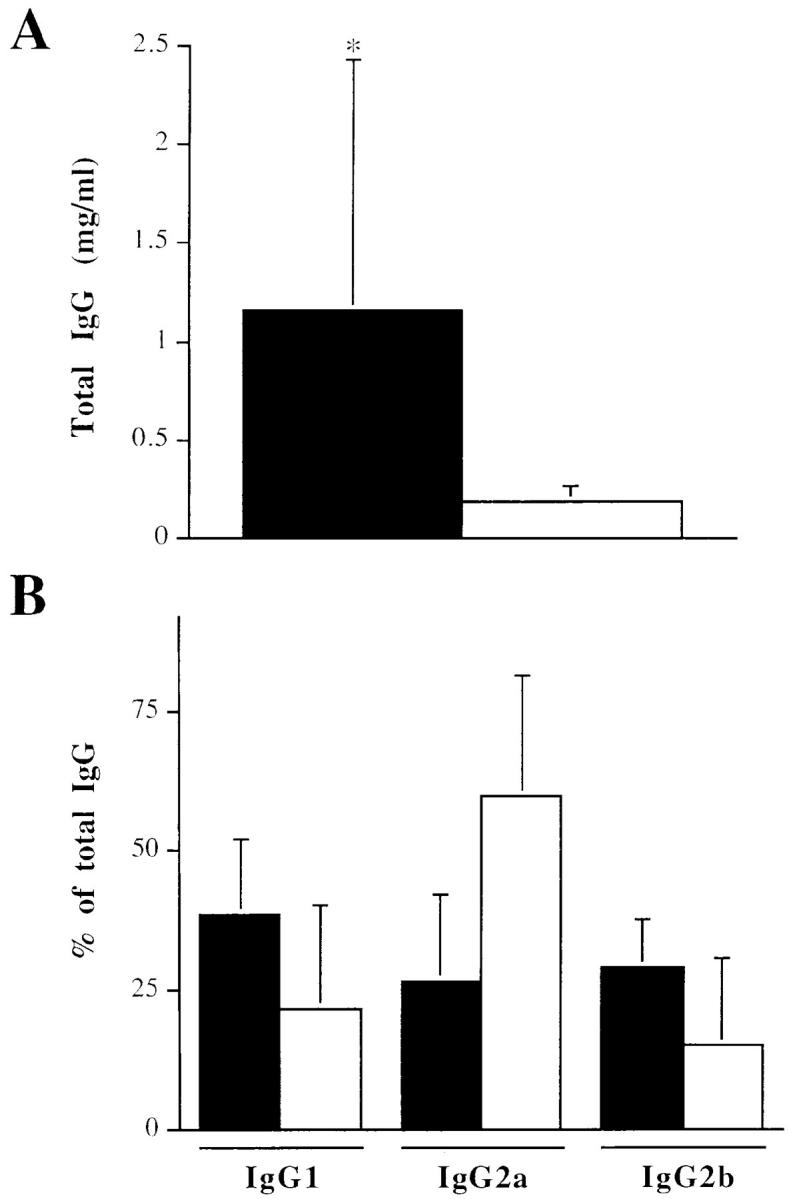
DIL-6−/− mice have abnormal humoral response to type II collagen. DIL-6+/+ (black bars; n = 9) and DIL-6−/− (white bars; n = 8) mice were bled 10 wk after the first immunization with CII, and levels of anti-CII antibodies were measured by ELISA. (A) Total IgG anti-CII antibodies were reported as mean ± SD (*P <0.02). (B) Isotype percentages of total IgG antibodies anti-CII were reported as mean ± SD. IgG isotypes are indicated.
Inflammatory Polyarthritis in TNF-α Transgenic Mice Is IL-6 Independent.
TNF-α transgenic mice overexpress a 3′–untranslated region–modified form of the human TNF-α messenger RNA that is responsible for the development of chronic inflammatory polyarthritis (19). Since TNF-α is known to be an inducing factor of IL-6 synthesis (28), we measured IL-6 levels in vivo to monitor possible correlation with disease development. As shown in Table 1, IL-6 serum levels were constantly elevated in TNF-α transgenic mice independently from their age, and did not increase with the development of the articular disease, in contrast with what we found in the CIA model.
Table 1.
IL-6 Serum Levels of TNF-α Transgenic and Wild-type Mice at Different Weeks of Age
| Mice | IL-6* | |||||
|---|---|---|---|---|---|---|
| 7 wk | 9 wk | 11 wk | ||||
| U/ml | ||||||
| TNF-α (n = 8) | 67.65 ± 11.74 | 56.44 ± 19.14 | 55.25 ± 5.28 | |||
| Wild type (n = 11) | 20.93 ± 4.40 | 20.01 ± 1.44 | 24.68 ± 2.20 | |||
| (P <0.01) | (P <0.02) | (P <0.001) | ||||
Mean values ± SE. IL-6 activity was measured by hybridoma growth assay and P values were calculated by ANOVA.
To assess if higher IL-6 production was critical for the development of arthritis, we have generated transgenic TIL-6−/− and TIL-6+/+ mice by genetic intercrosses. A total number of 16 TIL-6−/− and 11 TIL-6 +/+ mice were analyzed for timing and severity of arthritis. The results (Fig. 6) clearly show that both groups of animals displayed identical disease progression and severity. In agreement with these data, hind paw histological analysis showed comparable signs of arthritic lesions in mice belonging to the two groups (data not shown). Since IL-6 is known to downregulate TNF-α production in both in vitro and in vivo models of inflammation (29, 30), the development of arthritis in TIL-6−/− mice could be at least in part due to TNF-α upregulation in the absence of IL-6. However, at 8 wk of age, TIL-6−/− mice showed lower serum TNF-α levels when compared to those of the TIL-6+/+ mice, whereas 3 wk later, this difference had been eliminated (Table 2), indicating that TNF-α production is differently regulated during the LPS-induced acute inflammation and the chronic inflammation in TNF-α transgenic mice. Taken together, these data indicate that IL-6 does not play a pathogenic role in the onset and development of this TNF-α–dependent arthritis.
Figure 6.

The lack of IL-6 activity does not influence the onset and the development of arthritis in TNF-α transgenic mice. TIL-6−/− (squares; n = 16) and TIL-6+/+ (diamonds; n = 11) mice were scored for arthritic lesions as described in Materials and Methods. Arthritis index is expressed as mean values ± SD. The differences were not statistically significant.
Table 2.
Human TNF-α Serum Levels of TIL-6 Mice at Different Weeks of Age
| Mice | Human TNF-α (pg/ml)* | |||
|---|---|---|---|---|
| 8 wk | 11 wk | |||
| pg/ml | ||||
| TIL-6−/− (n = 6) | 150 ± 56 | 556 ± 134 | ||
| TIL-6+/+ (n = 4) | 268 ± 86 | 647 ± 81 | ||
| (P <0.01) | (P >0.05) | |||
Mean values ± SD. Human TNF-α serum levels were measured by ELISA and P values were calculated by ANOVA.
Discussion
The goal of the present study was to further understand the role played by IL-6 in the pathogenesis of different arthritic diseases by interfering with this cytokine's activity. In a first attempt we made use of mAb 15A7 directed against the IL-6–specific receptor subunit gp80. This particular antibody was chosen for its demonstrated efficacy in other IL-6–dependent murine models (20, 23–25) and because in contrast with anti–IL-6 mAbs it is not expected to stabilize the cytokine in vivo thus prolonging its half life (21, 31). Treatment with 15A7 effectively decreased CIA incidence and morbidity only at the dose of 1 mg/wk/ mouse, a dosage which has also been demonstrated as effective in several other models (20, 23–25). The requirement for these amounts of 15A7 mAb may be explained by the peculiar properties of the IL-6 receptorial system, with the soluble form of the IL-6Rα acting as a cytokine agonist and its levels increasing considerably during inflammatory diseases in both animals and humans (2, 8, 32). At this dosage the isotype-matched mAb used as a control showed a similar therapeutic effect, and even total rat IgGs exerted a protective activity, although less pronounced. The most likely interpretation of the effect of unrelated antibodies on CIA is that this relatively high dose of exogenous Ig may cause nonspecific deviation of the immune system, similar to what is observed in patients with autoimmune diseases treated with high dose intravenous Ig (33). Since these results did not provide unequivocal evidence of the role of IL-6 in CIA, we genetically ablated IL-6 activity, and found that this resulted in total protection from CIA and in the absence of inflammatory cell infiltrates in the joints.
Although knockout mice have previously been used to evaluate the role of a variety of immune response–related molecules, including cytokines, in CIA (34–39), this is the first time that total protection is reported. This result is therefore of particular relevance and should promote future investigation as to the role of this cytokine in autoimmune arthritis. It may be argued that the protection from CIA afforded by IL-6 gene inactivation could be at least partly due to a higher presence of non-DBA genes physically linked to the IL-6 locus in the DIL-6−/− mice. Although this possibility cannot be ruled out, it is extremely unlikely to account for total protection in 100% of the mice. In addition, none of the non-MHC genes that have been previously identified as contributing to genetic susceptibility to CIA map close to the IL-6 locus (22, 40, 41).
The multiple biological activities of IL-6 make it difficult to unravel the mechanism by which absence of this cytokine offers protection from CIA. We have found that although anticollagen IgG production had decreased in DIL-6−/− mice, it was predominantly the IgG2a subclass that exerts a pathogenic role in CIA (26, 27). In addition, as the production of IgG2a isotypes is driven by a polarized T helper 1 response, this finding indirectly suggests that a pathogenic polarized Th1 response to collagen still occurs in the absence of IL-6. It is therefore likely that the weaker anti-CII response in DIL-6−/− mice is but one of the factors contributing to protection from the disease, also in the light of results obtained using IL-12 p40−/− mice (35), which showed impaired anti-CII humoral response but still developed arthritis, albeit at a reduced level. Since IL-6 functions as a late-acting killer helper factor in the differentiation of CTLs (42), and CD8+ T cells play an important role in CIA initiation (39, 43), IL-6 ablation may, for example, provoke a decrease in MHC class I–restricted anti-CII cytotoxic activity. In addition, the defective chemokine production and leukocyte recruitment at inflammatory sites caused by IL-6 deficiency (11) may represent an important factor, as the continuous influx of activated cells to inflamed joints is known to be responsible for disease perpetuation through increased cytokine production, tissue destruction, and enhancement of the immune response. IL-6 inactivation may therefore exert its effects on CIA development by acting at several levels. Further studies are needed to clarify this issue.
Ablation of IL-6 production did not, in contrast, affect the development of arthritis in TNF-α transgenic mice. This difference to CIA might be due to a differentiated involvement of the immune system in the two models, as suggested by the finding that TNF-α transgenic mice bearing a RAG null mutation develop arthritis in the absence of functional lymphocytes (44). Moreover, it has been recently shown that most of the TNF-α activity in this model is mediated by its capacity to induce IL-1β and to synergize with it (45), and the constitutive expression of 3′–untranslated region–modified TNF-α may not easily come under the control of regulatory factors, hence leading to an unbalanced cytokine cascade (44). Taken together, our results demonstrate that a given cytokine can have varying relevance in different arthritis models, further corroborating previous observations showing different effects of the neutralization of TNF-α in CIA or streptococcal cell wall– induced arthritis (46), and of the IL-1 receptor antagonist in CIA and antigen-induced arthritis in rabbits or mice (47, 48). The same concept may also be true for human arthritides: although anti–TNF-α mAbs markedly ameliorate joint involvement in the majority of patients with RA (49), administration of the same antibody did not affect arthritis in one patient with severe systemic juvenile RA (50).
Our data support the idea that IL-6 blockade could be beneficial for the treatment of human autoimmune arthritis. Administration of an mAb to IL-6 in an open pilot trial of five patients with RA led to clinical and biological improvements (51). An alternative approach is the recently generated human IL-6 receptor superantagonist Sant7, an IL-6 variant that binds the human IL-6Rα receptor chain at high affinity but no longer binds to the gp130 receptor subunit. This molecule has been shown to inhibit the effects of IL-6 with great efficacy on a variety of human cell lines, including myeloma cells (for review see reference 52). In contrast to antibodies directed against IL-6, Sant7 is not expected to prolong IL-6 half-life. Its potential for use in therapy of chronic arthritis awaits further testing in suitable animal models, an approach that so far has not been implemented in the mouse and rat systems due to lack of binding to rodent IL-6Rα (Ciliberto, G., unpublished observation).
Footnotes
We wish to thank Mr. M. Aquilina and Mrs. S. Germoni (Istituto Ricerche di Biologia Molecolare; IRBM) for animal care, H. Bazin for the generous gift of LO-DNP-57 antibody, and Drs. A. Nicosia, M. Sollazzo, and C. Toniatti, and Mrs. J. Clench (IRBM) for critically reading the manuscript.
Address correspondence to Gennaro Ciliberto, IRBM P. Angeletti, Via Pontina Km 30,600, 00040 Pomezia, Rome, Italy. Phone: 39-6-91093205; Fax: 39-6-91093654; E-mail: ciliberto@irbm.it
The present address of T. Alonzi and V. Poli is Department of Biochemistry, Wellcome Trust Building, University of Dundee, Dundee DD1 4HN, UK.
Abbreviations used in this paper: CII, type II collagen; CIA, collagen-induced arthritis; DIL, DBA/1J, IL-6; DIL-6, DBA/1J mice crossed with IL-6–deficient mice; gp, glycoprotein; RA, rheumatoid arthritis; sIL-6Rα, soluble IL-6 receptor subunit α; TIL, TNF, IL-6; TIL-6, TNF-α transgenic mice crossed with IL-6–deficient mice.
References
- 1.Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 2.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 3.Guerne AP, Zuraw BL, Vaughan JH, Carson DA, Lotz M. Synovium as a source of interleukin-6 in vivo. Contribution to local and systemic manifestations of arthritis. J Clin Invest. 1989;83:585–592. doi: 10.1172/JCI113921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaines CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988;31:784–788. doi: 10.1002/art.1780310614. [DOI] [PubMed] [Google Scholar]
- 5.De Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A. Correlation of serum interleukin 6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34:1158–1163. doi: 10.1002/art.1780340912. [DOI] [PubMed] [Google Scholar]
- 6.De Benedetti F, Robbioni P, Massa M, Viola S, Albani S, Martini A. Serum interleukin 6 and joint involvement in polyarticular and pauciarticular in systemic juvenile chronic arthritis. Clin Exp Rheumatol. 1992;10:493–498. [PubMed] [Google Scholar]
- 7.Kotake S, Sato K, Takahashi N, Udagawa N, Nakamura I, Yamaguchi A, Kishimoto T, Suda T, Kashiwazaki T. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res. 1996;11:88–95. doi: 10.1002/jbmr.5650110113. [DOI] [PubMed] [Google Scholar]
- 8.De Benedetti F, Massa M, Pignatti P, Albani S, Novick D, Martini A. Serum soluble IL-6 receptor and IL-6/soluble IL-6 receptor complex in juvenile rheumatoid arthritis. J Clin Invest. 1994;93:2114–2119. doi: 10.1172/JCI117206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor: a paradigm for cytokines. Science. 1992;258:593–597. doi: 10.1126/science.1411569. [DOI] [PubMed] [Google Scholar]
- 10.Van Snick J. Interleukin-6 an overview. Annu Rev Immunol. 1990;8:253–278. doi: 10.1146/annurev.iy.08.040190.001345. [DOI] [PubMed] [Google Scholar]
- 11.Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 12.Mihara M, Moriya Y, Kishimoto T, Ohsugi Y. Interleukin-6 (IL-6) induces the proliferation of synovial fibroblastic cells in the presence of soluble IL-6 receptor. Br J Rheumatol. 1995;34:321–325. doi: 10.1093/rheumatology/34.4.321. [DOI] [PubMed] [Google Scholar]
- 13.Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, Koishihara Y, Ohsugi Y, Kumaki K, Taga T. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci USA. 1993;90:11924–11928. doi: 10.1073/pnas.90.24.11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chai Z, Gatti S, Toniatti C, Poli V, Bartfai T. IL-6 gene expression in the CNS is necessary for fever response to LPS or IL-1β: a study on IL-6–deficient mice. J Exp Med. 1996;183:311–316. doi: 10.1084/jem.183.1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poli, V., and G. Ciliberto. 1994. Transcriptional regulation of acute phase genes by IL-6 and related cytokines. In Liver Gene Expression. F. Tronche and M. Yaniv, editors. R.G. Landes Company Biomedical Publishers, Austin. 131–151.
- 16.Kopf M, Le Gros G, Coyle AJ, Kosco-Vilbois M, Brombacher F. Immune response of IL-4, IL-5, IL-6 deficient mice. Immunol Rev. 1995;148:45–70. doi: 10.1111/j.1600-065x.1995.tb00093.x. [DOI] [PubMed] [Google Scholar]
- 17.Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan GA, Costantini F. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO (Eur Mol Biol Organ) J. 1994;13:1189–1196. doi: 10.1002/j.1460-2075.1994.tb06368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lattanzio G, Libert C, Aquilina M, Cappelletti M, Ciliberto G, Musiani P, Poli V. Defective development of pristane oil-induced plasmacytomas in IL-6–deficient Balb/c mice. Am J Pathol. 1997;151:689–696. [PMC free article] [PubMed] [Google Scholar]
- 19.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumor necrosis factor: a predictive genetic model of arthritis. EMBO (Eur Mol Biol Organ) J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vink A, Coulie P, Warnier G, Renauld JC, Stevens M, Donckers D, Van Snick J. Mouse plasmacytoma growth in vivo: enhancement by interleukin-6 (IL-6) and inhibition by antibodies direct against IL-6 or its receptor. J Exp Med. 1990;172:997–1000. doi: 10.1084/jem.172.3.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heremans H, Dillen C, Put W, Van-Damme J, Billiau A. Protective effect of anti–interleukin (IL)-6 antibody against endotoxin, associated with paradoxally increased IL-6 levels. Eur J Immunol. 1992;22:2395–2401. doi: 10.1002/eji.1830220932. [DOI] [PubMed] [Google Scholar]
- 22.Holmdahl R, Andersson M, Goldschidt TJ, Gustafsson K, Jansson L, Mo JA. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 23.Libert C, Vink A, Coulie P, Brouckarter P, Everaerdt B, Van Snick J, Fiers W. Limited involvement of interleukin-6 in the pathogenesis of lethal septic shock as revealed by the effect of monoclonal antibodies against interleukin-6 or its receptors in various murine models. Eur J Immunol. 1992;22:2625–2630. doi: 10.1002/eji.1830221023. [DOI] [PubMed] [Google Scholar]
- 24.Giraudo E, Arese M, Toniatti C, Strasly M, Primo L, Mantovani A, Ciliberto G, Bussolino F. IL-6 is an in vitro and in vivo autocrine growth factor for middle T antigen-transformed endothelial cells. J Immunol. 1996;157:2618–2623. [PubMed] [Google Scholar]
- 25.De Benedetti F, Alonzi T, Moretta A, Lazzaro D, Costa P, Poli V, Martini A, Ciliberto G, Fattori E. IL-6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I: a model for stunted growth in children with chronic inflammation. J Clin Invest. 1997;99:1–8. doi: 10.1172/JCI119207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson WC, Townes AS. Genetic susceptibility to murine collagen II autoimmune arthritis. Proposed relationship to the IgG2 autoantibody subclass response, complement C5, major histocompatibility complex (MHC) and non-MHC loci. J Exp Med. 1985;162:1878–1891. doi: 10.1084/jem.162.6.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boissier MC, Carlioz A, Fournier C. Experimental autoimmune arthritis in mice. II. Early events in the elicitation of the autoimmune phenomenon induced by homologous type II collagen. Clin Immunol Immunopathol. 1988;48:225–237. doi: 10.1016/0090-1229(88)90086-4. [DOI] [PubMed] [Google Scholar]
- 28.Walther Z, May LT, Sehgal PB. Transcriptional regulation of the interferon-β2cell differentiation factor BSF-2/hepatocyte–stimulating factor gene in human fibroblasts by other cytokines. J Immunol. 1988;140:974–977. [PubMed] [Google Scholar]
- 29.Aderka D, Le J, Vilcek J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J Immunol. 1989;143:3517–3523. [PubMed] [Google Scholar]
- 30.Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, Faggioni R, Fantuzzi G, Ghezzi P, Poli V. Defective inflammatory response in IL-6–deficient mice. J Exp Med. 1994;180:1243–1250. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu ZY, Brailly H, Wijdenes J, Bataille R, Rossi JF, Klein B. Measurement of whole body interleukin-6 (IL-6) production: prediction of the efficacy of anti–IL-6 treatments. Blood. 1995;86:3123–3131. [PubMed] [Google Scholar]
- 32.Suzuki H, Yasukawa K, Saito T, Narazaki M, Hasegawa A, Taga T, Kishimoto T. Serum soluble interleukin-6 receptor in MRL/lpr mice is elevated with age and mediates the interleukin-6 signal. Eur J Immunol. 1993;23:1078–1082. doi: 10.1002/eji.1830230515. [DOI] [PubMed] [Google Scholar]
- 33.Sany J. Intravenous immunoglobulin therapy for rheumatic diseases. Curr Opin Rheumatol. 1994;6:305–310. doi: 10.1097/00002281-199405000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Mori L, Iselin S, De Libero G, Lesslauer W. Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG1–treated and TNFR1-deficient mice. J Immunol. 1996;157:3178–3182. [PubMed] [Google Scholar]
- 35.McIntyre KM, Shuster DJ, Gillooly KM, Warrier RR, Connaughton SE, Hall LB, Arp LH, Gately MK, Magram J. Reduced incidence and severity of collagen-induced arthritis in interleukin-12–deficient mice. Eur J Immunol. 1996;26:2933–2938. doi: 10.1002/eji.1830261219. [DOI] [PubMed] [Google Scholar]
- 36.Bullard DC, Hurley LA, Lorenzo I, Sly LM, Beaudet AL, Staite ND. Reduced susceptibility to collagen-induced arthritis in mice deficient in intracellular adhesion molecule–1. J Immunol. 1996;157:3153–3158. [PubMed] [Google Scholar]
- 37.Tada Y, Ho A, Matsuyama T, Mak TW. Reduced incidence and severity of antigen-induced autoimmune diseases in mice lacking interferon regulatory factor-1. J Exp Med. 1997;185:231–238. doi: 10.1084/jem.185.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffiths RJ, Smith MA, Roach ML, Stock JL, Stam EJ, Milici AJ, Scampoli DN, Eskra JD, Byrum RS, Koller BH, McNeish JD. Collagen-induced arthritis is reduced in 5-lipoxygenase–activating protein-deficient mice. J Exp Med. 1997;185:1123–1129. doi: 10.1084/jem.185.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tada T, Ho A, Koh DR, Mak TW. Collagen-induced arthritis in CD4- or CD8-deficient mice. J Immunol. 1996;156:4520–4526. [PubMed] [Google Scholar]
- 40.Remmers EF, Longman RE, Du Y, O'Hare A, Cannon GW, Griffiths MM, Wilder RL. A genome scan localizes five non-MHC loci controlling collagen-induced arthritis in rats. Nat Genet. 1996;14:82–85. doi: 10.1038/ng0996-82. [DOI] [PubMed] [Google Scholar]
- 41.Jansson L, Holmdahl R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin Exp Immunol. 1993;94:459–465. doi: 10.1111/j.1365-2249.1993.tb08218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada M, Kitahara M, Kishimoto S, Matsuda T, Hirano T, Kishimoto T. IL-6/BSF-2 functions as a killer helper factor in the in vitro induction of cytotoxic T cells. J Immunol. 1988;141:1543–1549. [PubMed] [Google Scholar]
- 43.Chioccia G, Boissier MC, Manoury B, Fournier C. T cell regulation of collagen-induced arthritis in mice. II. Immunomodulation of arthritis by cytototoxic T cell hybridomas specific for type II collagen. Eur J Immunol. 1993;23:327–332. doi: 10.1002/eji.1830230204. [DOI] [PubMed] [Google Scholar]
- 44.Douni E, Akassoglou K, Alexopoulou L, Georgopoulos S, Haralambous S, Hill S, Kassiotis G, Kontoyiannis D, Pasparakis M, Plows D, et al. Transgenic and knockout analyses of the role of TNF in immune regulation and disease pathogenesis. J Inflamm. 1996;47:27–38. [PubMed] [Google Scholar]
- 45.Probert, L., D. Plows, G. Kontogeorgos, and G. Kollias. The type I interleukin-1 receptor acts in series with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic mice. Eur. J. Immunol. 25:1794–1797. [DOI] [PubMed]
- 46.Joosten LAB, Helsen M, van de Loo FA, van den Berg WB. Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. Arthritis Rheum. 1996;39:797–809. doi: 10.1002/art.1780390513. [DOI] [PubMed] [Google Scholar]
- 47.Lewthwaite, J., S.M. Blake, T.E. Hardingham, P.J. Warden, and B. Henderson. The effect of recombinant human interleukin 1 receptor antagonist on the induction phase of antigen induced arthritis in the rabbit. J. Rheumatol. 21:467–472. [PubMed]
- 48.Wooley PH, Whalen JD, Chapman DL, Berger AE, Richard KA, Asparr DG, Staite ND. The effect of an interleukin-1 receptor antagonist protein on type II collagen-induced arthritis and antigen-induced arthritis in mice. Arthritis Rheum. 1993;36:1305–1314. doi: 10.1002/art.1780360915. [DOI] [PubMed] [Google Scholar]
- 49.Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H, Woody JN. Randomised double-blind comparison of chimeric monoclonal antibody to tumor necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 50.Elliott MJ, Woo P, Charles P, Long-Fox A, Woody JN, Maini RN. Suppression of fever and acute phase response in a patient with juvenile chronic arthritis treated with monoclonal antibody to tumour necrosis factor-α (cA2) Br J Rheumatol. 1997;36:589–593. doi: 10.1093/rheumatology/36.5.589. [DOI] [PubMed] [Google Scholar]
- 51.Wendling, A., E. Racadot, and J. Wijdenes. Treatment of severe rheumatoid arthritis by anti-interleukin 6 monoclonal antibody. J. Rheumatol. 20:259–262. [PubMed]
- 52.Ciliberto G, Cortese R. Interleukin-6-blocking agents. Drugs News & Perspectives. 1997;10:394–400. [Google Scholar]