

Abstract
Two novel synthetic tetrapeptides, VEID-CHO and DMQD-CHO, could selectively inhibit caspase-6 and caspase-3, respectively. We used these inhibitors to dissect the pathway of caspase activation in Fas-stimulated Jurkat cells and identify the roles of each active caspase in apoptotic processes. Affinity labeling techniques revealed a branched protease cascade in which caspase-8 activates caspase-3 and -7, and caspase-3, in turn, activates caspase-6. Both caspase-6 and -3 have major roles in nuclear apoptosis. Caspase-6 cleaves nuclear mitotic apparatus protein (NuMA) and mediates the shrinkage and fragmentation of nuclei. Caspase-3 cleaves NuMA at sites distinct from caspase-6, and mediates DNA fragmentation and chromatin condensation. It is also involved in extranuclear apoptotic events: cleavage of PAK2, formation of apoptotic bodies, and exposure of phosphatidylserine on the cell surface. In contrast, a caspase(s) distinct from caspase-3 or -6 mediates the disruption of mitochondrial membrane potential (permeability transition) and the shrinkage of cytoplasm. These findings demonstrate that caspases are organized in a protease cascade, and that each activated caspase plays a distinct role(s) in the execution of Fas-induced cell death.
Fas/APO-1/CD95 is a cell-surface receptor essential for the regulation of the immune system (1), especially for the termination of T cell–mediated responses, the maintenance of immune privilege (2), and the prevention of autoantibody production (3). Evidence is accumulating that a family of cysteine proteases, named caspases (4), play critical roles in Fas-induced apoptotic cell death. The inhibition of caspases by cowpox serpin CrmA or by synthetic peptide inhibitors such as zVAD-fmk (benzyloxycarbonyl-Val-Ala-Asp(OMe)- fluoromethylketone) and zDEVD-fmk (benzyloxycarbonyl-Asp-Glu-Val-Asp(OMe)-fluoromethylketone), allows Fas-stimulated cells to survive (5) and proliferate (6). Cross-linking of the Fas receptor with Fas ligand (7) or with anti-Fas mAb (8) recruits the death-inducing signaling complex (DISC)1 consisting of FADD/MORT1 and pro–caspase-8 (MACH/ FLICE/Mch5) (9, 10). Pro–caspase-8 recruited to the DISC is activated and released into the cytosol (11). The active caspase-8 activates multiple caspases (12–14), which elicit downstream biochemical changes such as mitochondrial permeability transition (PT; 15), DNA fragmentation (16), loss of nuclear lamina (17), and exposure of phosphatidylserine (PS) on the cell surface (18). Together, these changes culminate in the process of apoptosis (19). However, the activation of caspases downstream of caspase-8 is a poorly understood process. Nor is it clear which caspase(s) contributes to each apoptotic biochemical or morphological change.
Each caspase family protease is present in nonapoptotic cells as an inactive proenzyme. The enzyme becomes active when the precursor is cleaved into a large subunit with a molecular mass of ∼20 kD and a small subunit with a molecular mass of ∼10 kD, and forms a tetramer consisting of two large and two small subunits (20). Most well-known activation sites are immediately after aspartic acid (D) residues of pro-caspases (21), and the unique characteristic of caspases is that they cleave after aspartic acids within their substrates (22). This suggests that caspases are activated by autocatalysis or by mutual processing (23). Different cascade pathways for the activation of caspases have been proposed, based on the ability of certain recombinant caspases to cleave and activate a limited spectrum of recombinant pro-caspases in vitro and on the cleavage of endogenous pro-caspases within cells in which a caspase(s) is exogenously overexpressed (12, 24–26). However, these studies did not identify the cascades that are induced in cells in response to death-inductive stimuli. A study using YVAD-CHO, which inhibits caspase-1 (IL-1β converting enzyme; ICE), and DEVD-CHO, which inhibits caspase-3 (CPP32/Yama/ apopain), indicated that there is a protease cascade in which caspase-1–like activity is upstream of caspase-3–like activity in Fas-mediated apoptosis of mouse W4 cells (27). However, the caspases involved in this cascade, especially the identity of the caspase-1–like protease(s), remain unclear.
Affinity labeling techniques have been developed taking advantage of the ability of active caspases, not their inactive precursors, to bind their substrates. Derivatized peptides mimicking substrates for caspases enter the substrate-binding pockets of active caspases and irreversibly bind to the active site cysteine in the large subunits, allowing their detection on immunoblots (17, 28–30) or on histological sections (31). Application of this technique, combined with inhibitor studies (14), identified multiple active caspases including caspase-3–p20, caspase-3–p17, caspase-6 (Mch2), caspase-7 (Mch3/ ICE-LAP3/CMH-1), and caspase-8 in Fas-stimulated human Jurkat T cells. Stepwise appearances of active caspases (14) were highly suggestive of a cascade of caspase activation. However, a sufficiently detailed dissection of the activation pathway would have required inhibitors with high specificity for individual caspases, and the available caspase inhibitors such as DEVD-CHO had too broad of a spectrum for this purpose. Although CrmA is rather selective for caspase-1, -4, -5, and -8 (32–34), the inhibition of caspase-8 by CrmA completely suppresses subsequent activation of other caspases (35). This has made further analysis of downstream events difficult.
Here, we demonstrated that novel tetrapeptide inhibitors, VEID-CHO (acetyl-Val-Ileu-Asp-aldehyde) and DMQD-CHO (acetyl-Asp-Met-Gln-Asp-aldehyde), can work as specific inhibitors of caspase-6 and -3, respectively. These reagents were used to analyze the caspase activation induced by Fas ligation in Jurkat cells with that elicited by caspase-8 in cell-free Jurkat extracts. In each case, caspase-6 is activated downstream of caspase-3, forming a protease cascade. Moreover, we identified the caspases upstream of each caspase-dependent event in Fas-stimulated Jurkat cells.
Materials and Methods
Reagents.
Rabbit polyclonal antiserum against the large subunit of caspase-7 (36) was provided by Gerald M. Cohen (University of Leicester, Leicester, UK). DMQD-CHO (14), VEID-CHO (Peptide Institute, Osaka, Japan), and zVAD-fmk (Enzyme Systems, Dublin, CA) were dissolved at 10 mM in DMSO and stored at −80°C. DiOC6(3) (3, 3′-dihexyloxacarbocyanine iodide; Molecular Probes Inc., Eugene, OR) was dissolved at 0.5 mM in DMSO and stored at −20°C. Propidium iodide (Calbiochem Corp., La Jolla, CA) was dissolved in PBS at 100 μg/ml and stored at 4°C.
Inhibition of Caspases in Fas-stimulated Cells and in a Cell-free Reaction Triggered by Caspase-8.
Jurkat cells were pretreated with VEID-CHO or DMQD-CHO for 1 h at 37°C before stimulation with anti-Fas mAb (CH-11, 100 ng/ml; reference 8). Preparation of cytoplasmic extracts and the affinity labeling of active caspases using YV(bio)KD-aomk (provided by Nancy A. Thornberry, Merck, Rahway, NJ; reference 28) were performed as previously described (14). Cytoplasmic extracts from logarithmically growing Jurkat cells were preincubated with VEID-CHO or DMQD-CHO for 1 h at 37°C before the addition of purified recombinant active caspase-8 to trigger the stepwise activation of caspases (14).
Single-cell Analysis of Intracellular Caspase Activity in Fas-stimulated Jurkat Cells.
Jurkat cells (2.5 × 105) treated with anti-Fas mAb were gently centrifuged in a microfuge tube. The cell pellet was resuspended with 50 μl of 10 μM PhiPhiLux-G2D2 substrate solution (OncoImmunin, College Park, MD) in RPMI-1640 supplemented with 10% FCS. After incubation for 1 h at 37°C avoiding direct light, the sample was diluted with 0.5 ml of ice-cold flow cytometry dilution buffer (OncoImmunin) and filtered through a nylon mesh to remove cell aggregates and/or debris. Flow cytometric analysis was performed within 90 min of the end of the incubation period using FACScan® flow cytometer and LYSIS II software (Becton Dickinson, Mountain View, CA).
Immunoblotting for the Cleavage of Caspase Substrates.
Nuclei isolated from HeLa cells as described (37) were incubated with purified recombinant caspases, with Escherichia coli lysates containing recombinant caspases (14), or with the extract from preapoptotic chicken DU249 cells (S/M extract; reference 37). Whole cell extracts from Jurkat cells were obtained by adding SDS-PAGE sample buffer (50 mM Tris-HCl [pH 6.8], 15% sucrose, 3% SDS, and 0.01% bromophenol blue) to cell pellets and boiling for 3 min. Proteins were resolved in SDS-PAGE gels, transferred to nitrocellulose membranes (Hybond ECL, Amersham Corp., Arlington Heights, IL), and stained with rabbit polyclonal anti–nuclear mitotic apparatus protein (NuMA) antiserum (r240-C; reference 38), anti–poly (ADP-ribose) polymerase (PARP) mAb (C-2-10; BIOMOL, Plymouth Meeting, PA), or rabbit polyclonal anti-ste20-subdomain VI Ab (Upstate Biotechnology Inc., Lake Placid, NY). Signals were visualized by enhanced chemiluminescence reagents (Amersham Corp.) according to the manufacturer's instructions.
Flow Cytometric Analysis of DNA Fragmentation, Mitochondrial Membrane Potential, and Cell-surface Exposure of PS.
For the analysis of DNA fragmentation, Jurkat cells (106) were permeabilized by fixation with 70% ethanol for 2 h on ice, washed once with PBS, treated with 10 μg/ml RNase A in PBS for 30 min at 37°C, and incubated with 50 μg/ml propidium iodide for 30 min on ice protected from light. For simultaneous recording of mitochondrial PT and PS externalization, Jurkat cells (5 × 105) were incubated with 40 nM DiOC6 (3) (Molecular Probes) together with 1.5 μg/ml PE-conjugated annexin V (R&D Sys. Inc., Minneapolis, MN) in 10 mM Hepes/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2 for 15 min at room temperature. Stained cells were filtered through a nylon mesh and immediately subjected to flow cytometry.
Results
Affinity Labeling of Active Caspases in Fas-stimulated Jurkat Cells.
YV(bio)KD-aomk (28), an affinity-labeling reagent for active caspases (14), recognized four major polypeptides in Fas-stimulated Jurkat cells, designated F22, F20, F19, and F17 according to their apparent molecular mass in kilodaltons (Fig. 1 A, lane 1). Previous studies revealed that these correspond to caspase-7, caspase-3-p20, caspase-6, and caspase-3-p17, respectively (14, 30). Using rabbit antiserum raised against caspase-7 (36), we have confirmed that F22 corresponds to caspase-7 (Fig. 1 B, lane 1, and data not shown).
Figure 1.

(A) Affinity labeling of caspases activated in Fas-stimulated Jurkat cells and their inhibition with VEID-CHO. Jurkat cells (106/ml), preincubated for 1 h with indicated concentrations of VEID-CHO at 37°C in 5% CO2, were stimulated with anti-Fas mAb (CH-11, 100 ng/ ml) in serum-free RPMI-1640 for 4 h at 37°C. Cytoplasmic extracts from the cells were incubated with 10 μM YV(bio)KD-aomk for 5 min at 37°C, resolved in a 16% SDS-PAGE gel, and transferred to a nitrocellulose membrane. Labeled caspases were visualized with horseradish peroxidase–conjugated streptavidin and enhanced chemiluminescence. (B) Effect of VEID-CHO on processing of pro–caspase-7. The same blot as in A was reprobed with rabbit antiserum raised against the large subunit of caspase-7. The processing of pro–caspase-7 (precursor) into the mature large subunit of caspase-7 (p22), which comigrated with F22, is displayed.
Selective Inhibition of Caspase-6 by VEID-CHO.
VEID-CHO was synthesized based on the sequence within lamin A cleaved by caspase-6, but not by caspase-3 or -7 (17, 39). Caspase-3, -4, -6, -7, and -8 were expressed as recombinant proteins in E. coli and tested for their sensitivities to VEID-CHO. Caspase-4 (TX/ICH-2/ICErel-II) was included because it is involved in the Fas-mediated apoptosis of HeLa cells (40). As shown in Table 1, the activity of caspase-6 was inhibited by VEID-CHO at concentrations at least 100-fold lower than those required to inhibit caspase-3, -4, -7, and -8. The active caspases in Fas-stimulated Jurkat cells exhibited the sensitivities to VEID-CHO similar to the recombinant proteins: F19/caspase-6 was at least 100-fold more sensitive to VEID-CHO than were F22/ caspase-7, F20/caspase-3-p20, or F17/caspase-3-p17 (Table 1). Therefore, VEID-CHO at 100 nM can work as a selective inhibitor of caspase-6.
Table 1.
Effect of VEID-CHO on the Activity of Caspases
| VEID-CHO | F22 | F20 | F19 | F17 | Caspase-8 | Caspase-7 | Caspase-6 | Caspase-3 | Caspase-4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 nM | + | + | + | + | + | + | + | + | + | |||||||||
| 10 nM | + | + | ± ∼ − | + | + ∼ ± | + | ± ∼ − | + | + | |||||||||
| 100 nM | + | + | − | + ∼ ± | ± | + | − | + ∼ ± | + | |||||||||
| 1 μM | ± | + ∼ ± | − | ± ∼ − | ± ∼ − | ± | − | ± ∼ − | + | |||||||||
| 10 μM | − | ± ∼ − | − | − | − | − | − | − | + ∼ ± | |||||||||
| 100 μM | − | − | − | − | − | − | − | − | − |
Cytoplasmic extracts (30 μg total protein) from Jurkat cells treated with anti-Fas mAb for 3 h (F22, F20, F19, and F17), purified His-tagged caspase-8 (453 ng), or -6 (221 ng), and E. coli lysates containing caspase-7 (1.2 μg total protein), -3 (4.8 μg total protein), or -4 (2.1 μg total protein) were pretreated for 15 min at 37°C with VEID-CHO at concentrations indicated in 10-μl mixtures before labeling with 10 μM YV(bio)KD-aomk. +, no inhibition; ± ∼ −, <25% inhibition, ±, 25–75% inhibition; ± ∼ −, 75–99% inhibition; −, complete inhibition.
Studies in vitro have shown that caspase-6 can process pro-caspase-3 (26, 41). A hypothetical cascade pathway has been proposed in which caspase-3 and -7 are activated downstream of caspase-6 (24). Selective inhibition of caspase-6 activity by VEID-CHO should block the activation of those caspases activated downstream of caspase-6. Accordingly, Jurkat cells were exposed to VEID-CHO before stimulation with anti-Fas mAb. As shown in Fig. 1 A, the activity of F19/caspase-6 was inhibited by VEID-CHO partially at 1 μM (lane 4) and completely at 10 μM (lane 5). Concentrations of VEID-CHO in the culture medium required to inhibit caspase-6 was much higher than in vitro experiments (Table 1), most likely due to the impermeability of the plasma membrane to the peptide inhibitor. Despite the loss of F19/caspase-6 activity at 10 μM VEID-CHO, the labeling of F22/caspase-7 and F20/caspase-3-p20 was unaffected (Fig. 1 A, lane 5). Thus, caspase-3 and caspase-7 can be activated independent of caspase-6 activity. Suppression of F17/caspase-3-p17 labeling was observed at 10 μM (lane 5), possibly because the activity of caspase-3-p17 may be blocked at the effective intracellular concentration of VEID-CHO, perhaps around 1 μM (Table 1). The loss of all caspase labeling at 100 μM (lane 6) should reflect the ability of VEID-CHO to inhibit caspase-8 at high concentrations (Table 1), thereby preventing the processing of all downstream caspases. Indeed, the processing of pro–caspase-7 was blocked at 100 μM VEID-CHO, as discerned by reprobing the same blot with anti–caspase-7 Ab (Fig. 1 B, lane 6). Similar analysis was performed in cell-free conditions in which the stepwise activation of caspases similar to that in Fas-stimulated cells was induced by adding active recombinant caspase-8 to cytoplasmic extracts from normally growing Jurkat cells (14). VEID-CHO at 10 nM added before caspase-8 selectively blocked the activity of caspase-6 with minimal effects on the activation of caspase-7, caspase-3-p20, or caspase-3-p17 (data not shown). Taken together, these results indicate that caspase-3 and -7 are activated independently of caspase-6 in Fas-stimulated Jurkat cells.
Analysis of Caspase Cascade by Selective Inhibition of Caspase-3 with DMQD-CHO.
DMQD-CHO was synthesized based on the cleavage site within protein kinase C δ (PKCδ). Although PKCδ is cleaved by caspase-3, it is not a substrate for caspase-1, -2, -4, -5, -6, or -7 (42). Our previous study showed that F20/caspase-3-p20 is inhibited at lower concentrations than either F22/caspase-7 or F19/caspase-6 (14). Caspase-8 is also relatively insensitive to DMQD-CHO (data not shown). First, we investigated the effects of DMQD-CHO on the cell-free activation of caspases in Jurkat cytoplasmic extracts triggered by recombinant caspase-8 (14). YV(bio)KD-aomk labeling of F20/caspase-3-p20 was inhibited by DMQD-CHO at 1–2 μM (Fig. 2 A, lanes 4 and 5), the concentrations previously shown to block the activity of F20/caspase-3-p20 (14). The labeling of F17/ caspase-3-p17 was also suppressed, most likely because the cleavage of the prodomain of caspase-3-p20, which generates caspase-3-p17, is mediated by autocatalysis (25, 43). Interestingly, although the activity of F19/caspase-6 is insensitive to DMQD-CHO even at 100 μM (14), the appearance of F19/caspase-6 was prevented by the preincubation with 1–2 μM DMQD-CHO before the addition of caspase-8 (Fig. 2 A, lanes 4 and 5). This indicated that the activation of F19/caspase-6 in response to caspase-8 is dependent on the activity of F20/caspase-3-p20. This finding is supported by a previous report in which efficient activation of caspase-6 by caspase-8 in a cell-free system requires the presence of cytoplasmic extracts (13). This suggests that caspase-6 is activated indirectly via another protease(s) present in the extracts. In contrast, the labeling of F22/caspase-7 was unhindered up to 5 μM DMQD-CHO (lane 6). This indicated that caspase-7 is activated independent of caspase-3 activity. The loss of all labeling at 10 μM (lane 7) is due to the inhibition of caspase-8 (data not shown).
Figure 2.

(A) DMQD-CHO inhibits the caspase-8–triggered appearances of active caspase-3 and caspase-6 in Jurkat cell extracts. Cytoplasmic extracts (80 μg total protein) from normally growing Jurkat cells were preincubated with the indicated concentrations of DMQD-CHO before the addition of caspases-8 (680 ng). After incubation for 4 h at 37°C, the incubated mixtures were labeled with 10 μM YV(bio)KD-aomk (lanes 1–7). (B) Caspase-3 activates endogenous caspase-6 in Jurkat cytoplasmic extracts. Purified recombinant caspase-3 (10 ng) was added to extracts (70 μg total protein) from nonapoptotic Jurkat cells and incubated for the indicated time periods (lanes 1–4, and 7) at 37°C. Jurkat extracts alone (lane 5) or caspase-3 alone (lane 6) were also incubated for 4 h at 37°C. The samples were treated with (lanes 1–6) or without (lane 7) 10 μM YV (bio)KD-aomk. (C) Inhibition of caspase-6 activation in Fas-stimulated Jurkat cells by the blockade of caspase-3 with DMQD-CHO. Jurkat cells (106/ml) were pretreated with the indicated concentrations of DMQD-CHO before Fas stimulation. Cytoplasmic extracts prepared from the cells were incubated with (lanes 1–4) or without (lane 5) 10 μM YV(bio)KD-aomk. (D) Time course of Fas-induced caspase activation in the presence or absence of DMQD-CHO. Jurkat cells (106/ml), preincubated for 1 h with (lanes 1–5) or without (6–10) 100 μM DMQD-CHO, were treated with anti-Fas mAb for the time indicated, and their cytoplasmic extracts were affinity labeled with 10 μM YV(bio)KD-aomk. (A–D) Labeled active caspases were visualized with horseradish peroxidase–conjugated streptavidin after electrophoresis in 16% SDS-PAGE gels and transfer to nitrocellulose membranes.
We next tested whether caspase-3 can activate endogenous pro-caspase-6 present in the extracts from normally growing Jurkat cells. Although caspase-3 cleaves and activates recombinant pro-caspase-6 expressed in E. coli (26), Orth et al. could not detect processing of pro-caspase-6 when caspase-3 was added to extracts of nonapoptotic Jurkat cells (24). We added recombinant active caspase-3 to the extract and assessed the activity of caspase-6 by YV(bio)KD-aomk affinity labeling. As shown in Fig. 2 B, the addition of caspase-3 induced active F19/caspase-6 together with F20/ caspase-3-p20 (lanes 2–4). Thus, caspase-3 is able to activate endogenous pro-caspase-6 in Jurkat cytoplasmic extracts, supporting the existence of the protease cascade initiated by caspase-8, in which caspase-6 is activated downstream of caspase-3. Overexposure of the film showed that F22/ caspase-7 and F17/caspase-3-p17 are induced to a lesser degree (data not shown). This suggests that caspase-3 can also activate caspase-7 at much lower efficiency than caspase-8.
Are caspases organized in a similar cascade within Fas-stimulated Jurkat cells? Preincubation of Jurkat cells with 10–100 μM DMQD-CHO before Fas receptor cross-linking suppressed the labeling of F20/caspase-3-p20 and F17/ caspase-3-p17. Concomitant with the suppression of caspase-3 activity, the appearance of F19/caspase-6 was also blocked (Fig. 2 C, lanes 3 and 4). In contrast, the labeling of F22 persisted up to 100 μM DMQD-CHO (Fig. 2 C, lane 4). Reprobing of the same blot with anti–caspase-7 Ab confirmed that F22 in the presence of 1–100 μM DMQD-CHO corresponds to the large subunit of caspase-7 (data not shown). As the concentration of DMQD-CHO required to block the activity of caspase-6 is ∼10-fold higher than caspase-7 (14), the loss of caspase-6 labeling with preserved caspase-7 activity indicated that the suppression of caspase-3-p20 activity resulted in the loss of caspase-6 activation. Time course studies (Fig. 2 D) demonstrated that active F22/caspase-7 appeared at the same time point irrespective of the presence of DMQD-CHO. This result showed that activation of caspase-7 was uninterrupted when caspase-3 activity was blocked with 100 μM DMQD-CHO.
Demonstration of Caspase Activity in Intact Fas-stimulated Jurkat Cells Pretreated with DMQD-CHO.
We were concerned about the possibility that caspase-7 was inhibited by DMQD-CHO in Jurkat cells, whereas it was reactivated by reversible dissociation from the inhibitor when the cells were disrupted to prepare cytoplasmic extracts. Thus, we measured caspase activity in intact Fas-stimulated cells by flow cytometry, using PhiPhiLux-G2D2. This cell-permeable fluorescent substrate for caspases emits increased fluorescence when it is proteolyzed within its PARP cleavage site sequence (GDEVDGID). Since caspase-3 and -7 appear to be primarily responsible for PARP cleavage during apoptosis (21), the increase in fluorescence should mainly reflect the activities of caspase-3 and -7. Fig. 3 A depicts time course studies displaying the forward light scatter (FSC), an estimate of cell size, and the PhiPhiLux-G2D2 fluorescence of Jurkat cells (FL2). After Fas ligation, a gradual augmentation of the PhiPhiLux-G2D2 fluorescence was noted along with a decrease in FSC, suggesting the activation of caspases concomitant with the shrinkage of cytoplasm. The increase in fluorescence correlated well with the kinetics of caspase activation measured by the ability of Jurkat cytoplasmic extracts to cleave DEVD-MCA (14). The enhanced PhiPhiLux-G2D2 fluorescence and the decrease in FSC was not observed on incubation of the cells without anti-Fas mAb (data not shown). The increase in fluorescence was abolished by the presence of 100 μM zVAD-fmk (Fig. 3 A), a broad-spectrum caspase inhibitor that blocks the activation of PARP-cleaving caspases (44), supporting the notion that caspases are responsible for cleavage of PhiPhiLux-G2D2. Lower fluorescence was observed in the population of cells with the lowest FSC, possibly due to the loss of the fluorescent molecules by budding out of apoptotic bodies. In the presence of DMQD-CHO at 100 μM, only minimal suppression of protease activity was noted, with the PhiPhiLux-G2D2 fluorescence significantly enhanced (Fig. 3 B, DMQD 100 μM) compared with untreated cells (Fig. 3 B, control). This was consistent with the persistence of caspase-7 activity demonstrated by affinity labeling (Fig. 2 C, lane 4). As expected, incubation with 100 μM DMQD-CHO alone without Fas simulation did not induce the increase in PhiPhiLux-G2D2 fluorescence or cell shrinkage (data not shown).
Figure 3.

Flow cytometric analysis of caspase activity in Fas-stimulated Jurkat cells. (A) Time course of Fas-induced caspase activation and cell shrinkage. Jurkat cells (2.5 × 105) were treated with anti-Fas Ab (CH-11, 100 ng/ml) at 37°C for the time indicated, followed by incubation with 10 μM PhiPhiLux-G2D2 substrate for 1 h. Three-dimensional display of PhiPhiLux-G2D2 fluorescence (FL2) on a logarithmic scale, FSC on a linear scale, and relative cell number on a linear scale are shown for each time point. Arrowheads, baseline fluorescence of substrate-loaded cells in the absence of caspase activation; zVAD-fmk, Jurkat cells were preincubated with 100 μM zVAD-fmk for 1 h at 37°C before the stimulation with anti-Fas mAb for 4 h. (B) Effect of DMQD-CHO on caspase activity and cell size. Jurkat cells were preincubated in the absence or presence of the indicated concentrations of DMQD-CHO before Fas stimulation for 4 h. control, Jurkat cells before Fas stimulation. Dot plots showing fluorescence intensity (FL2) on a logarithmic scale as a function of FSC on a linear scale are presented. Dotted lines, average fluorescence of substrate-loaded nonapoptotic cells. Histogram shows fluorescence intensity (FL2) of the gated cells.
Dissection of Fas Signaling Pathways by Selective Caspase Inhibitors.
Our results above indicated that VEID-CHO up to 10 μM can selectively inhibit the activity of caspase-6, and that 100 μM DMQD-CHO can selectively suppress the activities of caspase-3 and -6 in Fas-stimulated Jurkat cells (Fig. 4). We extended these findings to dissect the events downstream of these caspases. In particular, events downstream of caspase-6 should be blocked both by 10 μM VEID-CHO and by 100 μM DMQD-CHO, whereas events downstream of caspase-3 should be blocked only by 100 μM DMQD-CHO. Processes unaffected by 10 μM VEID-CHO or by 100 μM DMQD-CHO should be downstream of a caspase(s) distinct from caspase-3 and -6, or caspase independent. Finally, all caspase-8–dependent events should be suppressed by 100 μM VEID-CHO.
Figure 4.

Protease cascade in Fas-stimulated Jurkat cells and the targets of tetrapeptide inhibitors.
Cleavages of Nuclear Substrates by Caspases.
NuMA, a structural protein localized in the nuclear matrix, is cleaved in Fas-stimulated Jurkat cells (45, 46). Although NuMA cleavage is blocked by broad-spectrum caspase inhibitors such as DEVD-CHO and zVAD-fmk (45), it remained unsettled whether the cleavage is directly catalyzed by a caspase(s), or is due to a noncaspase protease(s) activated downstream of a caspase(s) (47). We have found that recombinant and HeLa nuclear NuMA proteins are cleaved at multiple sites by multiple distinct caspases active in a cell-free system (data not shown) using extracts from preapoptotic chicken DU249 cells (S/M extracts; reference 37). As shown in Fig. 5 A, recombinant caspase-3 (lane 5), caspase-7 (lane 4), and caspase-4 (lane 3) cleaved NuMA in nuclei isolated from HeLa cells, yielding fragments with an Mr of ∼ 190 and 180 kD. Time course studies revealed that the 190-kD fragment was the first to be detected, then disappeared as the 180-kD fragment gradually appeared (data not shown). This suggested that the 180 kD fragment was derived from proteolysis of the 190-kD fragment. Caspase-6 also yielded a fragment with Mr of ∼160 kD in addition to the 190- and 180-kD fragments (Fig. 5 A, lane 6). These fragments comigrated with those detected in the cell-free system composed of HeLa nuclei and S/M extracts (Fig. 5 A, lane 7). Jurkat cells undergoing apoptosis in response to 1 μM staurosporine (14) showed two distinct fragments of NuMA with Mr of ∼180 and 160 kD (Fig. 5 A, lane 8) comigrating with those of HeLa nuclear NuMA generated by recombinant caspases or by caspases in S/M extracts. Similar results were obtained using purified recombinant human NuMA and recombinant caspase-3, -4, -6, -7, -8, or S/M extracts (data not shown). Thus, these caspases can cleave NuMA protein directly.
Figure 5.

(A) Cleavage of HeLa nuclear NuMA by recombinant caspases. HeLa nuclei (1.3 ×105) were incubated for 2 h at 37°C with mitotic dilution buffer (37; lane 1), 884 ng purified His-tagged active caspase-8 (lane 2), 70 μg E. coli lysate expressing caspase-4 (lane 3), 40 μg E. coli lysate expressing caspase-7 (lane 4), 2.7 μg purified His-tagged active caspase-3 (lane 5), 289 ng purified His-tagged active caspase-6 (lane 6), or 76 μg S/M extracts. Lane 8, whole cell extracts from Jurkat cells (2 × 105) treated with 1 μM staurosporine for 3 h. (B–D) Effects of DMQD-CHO and VEID-CHO on the cleavage of NuMA (B), PARP (C), and PAK2 (D) in Fas-stimulated Jurkat cells. Whole cell extracts from Fas-stimulated Jurkat cells preincubated with indicated concentrations of DMQD-CHO or VEID-CHO were resolved in 5% (B) or 10% (C and D) SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were probed with rabbit polyclonal anti-NuMA Ab (r240-C; A and B), anti-PARP mAb (C-2-10; C), and rabbit polyclonal anti-ste20-subdomain VI Ab that stains PAK2 (D).
Fas-stimulated Jurkat cells also showed two fragments of NuMA with Mr of ∼180 and 160 kD (Fig. 5 B, lane 1). In the presence of 1–10 μM VEID-CHO, the production of the 160-kD fragment in Fas-stimulated Jurkat cells was suppressed (Fig. 5 B, lanes 7 and 8), in parallel with the loss of caspase-6 activity (Fig. 1 A, lanes 4 and 5). This is consistent with the unique ability of recombinant caspase-6 to generate the 160-kD fragment, and indicates that caspase-6 is responsible for the cleavage producing this fragment in Fas-stimulated Jurkat cells. In contrast, the proteolysis yielding the 180-kD fragments was not affected at 1 μM (Fig. 5 B, lane 7). Although recombinant caspase-6 is able to produce the 190- and 180-kD fragments in vitro, it is not the major protease responsible for the production of those fragments in apoptotic Jurkat cells. The moderate decrease in the 180-kD fragment at 10 μM VEID-CHO (Fig. 5 B, lane 8) was considered to reflect the inhibition of the activities of caspase-3-p17 as well as caspase-6 at this concentration (Fig. 1 A, lane 5). Interestingly, 100 μM DMQD-CHO blocked the generation of the 180-kD fragment as well as the 160-kD fragment. These results indicated that caspase-3 is responsible for the production of the 180-kD fragment in Fas-stimulated Jurkat cells. Although caspase-7 is able to cleave HeLa nuclear and recombinant NuMA in vitro (Fig. 5 A, lane 4, and data not shown), the presence of caspase-7 activity alone was not sufficient for the cleavage of NuMA in Fas-stimulated Jurkat cells. Thus, there is a discrepancy between the cell-free and cellular results.
This discrepancy may be explained by the unique intracellular localization of caspase-7 within intact cells. Caspase-7 has been detected solely in the cytoplasm of Jurkat cells (48), whereas caspase-3 can be detected in the nuclei of some cell types (49) and of regressing, apoptotic neuroblastoma cells (50). In cell-free systems, nuclei may be permeabilized to some extent, allowing free entry of proteins normally inaccessible to the intranuclear compartment. To test this possibility, we examined whether the cleavage of another nuclear substrate, PARP, is mediated solely by caspase-3 in Jurkat cells. In contrast to NuMA, 100 μM DMQD-CHO only weakly inhibited the PARP cleavage (Fig. 5 C, lane 4). Suppression of caspase-6 activity with 1 μM VEID-CHO did not affect the PARP cleavage (Fig. 5 C, lane 6). Partial blockade of the cleavage with 10 μM VEID-CHO (Fig. 5 C, lane 7) was likely to reflect the loss of caspase-3-p17 activity (Fig. 1 A, lane 5). PARP cleavage was markedly suppressed by 100 μM VEID-CHO (Fig. 5 C, lane 8). These indicated that caspase-3 is only partially responsible for PARP cleavage within Fas-stimulated Jurkat cells. Thus, another caspase(s), most likely caspase-7, cleaves PARP in the cells. This suggests that caspase-7 can enter the nucleus and cleave its nuclear substrate(s). The basis for the difference between caspase-3 and -7 in the ability to cleave NuMA in Jurkat cells remained unclear.
Role of Caspase-3 and -6 in Nuclear Apoptotic Changes.
The inhibition of caspase-3 plus -6 by 100 μM DMQD-CHO completely blocked the nuclear morphological changes as revealed by staining of chromatin structures with 4′,6-diamidino-2-phenylindole (DAPI; Fig. 6 h). In contrast, the inhibition of caspase-6 alone with 10 μM VEID-CHO resulted in the blockade of apoptotic morphology halfway after the nuclear chromatin condensation into the nuclear periphery (“rim collapse”; Fig. 6 d). These indicate that caspase-3 is essential for the early morphological changes of nuclei up to the rim collapse, whereas caspase-6 plays an essential role in the late morphological changes such as the shrinkage and fragmentation of apoptotic nuclei. This is consistent with previous results in a cell-free system that a lamin-cleaving caspase is responsible for the late nuclear events, whereas a different caspase mediates the initiation of nuclear morphological changes (51–53).
Figure 6.
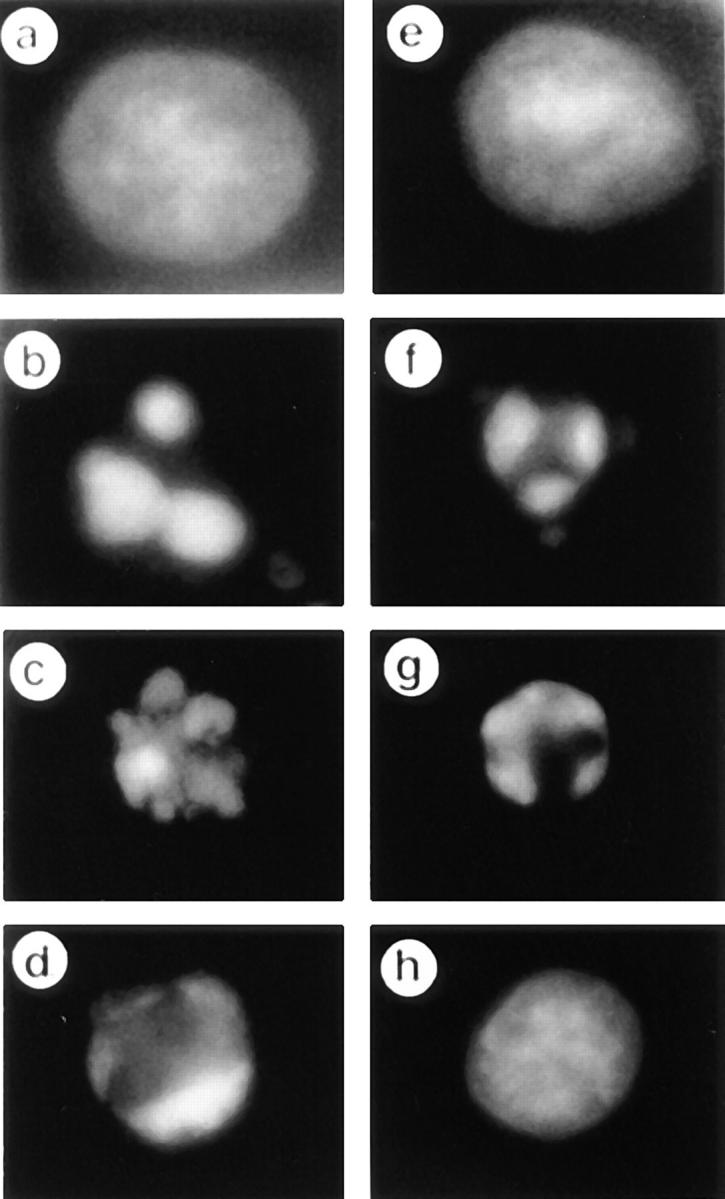
Effects of DMQD-CHO and VEID-CHO on chromatin condensation, nuclear shrinkage, and nuclear fragmentation. Jurkat cells were preincubated for 1 h at 37°C with 1% DMSO (b), with 1 μM (c), 10 μM (d), or 100 μM (e) VEID-CHO, or with 1 μM ( f ), 10 μM (g), or 100 μM (h) DMQD-CHO before stimulation with anti-Fas mAb for 4 h. Jurkat cells were also incubated for 4 h without Fas stimulation (a). Cells were fixed by 1% glutaraldehyde and stained with 2 μg/ml DAPI as previously described (14).
The principal role of caspase-3 and -6 in nuclear morphological changes prompted us to examine whether these proteases are upstream of DNA fragmentation, one of the major biochemical changes in apoptotic nuclei (19). Flow cytometric analysis of Fas-stimulated Jurkat cells stained with propidium iodide displayed time-dependent increase in the population of cells with subdiploid DNA contents and with decreased FSC (Fig. 7 A), representing apoptotic cells with DNA fragmentation and shrunken cytoplasm. Preincubation with VEID-CHO up to 10 μM did not prevent the DNA fragmentation (Fig. 7 B), suggesting that the activity of caspase-6 is not required. This is reminiscent of previous results in a cell-free system where DNA fragmentation was not affected by the inhibition of the lamin-cleaving caspase (51, 52). When Jurkat cells were stimulated with anti-Fas mAb in the presence of 100 μM DMQD-CHO, DNA fragmentation was completely inhibited (Fig. 7 B). Thus, fragmentation of DNA is an event downstream of caspase-3. It is interesting to note that the addition of caspase-7 to a cell-free Jurkat extract induced fragmentation of nuclear DNA without activation of caspase-3 or -6 (24). In contrast, the activity of caspase-7 alone, preserved at 100 μM DMQD-CHO (Fig. 2 C, lane 4), was not sufficient for the induction of DNA fragmentation in Fas-stimulated cells (Fig. 7 B).
Figure 7.

Flow cytometric analysis of DNA fragmentation and cell shrinkage. Dot plots depict DNA content quantitated by means of propidium iodide staining of ethanol-permeabilized cells on a logarithmic scale (PI) as a function of FSC on a linear scale. (A) Time course of DNA fragmentation and cell shrinkage in response to Fas ligation. (B) DMQD-CHO at 100 μM inhibits DNA fragmentation without affecting cell shrinkage, whereas100 μM VEID-CHO suppresses both. Jurkat cells were pretreated in the absence or presence of the indicated concentrations of VEID-CHO or DMQD-CHO before Fas stimulation for 4 h. control, Jurkat cells before stimulation with anti-Fas mAb; dotted lines, the lower border of the distribution of FSC among normally growing Jurkat cells.
Involvement of Caspase-3 in the Cleavage of p21-activated Kinase 2 and Apoptotic Body Formation.
DMQD-CHO at 100 μM suppressed the appearance of the cell population with the lowest FSC (Figs. 3 B and 7 B), whereas VEID-CHO at 10 μM had no effect (Fig. 7 B). This raised the possibility that caspase-3 has a role in cytoplasmic process(es), most likely in the formation of apoptotic bodies. Phase contrast microscopy revealed that Fas-stimulated Jurkat cells formed no apoptotic bodies in the presence of 100 μM DMQD-CHO (Fig. 8 h), whereas VEID-CHO up to 10 μM did not prevent apoptotic body formation (Fig. 8, c and d). These indicated that caspase-3 is upstream of the apoptotic body formation.
Figure 8.
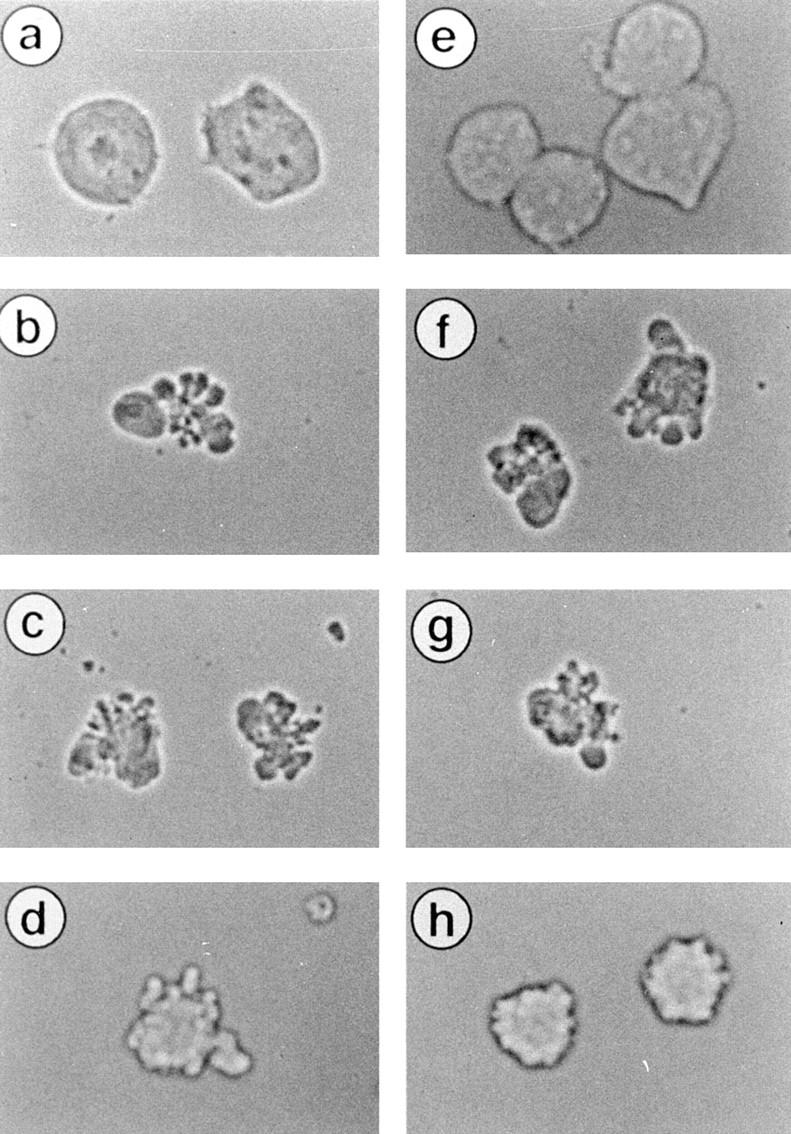
Effect of DMQD-CHO and VEID-CHO on cell shrinkage and apoptotic body formation. Jurkat cells treated as described in Fig. 6 were visualized for cell morphology under phase contrast microscope.
We therefore tested whether caspase-3 is responsible for the cleavage of p21-activated kinase (PAK)2, which mediates the formation of apoptotic bodies in Fas-stimulated Jurkat cells (54). Immunoblotting using Ab raised against the conserved region within kinase domains of PAK family kinases showed that the 65-kD PAK2 protein is cleaved in Fas-stimulated Jurkat cells, yielding a 34-kD COOH-terminal fragment (Fig. 5 D, lane 2) as previously described (54). The cleavage of PAK2 was suppressed by 100 μM DMQD-CHO (lane 4). VEID-CHO up to 10 μM failed to affect the PAK2 cleavage (lanes 5–7), suggesting that caspase-6 is not involved. Taken together, these results suggested that caspase-3 is mainly responsible for the apoptotic cleavage of PAK2 in Fas-stimulated Jurkat cells.
Identification of Caspases Upstream of Extranuclear Morphological and Biochemical Changes.
The shrinkage of cytoplasm in Fas-stimulated Jurkat cells proceeded to a significant degree despite the blockade of both caspase-3 and -6 by 100 μM DMQD-CHO (Figs. 3 B and 7 B). Although the selective blockade of caspase-6 with 10 μM VEID-CHO had no effect on cell size (Fig. 7 B), the broad-spectrum blockade of all caspases at 100 μM VEID-CHO markedly suppressed the decrease in cell size (Fig. 7 B). The shapes of cells observed by phase contrast microscopy were consistent with the flow cytometric data. Neither 100 μM DMQD-CHO (Fig. 8 h) nor 10 μM VEID-CHO (Fig. 8 d) affected the Fas-induced shrinkage of Jurkat cells. In contrast, cytoplasmic shrinkage was completely blocked with 100 μM VEID-CHO (Fig. 8 e). Taken together, a caspase(s) other than caspase-3 or -6 plays a critical role(s) in the Fas-induced shrinkage of cytoplasm.
To delineate further the involvement of each caspase in extranuclear processes, we examined apoptotic biochemical changes in mitochondria and the plasma membrane. Breakdown of the inner mitochondrial membrane potential (mitochondrial PT) is one of the earliest biochemical changes in cells undergoing apoptosis (55, 56). Although recent reports indicate that Fas-induced PT is dependent on the activity of caspases (5, 15, 57), it remained to be determined which caspase(s) is upstream of PT in the cells. The loss of plasma membrane asymmetry, resulting in the surface exposure of PS (58) and phosphatidylethanolamine (59), is another characteristic apoptotic change. Fas-induced PS externalization depends on the activity of caspases (18). We measured PT and PS externalization by double staining with DiOC6(3), the fluorescent probe that is sequestered to mitochondria depending on the membrane potential (60), and PE-conjugated annexin V, a natural protein that binds PS exposed to the cell surface with high affinity (61). As shown in Fig. 9 A, a cell population with decreased DiOC6(3) uptake and increased annexin V binding was first noted at 1 h after Fas ligation. This population with collapsed mitochondrial membrane potential and with externalized PS increased over time. Neither PT nor PS externalization were affected by 10 μM VEID-CHO (Fig. 9 B). The blockade of all caspases with 100 μM VEID-CHO markedly inhibited both processes (Fig. 9 B), confirming previous reports that those events are downstream of caspases. DMQD-CHO at 100 μM suppressed the increase in annexin V binding (Fig. 9 B). Thus, the activity of caspase-3 is essential for PS externalization. In contrast, PT was not affected by 100 μM DMQD-CHO, indicating that a caspase(s) other than caspase-3 or -6 is involved in the disruption of mitochondrial membrane potential in Fas-stimulated Jurkat cells.
Figure 9.
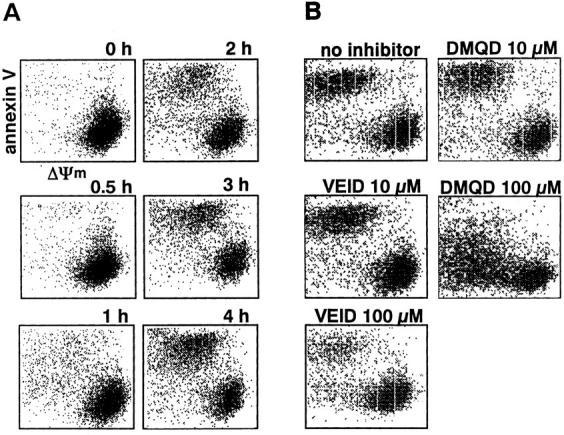
(A) Time course of cell surface exposure of PS and mitochondrial PT. Jurkat cells (5 × 105) were treated with anti-Fas Ab for the time indicated, and double stained with PE–annexin V and DiOC6(3). (B) Effect of DMQD-CHO and VEID-CHO on PS externalization and on mitochondrial PT. Jurkat cells were pretreated for 1 h with the indicated concentrations of the inhibitors before Fas stimulation for 4 h.
Discussion
The present study established the existence of a protease cascade, in which caspase-3 mediates the activation of caspase-6 triggered by Fas or by caspase-8 in the cytoplasm of Jurkat cells. Selective inhibition of caspase-3 by 100 μM DMQD-CHO blocked the activation of caspase-6. It is formally possible that pro-caspase-6 is processed by another noncaspase protease(s) sensitive to DMQD-CHO. However, the stepwise activation of caspases triggered by recombinant caspase-8 was observed in the Jurkat cytoplasmic extracts in the presence of protease inhibitors including PMSF, aprotinin, chymostatin, leupeptin, antipain, pepstatin, and EGTA (14). Even in this condition (unfavorable for most serine-, cysteine-, and metalloproteinases), pro-caspase-6 was processed to the active form unless the activation was blocked by 100 μM DMQD-CHO. Thus, it is likely that caspase-3 directly processes and activates caspase-6 in the cytoplasm of Jurkat cells.
The ability of caspase-8 to induce stepwise activation of caspases in organelle-free cytoplasmic extracts of Jurkat cells (14) suggests that mitochondria are dispensable for the organization of this protease cascade in Jurkat cells. However, this does not rule out the possibility that caspase-8 indirectly activates caspase-3 by inducing mitochondrial release of cytochrome c (62, 63) and/or AIF (57) in other cell types. In cells with more cytoplasm and a lower concentration of pro-caspase-3, caspase-8 activated in the DISC on the plasma membrane and released into the cytoplasm might not process cytoplasmic pro-caspase-3 efficiently. The sequestration of caspase-8 to the mitochondria (64) and the subsequent release of cytochrome c and AIF could well facilitate the activation of caspase-3. This pathway should be particularly active in cells rich in mitochondria. The existence of both direct and indirect pathways for the activation of caspase-3 by caspase-8 would explain the differences among cell types in the protective effect of Bcl-2 and Bcl-xL on Fas-induced cell death (15, 35). In cells that predominantly use the indirect pathway for caspase-3 activation, Bcl-2 family proteins may inhibit Fas-induced apoptosis by blocking the release of cytochrome c and/or AIF from mitochondria.
It seems unlikely that caspase-8 activates caspase-3 via other caspases. Affinity labeling displayed three other caspases, caspase-7, caspase-6, and F25, in Fas-stimulated Jurkat cells (14). Inhibitor studies with VEID-CHO showed that caspase-6 activity is not required for the activation of caspase-3. However, recent evidence suggests that different caspase cascades may be used depending on cell types and death-inductive stimuli (65). In some cell death events, caspase-6 may act upstream of caspase-3 (24, 41). The appearance of F25 is a delayed event, much later than caspase-3, -7, and -6 (14). Therefore, it seems unlikely that F25 is upstream of other caspases. Previous studies indicated that recombinant caspase-7 cannot process recombinant pro-caspase-3 (66). Indeed, the addition of recombinant active caspase-7 to the extracts from normally growing Jurkat cells failed to induce F20/ caspase-3-p20 (data not shown). Overall, our findings strongly suggested that there is a direct two-step protease cascade in Fas-stimulated Jurkat cells, in which caspase-8 activated via FADD/MORT1 cleaves pro-caspase-3, and the activated caspase-3 in turn activates caspase-6 by directly cleaving pro-caspases-6 (Fig. 10).
Figure 10.

Schematic representation of Fas-induced caspase activation and signaling pathways downstream of caspases. Solid arrows, direct interactions; dashed arrows, indirect or incompletely defined interactions.
In contrast to caspase-6, caspase-7 is activated independent of caspase-3, since caspase-7 remained active despite the complete blockade of caspase-3 and -6 by 100 μM DMQD-CHO. It is unlikely that caspase-7 is activated independent of caspase-8. Caspase-8 is the only active caspase highly sensitive to cowpox CrmA in Fas-stimulated Jurkat cells (14) and the inhibition of caspase-8 by CrmA completely prevents the processing of pro–caspase-7 (35). The addition of caspase-8 to the extracts from proliferating Jurkat cells led to the appearance of both active caspase-7 and caspase-3-p20 (14). These findings further support the idea that pro-caspase-7 is directly cleaved and activated by caspase-8 (12, 13) in Fas-stimulated Jurkat cells (Fig. 10). It should be noted, however, that the affinity labeling method may not be able to detect a certain active caspase(s) (14). The possibility has not been excluded that such an undetectable caspase(s) plays a functional role(s) in Fas-induced apoptosis. Therefore, the Fas signaling pathway depicted in Fig. 10 is better viewed as a framework for future studies.
Detailed analyses of Fas-induced signaling using VEID-CHO and DMQD-CHO provided evidence that the protease cascade bifurcates into a caspase-7 arm and a caspase-3 arm, each inducing distinct downstream intracellular events (Fig. 10). There is a point of ramification in the Fas signaling pathway at the level of the caspase cascade.
Caspase-3 Arm.
The caspase-3 arm seems to play major roles in the execution of nuclear apoptosis. Caspase-3 activates caspase-6, which in turn plays an essential role in the induction of nuclear shrinkage and nuclear fragmentation. Cleavage of the nuclear structural protein NuMA is mediated exclusively by the caspases of this branch. DNA fragmentation, a hallmark of nuclear apoptosis (67), is downstream of caspase-3. This is consistent with the result in a cell-free system showing that caspase-3 is a direct activator of DNA fragmentation factor (DFF; reference 16). DFF can induce DNA ladder in isolated nuclei only after its 45-kD subunit is cleaved by caspase-3. Our present data support the idea that caspase-3 mediates DNA fragmentation by cleaving DFF in Fas-stimulated Jurkat cells.
Microscopic analysis of DAPI-stained nuclei indicated that caspase-3 has a role in the initiation of chromatin condensation. PKCδ is cleaved and activated by caspase-3, not by caspase-6 or -7 (42). By undetermined mechanisms, introduction of cleaved active PKCδ fragment to HeLa cells induces nuclear apoptotic changes, including chromatin condensation (42). This may underlie the ability of caspase-3 to initiate the nuclear apoptotic morphological changes.
Although caspase-3 plays major roles in nuclear apoptotic events, it is also involved in extranuclear processes. PS exposed on the cell surface is recognized by macrophages and triggers the engulfment of apoptotic cells by phagocytes (58). Our results indicated that this PS externalization in response to Fas ligation is downstream of caspase-3. The molecular mechanism by which caspase-3 mediates PS externalization remains to be determined. It has been suggested that PS exposure results from the inactivation of aminophospholipid translocase and the enhanced activity of scramblase (68). In the recently reported amino acid sequence of scramblase (69), however, no canonical consensus sequence for caspase-3 cleavage (22) was identified. We found that caspase-3 is a principal mediator of the apoptotic body formation. This may reflect the fact that PAK2 cleavage in Fas-stimulated Jurkat cells is mainly catalyzed by caspase-3. Alternatively, the apoptotic body formation may be coupled to nuclear events, as suggested by the exclusion of Ro, La, and small nuclear ribonucleoproteins (snRNPs) from the nucleoplasms and the clustering of these ribonucleoproteins in apoptotic bodies (70).
Caspase-7 Arm.
The caspase-7 arm is involved in the Fas-induced signaling in a manner distinct from caspase-3 and -6. Our results suggested that caspase-7 catalyzes, together with caspase-3, the cleavage of a nuclear substrate, PARP. This result accords well with the analysis of caspase-3 knockout mice showing that the PARP cleavage in apoptotic thymocytes is seemingly unaffected by the loss of caspase-3 (65). Thus, caspase-7 can compensate for some of the apoptotic biochemical events mediated by caspase-3.
Our observations suggest that caspase-7 also has a role(s) in the Fas-induced signaling that are not redundant with caspase-3. Apoptotic shrinkage of cytoplasm (19) persisted in the presence of 100 μM DMQD-CHO, which blocked caspase-3 and -6. A recent report described a similar partial blockade of apoptotic events in KB epidermal carcinoma cells. zDEVD-FK at 50 μM abolished Fas-induced chromatin condensation and apoptotic body formation, whereas surface blebbing, cell shrinkage, the disruption of actin network, and the activation of stress-activated MAP kinases, c-Jun NH2-terminal kinase (JNK)/stress-activated protein kinase (SAPK) and p38, remained uninterrupted (71). In contrast, the blockade of all caspases with 100 μM VEID-CHO abolished the cell shrinkage. Caspase-7, active in the presence of DMQD-CHO, may thus mediate cell shrinkage. Although caspase-8 might mediate cell shrinkage, active caspase-8 was not detected in staurosporine-induced apoptosis of Jurkat cells (14) in which cytoplasmic shrinkage is prominent. Further, although caspase-3 and -7 have closely related structures (66) and almost identical preferences for tetrapeptide substrates (72, 73), caspase-7 may play a role(s) distinct from caspase-3. In this regard, it is of interest that caspase-3 and -7 have different optimal pH ranges (74). Further elucidation of cytoplasmic substrates specific for caspase-7 may clarify its role(s) in the cytoplasmic shrinkage induced by Fas ligation.
Can Caspase-7 Substitute for Caspase-3?
A controversial issue is whether caspase-7 can substitute for caspase-3. The substrate specificity of caspase-7 is similar to caspase-3 (72, 73), although caspase-7 cannot cleave some caspase-3 substrates such as PKCδ (42) and pro-caspase-6 (26). This study demonstrated that many biochemical changes of Fas-induced apoptosis in Jurkat cells are mediated solely by caspase-3 (Fig. 10). However, in knockout mice deficient in caspase-3, most apoptotic events are not interrupted, except for those in neuronal development (65). One attractive possibility is that caspase-3 and -7 compete for a common cofactor(s) to fulfill their intracellular functions. In Jurkat cells, caspase-3 inactivated by 100 μM DMQD-CHO may still compete with caspase-7 for the cofactor(s). In the absence of caspase-3 protein (65), however, caspase-7 may be free to use the cofactor(s) and thus to compensate for the loss of caspase-3 protein.
Mitochondrial PT.
Analysis of the mitochondrial transmembrane potential in Fas-stimulated cells showed that a caspase(s) other than caspase-3 or -6 is required for PT. Since the cell shrinkage is also mediated by a caspase(s) other than caspase-3 or -6, PT may be upstream of Fas-induced cytoplasmic shrinkage. It is of interest in this regard that the caspase-independent cell death induced by Bax, which may be a direct trigger of PT (75), is accompanied by PT and cell shrinkage (5). Because AIF is released on PT and activates DNase involved in internucleosomal DNA cleavage (76), it is surprising that DNA fragmentation was not observed despite the occurrence of PT in the presence of 100 μM DMQD-CHO. It is unclear whether the release of AIF is inhibited in the presence of DMQD-CHO, whether the activity of AIF is suppressed by the inhibitor, or whether the activity of AIF alone is not sufficient for the induction of DNA fragmentation.
We could not unequivocally determine which caspase is upstream of PT in Fas-stimulated Jurkat cells. However, it has been reported that caspase-8 transfected to 293T cells can form a complex with cotransfected Bcl-xL (64). If caspase-8 is recruited to mitochondria via complex formation with such Bcl-2 family proteins, it might act on mitochondrial substrates and disrupt the membrane potential. The development of an inhibitor(s) selective for caspase-7 should allow the identification of a caspase(s) responsible for such extranuclear events as PT and cytoplasmic shrinkage.
Cell-free versus Cellular Apoptosis.
A constant concern of in vitro studies is their applicability to intact cells. Although caspase-7 cleaves recombinant and HeLa nuclear NuMA in vitro, the endogenous NuMA of Jurkat cells remained uncleaved despite caspase-7 activation. Despite the ability of caspase-6 to generate the 180-kD fragment in vitro, only caspase-3 was responsible for this cleavage in Fas-stimulated Jurkat cells. Similarly, caspase-7 combined with extracts from nonapoptotic cells can induce DNA fragmentation in isolated nuclei, without activation of caspase-3 (24). However, a blockade of caspase-3 abolished DNA fragmentation within Fas-stimulated Jurkat cells despite persistent caspase-7 activity. In contrast, it is worth noting that the role of a lamin-cleaving protease(s), such as caspase-6, in the shrinkage and fragmentation of nuclei (Fig. 10) was first established in a cell-free system (51, 52). Cell-free systems have their own roles in the development of new tools and concepts, which should be explored at the level of cells and organisms (77).
Our approach exemplified the use of synthetic inhibitors that are relatively selective for each caspase. Recognition of substrates that are cleaved by one or a few caspases (17, 42) is a vital step in the design of such selective caspase inhibitors. The application of these reagents leads to effective approaches for dissecting the signaling pathways of apoptotic cell death. Inhibitors are being designed as the results of systematic screening of defined sequence variants of preferred substrates (72) and by use of the positional scanning synthetic combinatorial library (73). Detailed analysis of each cell death event using an array of inhibitors may identify targets for therapeutic interventions of the pathological processes resulting from excessive (78) or inappropriately suppressed (79) apoptosis.
Overall, the present study shows that the Fas-induced apoptotic cell death process is an integration of multiple, parallel biochemical events that occur downstream of the activation of particular caspases. The apparent heterogeneity in the biochemistry of apoptosis in different systems may be the result of the activation of different combinations of caspases, and consequently from their organization into various different protease cascades.
Footnotes
1 Abbreviations used in this paper: DAPI, 4′,6-diamidino-2-phenylindole; DFF, DNA fragmentation factor; DISC, death-inducing signaling complex; DMQD-CHO, acetyl-Asp-Met-Gln-Asp-aldehyde; FSC, forward light scatter; ICE, IL-1β converting enzyme; NuMA, nuclear mitotic apparatus protein; PAK, p21-activated kinase; PARP, poly (ADP-ribose) polymerase; PKCδ, protein kinase C δ; PS, phosphatidylserine; PT, permeability transition; VEID-CHO, acetyl-Val-Ileu-Asp-aldehyde; zVAD-fmk, benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone.
We thank Emad S. Alnemri for the generous gift of cDNAs for caspases; Nancy A. Thornberry for providing YV(bio)KD-aomk; Gerald M. Cohen for anti–caspase-7 Ab; Charles H. Yang for anti-NuMA Ab; Alastair Mackay for critical review of the manuscript and creative suggestions; Yuri A. Lazebnik, Scott H. Kaufmann, William C. Earnshaw, Eisuke Nishida, Fumiko Toyoshima, and Katsumi Takada for helpful discussion and thoughtful suggestions; and Akira Komoriya, Kuniko Takano, Akinori Maeda, and Kouhei Yamashita for help in flow cytometric analysis.
A. Takahashi is a Research Resident of the Japanese Foundation of Aging and Health.
Address correspondence to Atsushi Takahashi, The First Division, Department of Internal Medicine, Kyoto University Hospital, 54 Shogoin Kawara-cho, Sakyo-ku, Kyoto 606, Japan. Phone: 81-75-751-4290; Fax: 81-75-751-4221; E-mail: atakahas@kuhp.kyoto-u.ac.jp
References
- 1.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 2.Abbas AK. Die and let live: eliminating dangerous lymphocytes. Cell. 1996;84:655–657. doi: 10.1016/s0092-8674(00)81042-9. [DOI] [PubMed] [Google Scholar]
- 3.Rathmell JC, Townsend SE, Xu JC, Flavell RA, Goodnow CC. Expansion or elimination of B cells in vivo: dual roles for CD40- and Fas (CD95)-ligands modulated by the B cell antigen receptor. Cell. 1996;87:319–329. doi: 10.1016/s0092-8674(00)81349-5. [DOI] [PubMed] [Google Scholar]
- 4.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 5.Xiang J, Chao DT, Korsmeyer SJ. BAX-induced cell death may not require interleukin 1β–converting enzyme– like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longthorne VL, Williams GT. Caspase activity is required for commitment to Fas-, mediated apoptosis. EMBO (Eur Mol Biol Organ) J. 1997;16:3805–3812. doi: 10.1093/emboj/16.13.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagata S, Goldstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 8.Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/ FADD-interacting protease in Fas/APO-1– and TNF receptor–induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 10.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3–like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 11.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO (Eur Mol Biol Organ) J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES. Molecular ordering of the Fas-apoptotic pathway: the Fas/APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci USA. 1996;93:14486–14491. doi: 10.1073/pnas.93.25.14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muzio M, Salvesen GS, Dixit VM. FLICE induced apoptosis in a cell-free system – cleavage of caspase zymogens. J Biol Chem. 1997;272:2952–2956. doi: 10.1074/jbc.272.5.2952. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi A, Hirata H, Yonehara S, Imai Y, Lee KK, Moyer RW, Turner PC, Mesner PW, Okazaki T, Sawai H, et al. Affinity labeling displays the stepwise activation of ICE-related proteases by Fas, staurosporine, and CrmA-sensitive caspase-8. Oncogene. 1997;14:2741–2752. doi: 10.1038/sj.onc.1201131. [DOI] [PubMed] [Google Scholar]
- 15.Boise LH, Thompson CB. Bcl-x (L) can inhibit apoptosis in cells that have undergone Fas-induced protease activation. Proc Natl Acad Sci USA. 1997;94:3759–3764. doi: 10.1073/pnas.94.8.3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu XS, Zou H, Slaughter C, Wang XD. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamin A by Mch2α but not CPP32: multiple ICE-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci USA. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin SJ, Finucane DM, Amarante-Mendes GP, O'Brien GA, Green DR. Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem. 1996;271:28753–28756. doi: 10.1074/jbc.271.46.28753. [DOI] [PubMed] [Google Scholar]
- 19.Webb, S.J., D.J. Harrison, and A.H. Wyllie. 1997. Apoptosis: an overview of the process and its relevance in disease. In Apoptosis: Pharmacological Implications and Therapeutic Opportunities. S.H. Kaufmann, editor. Academic Press, San Diego. 1–34. [DOI] [PubMed]
- 20.Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP, Rasper DM, Ruel R, Vaillancourt JP, et al. The three-dimensional structure of apopain/CPP32, a key mediator of apoptosis. Nat Struct Biol. 1996;3:619–625. doi: 10.1038/nsb0796-619. [DOI] [PubMed] [Google Scholar]
- 21.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholson DW, Thornberry NA. Caspases: killer proteases. TIBS (Trends Biochem Sci) 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 23.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? . Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 24.Orth K, O'Rourke K, Salvesen GS, Dixit VM. Molecular ordering of apoptotic mammalian CED-3/ ICE-like proteases. J Biol Chem. 1996;271:20977–20980. doi: 10.1074/jbc.271.35.20977. [DOI] [PubMed] [Google Scholar]
- 25.Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli K, Litwack G, Alnemri ES. In vitroactivation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA. 1996;93:7464–7469. doi: 10.1073/pnas.93.15.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, Robertson N, Armstrong R, Wang L, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. The Ced-3/interleukin 1β converting enzyme–like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- 27.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 28.Thornberry NA, Peterson EP, Zhao JJ, Howard AD, Griffin PR, Chapman KT. Inactivation of interleukin-1β converting enzyme by peptide (acyloxy)methyl ketones. Biochemistry. 1994;33:3934–3940. doi: 10.1021/bi00179a020. [DOI] [PubMed] [Google Scholar]
- 29.Martins LM, Kottke T, Mesner PW, Basi GS, Sinha S, Frigon NJ, Tatar E, Tung JS, Bryant K, Takahashi A, et al. Activation of multiple interleukin-1β converting enzyme homologues in cytosol and nuclei of HL-60 human leukemia cells during etoposide-induced apoptosis. J Biol Chem. 1997;272:7421–7430. doi: 10.1074/jbc.272.11.7421. [DOI] [PubMed] [Google Scholar]
- 30.Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y. Multiple species of CPP32 and Mch2 are the major active caspases present in apoptotic cells. EMBO (Eur Mol Biol Organ) J. 1997;16:2271–2281. doi: 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clayton LK, Ghendler Y, Mizoguchi E, Patch RJ, Ocain TD, Orth K, Bhan AK, Dixit VM, Reinherz EL. T-cell receptor ligation by peptide/MHC induces activation of a caspase in immature thymocytes: the molecular basis of negative selection. EMBO (Eur Mol Biol Organ) J. 1997;16:2282–2293. doi: 10.1093/emboj/16.9.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA – analysis of five caspases. J Biol Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 33.Datta R, Kojima H, Banach D, Bump NJ, Talanian RV, Alnemri ES, Weichselbaum RR, Wong WW, Kufe DW. Activation of a CrmA-insensitive, p35-sensitive pathway in ionizing radiation-induced apoptosis. J Biol Chem. 1997;272:1965–1969. doi: 10.1074/jbc.272.3.1965. [DOI] [PubMed] [Google Scholar]
- 34.Kamada S, Funahashi Y, Tsujimoto Y. Caspase-4 and caspase-5, members of the ICE/CED-3 family of cysteine proteases, are CrmA-inhibitable proteases. Cell Death Diff. 1997;4:473–478. doi: 10.1038/sj.cdd.4400268. [DOI] [PubMed] [Google Scholar]
- 35.Chinnaiyan AM, Orth K, O'Rourke K, Duan H, Poirier GG, Dixit VM. Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3–like apoptotic proteases. J Biol Chem. 1996;271:4573–4576. doi: 10.1074/jbc.271.9.4573. [DOI] [PubMed] [Google Scholar]
- 36.MacFarlane M, Cain K, Sun XM, Alnemri ES, Cohen GM. Processing/activation of at least four interleukin-1 beta converting enzyme–like proteases occurs during the execution phase of apoptosis in human monocytic tumor cells. J Cell Biol. 1997;137:469–479. doi: 10.1083/jcb.137.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell-free mitotic extracts: a model system for analysis of the active phase of apoptosis. J Cell Biol. 1993;123:7–22. doi: 10.1083/jcb.123.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang CH, Lambie EJ, Snyder M. NuMA: an unusually long coiled-coil related protein in the mammalian nucleus. J Cell Biol. 1992;116:1303–1317. doi: 10.1083/jcb.116.6.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi, A., E.S. Alnemri, T. Fernandes-Alnemri, Y.A. Lazebnik, R.D. Moir, R.D. Goldman, G.G. Poirier, S.H. Kaufmann, and W.C. Earnshaw. 1997. Biochemical dissection of nuclear events in apoptosis. In Cell Cycle Regulation. B. Metcalf, R.R.J. Ruffolo, and G. Poste, editors. Harwood Academic Publishers, Berks, UK. 131–149.
- 40.Kamada S, Washida M, Hasegawa J, Kusano H, Funahashi Y, Tsujimoto Y. Involvement of caspase-4 (–like) protease in Fas-mediated apoptotic pathway. Oncogene. 1997;15:285–290. doi: 10.1038/sj.onc.1201192. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Kim CN, Pohl J, Wang X. Purification and characterization of an interleukin-1β–converting enzyme family protease that activates cysteine protease P32 (CPP32) J Biol Chem. 1996;271:13371–13376. [PubMed] [Google Scholar]
- 42.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang YY, Kharbanda S, et al. Proteolytic activation of protein kinase C delta by an ICE/CED 3–like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han ZY, Hendrickson EA, Bremner TA, Wyche JH. A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J Biol Chem. 1997;272:13432–13436. doi: 10.1074/jbc.272.20.13432. [DOI] [PubMed] [Google Scholar]
- 44.Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J. 1996;315:21–24. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greidinger EL, Miller DK, Yamin TT, Casciola RL, Rosen A. Sequential activation of three distinct ICE-like activities in Fas-ligated Jurkat cells. FEBS Lett. 1996;390:299–303. doi: 10.1016/0014-5793(96)00678-3. [DOI] [PubMed] [Google Scholar]
- 46.Casiano CA, Martin SJ, Green DR, Tan EM. Selective cleavage of nuclear autoantigens during CD95 (Fas/ APO-1)–mediated T cell apoptosis. J Exp Med. 1996;184:765–770. doi: 10.1084/jem.184.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gueth-Hallonet C, Weber K, Osborn M. Cleavage of the nuclear matrix protein NuMA during apoptosis. Exp Cell Res. 1997;233:21–24. doi: 10.1006/excr.1997.3557. [DOI] [PubMed] [Google Scholar]
- 48.Duan H, Chinnaiyan AM, Hudson PL, Wing JP, He W-W, Dixit VM. ICE-LAP3, a novel mammalian homologue of the Caenorhabditis eleganscell death protein Ced-3 is activated during Fas- and tumor necrosis factor– induced apoptosis. J Biol Chem. 1996;271:1621–1625. doi: 10.1074/jbc.271.3.1621. [DOI] [PubMed] [Google Scholar]
- 49.Krajewska M, Wang HG, Krajewski S, Zapata JM, Shabaik A, Gascoyne R, Reed JC. Immunohistochemical analysis of in vivo patterns of expression of CPP32 (Caspase-3), a cell death protease. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- 50.Nakagawara A, Nakamura Y, Ikeda H, Hiwasa T, Kuida K, Su MS-S, Zhao H, Cnaan A, Sakiyama S. High levels of expression and nuclear localization of interleukin-1β converting enzyme (ICE) and CPP32 in favorable human neuroblastomas. Cancer Res. 1997;57:4578–4584. [PubMed] [Google Scholar]
- 51.Lazebnik YA, Takahashi A, Moir R, Goldman R, Poirier GG, Kaufmann SH, Earnshaw WC. Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA. 1995;92:9042–9046. doi: 10.1073/pnas.92.20.9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi A, Musy P-Y, Martins LM, Poirier GG, Turner PC, Moyer RW, Earnshaw WC. CrmA/ SPI-2 inhibition of an endogenous ICE-related protease responsible for lamin A cleavage and apoptotic nuclear fragmentation. J Biol Chem. 1996;271:32487–32490. doi: 10.1074/jbc.271.51.32487. [DOI] [PubMed] [Google Scholar]
- 53.Takahashi A, Goldschmidt-Clermont PJ, Alnemri ES, Fernandes-Alnemri T, Yoshizawa-Kumagaya K, Nakajima K, Sasada M, Poirier GG, Earnshaw WC. Inhibition of ICE-related proteases (caspases) and nuclear apoptosis by phenylarsine oxide. Exp Cell Res. 1997;231:123–131. doi: 10.1006/excr.1996.3459. [DOI] [PubMed] [Google Scholar]
- 54.Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- 55.Kroemer G, Petit P, Zamzami N, Vayssiere JL, Mignotte B. The biochemistry of programmed cell death. FASEB J. 1995;9:1277–1287. doi: 10.1096/fasebj.9.13.7557017. [DOI] [PubMed] [Google Scholar]
- 56.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 57.Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95– and ceramide–induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 59.Emoto K, Toyama-Sorimachi N, Karasuyama H, Inoue K, Umeda M. Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp Cell Res. 1997;232:430–434. doi: 10.1006/excr.1997.3521. [DOI] [PubMed] [Google Scholar]
- 60.Vayssiere JL, Petit PX, Risler Y, Mignotte B. Commitment to apoptosis is associated with changes in mitochondrial biogenesis and activity in cell lines conditionally immortalized with simian virus 40. Proc Natl Acad Sci USA. 1994;91:11752–11756. doi: 10.1073/pnas.91.24.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 63.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 64.Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122–1126. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 65.Kuida K, Zheng TS, Na S, Kuan C-Y, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 66.Fernandes-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Yu Z, Croce CM, Salveson G, et al. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res. 1995;55:6045–6052. [PubMed] [Google Scholar]
- 67.Earnshaw WC. Nuclear changes in apoptosis. Curr Opin Cell Biol. 1995;7:337–343. doi: 10.1016/0955-0674(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 68.Verhoven B, Schlegel RA, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou QS, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ. Molecular cloning of human plasma membrane phospholipid scramblase— a protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem. 1997;272:18240–18244. doi: 10.1074/jbc.272.29.18240. [DOI] [PubMed] [Google Scholar]
- 70.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toyoshima F, Moriguchi T, Nishida E. Fas induces cytoplasmic apoptotic responses and activation of the MKK7-JNK/SAPK and MKK6-p38 pathways independent of CPP32-like protease. J Cell Biol. 1997;139:1005–1015. doi: 10.1083/jcb.139.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 73.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B – functional, relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 74.Pai J-T, Brown MS, Goldstein JL. Purification and cDNA cloning of a second apoptosis-related cysteine protease that cleaves and activates sterol regulatory element binding proteins. Proc Natl Acad Sci USA. 1996;93:5437–5442. doi: 10.1073/pnas.93.11.5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Antosson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J-J, Mazzei G, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 76.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takahashi, A., and W.C. Earnshaw. 1997. In vitro systems for the study of apoptosis. In Apoptosis: Pharmacological Implications and Therapeutic Opportunities. S.H. Kaufmann, editor. Academic Press, San Diego. 89–106. [DOI] [PubMed]
- 78.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 79.Fisher DE. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]