

Abstract
Treatment of some inflammatory conditions with tumor necrosis factor-α (TNF-α) antagonists is efficacious, but such treatments are associated with infections with intracellular pathogens, including Histoplasma capsulatum. We explored protective immunity to H. capsulatum in mice given a fusion protein consisting of TNF-α receptor 2 (TNFR2) bound to the Fc portion of mouse IgG1. Intraperitoneal administration of this inhibitor exacerbated primary or secondary pulmonary infection at dosages ranging from 1 to 5 mg/kg. All mice with primary infection given the inhibitor succumbed to infection within 10–21 days of treatment. In secondary histoplasmosis, mice receiving 1, but not 5, mg/kg survived treatment. Fungal burden was increased even if treatment with the inhibitor was initiated after the onset of infection. The inflammatory response of the lungs of mice given the inhibitor did not differ from that of mice given control vehicle. Susceptibility was not associated with major alterations in cytokines known to protect or exacerbate infection. However, expression of nitric oxide synthase 2 (NOS2) was depressed early in primary infection. These results demonstrate that antagonism of endogenous TNF-α by this fusion protein modulates susceptibility. Impaired immunity is not a result of altered cytokine responses or changes in the inflammation and may not be demonstrable in other murine strains.
INTRODUCTION
In recent years, there has been an enormous increase in the number of biologic response modifiers (BRMs) available for clinical use. These agents exert a host of biologic activities that range from boosting immunity to dampening inflammation. Among those that are clinically available, the tumor necrosis factor-α (TNF-α) antagonists have gained wide acceptance as treatment for a number of aggressive inflammatory diseases, including rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, and Crohn’s disease.1 The expanding indications for this class of therapeutic agents have increased the numbers of patients who are receiving this type of therapy. TNF-α not only is a key proinflammatory mediator, but it also is an important host defense molecule. This latter action is independent on the ability of this cytokine to mobilize inflammatory cells.2 Thus, one of the consequences of TNF-α antagonism has been a surge in the infectious complications, especially intracellular pathogens.3–5 Among these infectious complications has been the pathogenic fungus, Histoplasma capsulatum.4,5
Human infection with H. capsulatum occurs worldwide but is particularly prevalent in the southeastern and midwestern United States. Exposure to airborne mycelia and microconidia either produces no symptoms or may cause a mild influenza-like illness that spontaneously resolves. Once these fungal elements have reached lung parenchyma, they transform into yeasts, and this morphotype replicates within phagocytes until cellular immunity is activated. Growth of the fungus is halted, but it can persist within tissues for many years in a dormant state.6
The major mediators involved in protective immunity are T cells and several cytokines. Interferon-γ (IFN-γ), granulocyte macrophage colony-stimulating factor (GM-CSF), and TNF-α are all necessary for the induction of a protective immune response to this fungus.7–12 Among these cytokines, TNF-α may be the most important, based on studies in experimental animals. Neutralization of it dramatically impairs protective immunity in naïve mice and in those that possess preexisting immunity.7 This effect is not observed for either IFN-γ or GM-CSF. Although both of these cytokines are necessary for survival in primary histoplasmosis, their absence in secondary does not reduce survival in mice challenged with a sublethal inoculum.8,11
Given the requirement of TNF-α for optimal host defenses against this fungus, we explored the influence of antagonism of this cytokine on the course of primary and secondary histoplasmosis in mice. Although there is compelling evidence that neutralization of endogenous murine TNF-α with either a polyclonal or monoclonal antibody (mAb) exacerbates infection,7,9–11 there are no data regarding the influence of another antagonist, a fusion protein consisting of TNF-α receptor 2 (TNFR2) linked to the Fc portion of mouse IgG1.
MATERIALS AND METHODS
Mice
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were housed in isolator cages and were maintained by the Department of Laboratory Animal Medicine, University of Cincinnati, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All animal experiments were performed in accordance with the Animal Welfare Act guidelines of the National Institutes of Health (NIH), and all protocols were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Preparation of H. capsulatum and infection of mice
H. capsulatum yeast (strain G217B) was grown in HMM medium for 60 h, washed, and enumerated.13 To produce infection in naïve mice, animals were inoculated intranasally (i.n.) with 2 × 106 H. capsulatum yeast cells in a 30-μL volume of HBSS. Secondary infection was produced by infecting mice with 104 yeasts i.n and, 8 weeks later, infecting them with 2 × 106 yeasts.
Organ culture for H. capsulatum
Recovery of H. capsulatum was performed as follows. Organs were homogenized in sterile saline and serially diluted. Homogenate (100 μL) was plated onto brain heart infusion agar plates containing 5% sheep blood and 5% glucose. Fungal burden was expressed as mean log10 colony-forming units (CFU) per whole organ ± SEM. The limit of detection was 102 CFU.
Treatment of mice with murine TNFR2-Fc fusion protein (p75-Fc) or with mAb to TNF-α
Mice were injected intraperitoneally (i.p.) with 1 mg/kg, 3 mg/kg, or 5 mg/kg mouse murine p75 fused to the Fc portion of mouse IgG1 (provided by Dr. H. Kohno, Amgen Corporation, Thousand Oaks, CA) dissolved in phosphate-buffered saline (PBS). Control animals were given an equal volume of PBS. Rat antimouse TNF-α (from cell line XT-22.1) was purchased the National Cell Culture Center (Minneapolis, MN) and purified. The cell line was obtained from Dr. J. Abrams (DNAX, Palo Alto, CA). Mice were injected i.p. with 1 mg mAb to TNF-α.
Isolation of lung leukocytes
Lung leukocytes were isolated as described.14 The homogenates were filtered through a 60-μm nylon mesh (Spectrum Laboratories Inc., Rancho Dominguez, CA) and washed three times with HBSS. Leukocytes were isolated by separation on a 40–70% Percoll (Pharmacia, Piscataway, NJ) gradient. Cells were washed three times with HBSS prior to examination.
Flow cytometry
The following antibodies were purchased from BD Biosciences: fluorescein isothiocyanate-conjugated CD4 and CD8-FITC, phycoerythrin (PE)-conjugated Mac-3 (macrophages), and PE-conjugated Gr-1 (neutrophils). Lungs were teased apart with the frosted ends of two glass slides and washed with HBSS; 2 × 106 cells were incubated with 0.5 μg antibody in staining buffer (1% bovine serum albumin [BSA] in PBS) for 10 min at 4°C. The cells were washed in staining buffer, and fluorescence was measured using a FACScalibur flow cytometer (BD Pharmingen). The number of cells was calculated by multiplying the total number of leukocytes by the percentage of cells bearing a specific surface receptor.
RNA isolation and cDNA synthesis
RNA was isolated from mouse lungs using trizol, and cDNA was synthesized as previously described.14
Real-time polymerase chain reaction (qRT-PCR)
qRT-PCR for nitric oxide synthase 2 (NOS2) was performed using the Taq-Man kit and primers from Applied Biosystems (Foster City, CA). Samples were analyzed on an ABI Prism 7500 from Applied Biosystems. Transcript quantification was standardized to expression from an uninfected lung. In each experiment, the housekeeping gene hypoxanthine phosphoribosyl transferase was analyzed. The data were expressed in log10 of relative quantification. The conditions for amplification were 50 EC for 2 min, 95 EC for 10 min, followed 40 cycles of 95 EC for 15 sec and 60 EC for 1 min.
Statistical analysis
ANOVA was used to compare groups. p < 0.05 was considered statistically significant. Survival was analyzed using log rank.
RESULTS
Treatment of naïve mice with p75-Fc exacerbates infection
In our initial experiments, naïve mice were treated with 1, 3, or 5 mg/kg of murine p75-Fc every other day beginning on the day of infection. They were killed on day 7 of infection. Lungs and spleens were cultured for H. capsulatum. As seen in Figure 1, the fungal burden was increased in each of the groups (p < 0.01). As each of the dosages caused indistinguishable increases in the fungal burden, we assessed if the highest and lowest dosage would impact survival of mice. Mice were infected with 2 × 106 yeast cells and treated with p75-Fc or PBS every 2 days. Both dosages caused mice to succumb to infection (Fig. 1C). Thus, TNF-α antagonism by this agent impaired host defenses severely.
FIG. 1.

Treatment with p75-Fc exacerbates primary histoplasmosis in mice. Groups of mice were infected with 2 × 106 yeasts i.n. and treated with p75-Fc or PBS on the day of infection and every 2 days thereafter. Mice were killed on day 7, and lungs (A) and spleens (B) were assessed for fungal burden. Results are expressed as means ± SEM of 6–8 animals. (C) Survival curve of mice treated with 1 or 5 mg/kg of p75-Fc or PBS. (D) Mice (6–8 per group) were treated with p75-Fc or PBS every 4 days. Animals were followed for survival. One of two experiments is shown. **p < 0.01 vs. controls.
Administration of the fusion protein created for human use is less frequent than every 2 days and often is just once or twice a week. Therefore, we analyzed whether p75-Fc given to naïve mice twice weekly was associated with impaired immunity. Mice were infected with H. capsulatum and given 1, 3, or 5 mg/kg of p75-Fc on the day of infection and every 4 days thereafter. All mice administered the TNF-α antagonist succumbed to infection (Fig. 1D). Thus, decreasing the frequency of administration did not dramatically alter the outcome of infection.
Mice with preexisting immunity and p75-Fc
Residents of an endemic area may be repeatedly exposed to H. capsulatum and thus develop immunity. We sought to determine if p75-Fc altered the course of infection in mice with preexisting immunity. Mice were exposed to 104 yeasts i.n., an inoculum that confers subsequent protection15; 8 weeks later, they were challenged with 2 × 106 yeasts and treated with p75-Fc or given PBS, and survival was followed. Mice given 1 mg/kg of p75-Fc every 2 days survived infection, whereas those given 3 mg/kg or 5 mg/kg every 2 or 4 days did not (Fig. 2A). To determine if the failure to survive was a result of an increase in CFU, we analyzed fungal burden in mice that received 3 mg/kg or 5 mg/kg every 4 days. In those mice, the fungal burden was increased compared with controls (Fig. 2B,C).
FIG. 2.

Effect of p75-Fc on secondary histoplasmosis. Mice were infected with 104 yeasts i.n. and 8 weeks later challenged with 2 × 106 yeasts i.n. and given 1 or 5 mg/kg of p75-Fc every 2 days. (A) Survival was assessed. (B and C) Mice were treated with 5 mg/kg of p75-Fc every 2 days, and fungal burden was assessed in lungs (B) or spleens (C) at day 7 of infection. Results represent mean ± SEM of 6–8 per group. **p < 0.01.
Does p75-Fc impact fungal burden if initiated during infection?
Naïve and immune mice were infected with H. capsulatum yeasts, and 5 mg/kg of p75-Fc was begun on the day of infection or on day 2 or 4 after infection. Mice were treated every 2 days i.p. with this antagonist or with PBS. The fungal burden in lungs and spleens was elevated in each of the groups receiving p75-Fc and significantly exceeded (p < 0.01, p < 0.05, spleens) that of controls (Fig. 3A,B). There were no significant differences (p > 0.05) among the groups of treated mice. In secondary histoplasmosis, treatment initiated after infection also blunted the protective immune response (Fig. 3C,D).
FIG. 3.
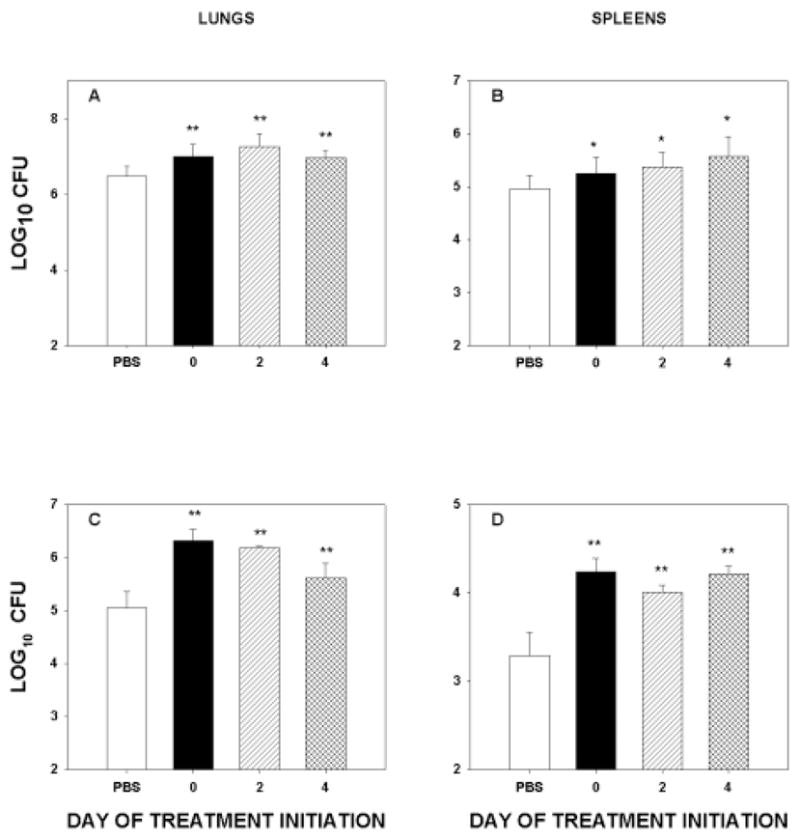
p75-Fc alters fungal burden when given in active infection. Naïve mice (A and B) or immune mice (C and D) were infected with 2 × 106 yeasts i.n. and treated on the day of infection or on day 2 or 4 after infection. The biological was administered every 2 days thereafter. Results represent mean ± SEM of 6 mice. *p < 0.05 vs. controls; **p < 0.01 vs. controls.
Inflammatory response in mice given p75-Fc
Because administration of p75-Fc altered the protective immune response to H. capsulatum, we queried if it modulated the nature of the inflammatory response to this fungus. Naïve and immune mice were infected with 2 × 106 yeasts and given 5 mg/kg every 4 days. We chose this regimen because it was the most potent dosage to dampen the protective immune response in both primary and secondary infection. Lungs were removed on days 3 and 7 of infection, and lung leukocytes were isolated. We analyzed the number of macrophages (Mac-3+), neutrophils (Gr-1+), and CD4+ and CD8+ cells, as these are the principal regulators of immunity to this fungus. At day 7 of infection, the numbers of macrophages and neutrophils were increased (p < 0.05) in animals receiving p75-Fc (Fig. 4A,B). In secondary infection, the numbers of each population were similar at days 3 and 7 of infection (Fig. 4C,D).
FIG. 4.

Inflammatory cells in lungs of mice administered p75-Fc. Mice were treated with 5 mg/kg p75-Fc every 4 days beginning on the day of infection. Lung leukocytes (>95% CD45+) were analyzed on (A and C) day 3 and (B and D) day 7 for several cell subpopulations. Results represent mean ± SEM of 4–8 animals. *p < 0.05.
Cytokine response
The lungs of mice were analyzed for alterations in mediators that are known to regulate the host response to this fungus. We analyzed by qRT-PCR transcription of interleukin-4 (IL-4) and IL-10, which inhibit protective immunity, IFN-γ, which promotes protective immunity, and NOS2, which induces the synthesis of NO, a key mediator of protection. In primary infection, there were no differences in transcription of IL-4, IL-10, or IFN-γ at day 3 or 7 (Fig. 5). However, there was a modest decline in NOS2 at day 3, but by day 7, this value was similar to that of controls. In secondary infection, the results for the cytokines and NOS2 were similar (Fig. 5).
FIG. 5.

qRT-PCR of cytokines and NOS2 for primary and secondary histoplasmosis. RNA from lungs of mice was prepared, and cDNA was synthesized. qRT-PCR was performed on lungs using commercially available primers. Expression in primary infection was compared with uninfected lungs and in secondary infection with lungs from mice that had received 104 yeasts 8 weeks before rechallenge. Results represent mean ± SEM of 4–6 animals per group. *p < 0.05.
Is effect of p75-Fc similar to that of mAb to TNF-α?
As mentioned in the Introduction, much of the literature on the influence of TNF-α and H. capsulatum has been derived from studies using polyclonal antibody or an mAb. We infected naïve mice with 2 × 106 yeasts and treated them with a single dose of 1 mg/kg or 5 mg/kg of p75-Fc every 4 days or with 1 mg of anti-TNF-α. Mice were killed at day 7 of infection and assessed for fungal burden in lungs and spleens. CFUs in lungs of mice given either p75-Fc or mAb to TNF-α were elevated (p < 0.01) compared with controls (Fig. 6). Analysis of the treated groups showed that CFUs in lungs of mice given 1 mg/kg but not 5 mg/kg of p75-Fc were modestly less (~0.7 log10 CFU, p < 0.05) than those of mAb recipients.
FIG. 6.
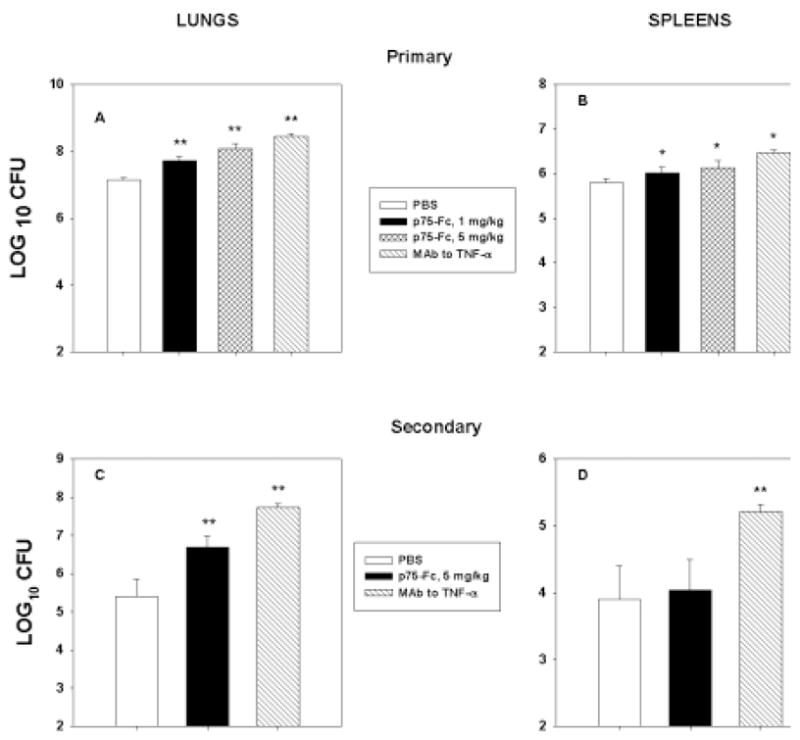
Comparison of p75-Fc and mAb to TNF-α. Mice were infected with H. capsulatum and given p75-Fc every 4 days or 1 mg mAb to TNF-α at the time of infection. On day 7, mice were killed, and CFUs were determined in lungs (A and C) and spleens (B and D) of mice with primary (A and B) or secondary (C and D) histoplasmosis. Results represent mean ± SEM of 4–8 animals. *p < 0.05; **p < 0.01.
We conducted the same experiment in immune mice, with the exception that we used only 5 mg/kg every 4 days, as the 1 mg/kg dose did not impact survival. As shown in Figure 6, the effect of the mAb was far more pronounced when compared with p75-Fc (p < 0.01).
DISCUSSION
A number of risk factors for progressive disseminated histoplasmosis have been identified from clinical studies.16 These include but are not limited to AIDS, immunosuppressive medications, and age. Among the many immunosuppressive agents available, one of the current classes of agents that are known to enhance susceptibility to disseminated histoplasmosis is the TNF-α antagonists.4,5 A recent report suggests that treatment with infliximab is associated with a higher risk of developing histoplasmosis than is treatment with etanercept.5 The reasons for these differences remain unknown. In this study, we endeavored to determine if the protein consisting of the TNFR2 receptor fused with IgG1 Fc molecule exacerbated infection with H. capsulatum in mice.
Using several different dosages, we found that naïve mice manifested increased CFU and did not survive this treatment. The increased susceptibility to infection was noted if p75-Fc was administered every other day or every 4 days. The enhanced susceptibility of mice given p75-Fc was not associated with marked decrements in the inflammatory cells in the lungs. In fact, two effector cell populations, macrophages and neutrophils, were increased in recipients of p75-Fc. The latter are known to inhibit the intracellular growth of H. capsulatum.17,18 Despite this effect, the mice still succumbed to infection.
The increased number of macrophages present in lungs of mice with primary infection might have added to the aggressive nature of the infection, as this cell population often serves as an intracellular haven for the fungus.19–21 Unless the cells are activated, the organism can replicate unimpeded within this population of phagocytes. IFN-γ is known to activate peritoneal macrophages but does not enhance the anti-Histoplasma activity of lung macrophages.7,22 Colony-stimulating factors (CSF) are known to augment short-term (within 2 h) killing of H. capsulatum within alveolar macrophages in vitro, but their effect on fungicidal activity in vivo is unknown.23 Antagonism of TNF-α activity in vivo by mAb is not known to depress GM-CSF levels, and thus it seems unlikely that p75-Fc would modulate the production of this family of cytokines.
In secondary infection, susceptibility to infection was clearly dependent on the dosage of p75-Fc. This distinction was not observed in naïve mice, although those that received a lower dose did survive longer. Administration of 1 mg/kg of p75-Fc every 2 days did not impact survival of immune mice, whereas the higher dosage did whether it was administered every 2 days or every 4 days. These results signify that preexisting immunity may modulate susceptibility to the severity of infection. Moreover, administration of p75-Fc to immune mice did not cause a modulation of either the inflammatory response or the cytokines that were analyzed. This finding is in contrast to the results with the mAb to TNF-α, where IL-4 and IL-10 are both elevated following neutralization in vivo in secondary histoplasmosis.7
In experimental murine histoplasmosis, the first week of infection is the crucial time for the subsequent development of immunity.14 During this time frame, such mediators as IL-12, IFN-γ, GM-CSF, and TNF-α are synthesized, and their release is necessary for protective immunity.7,9,14,15,24 TNF-α appears to be one of the earliest cytokines synthesized, followed by engagement of the IL-12-IFN-γ axis and GM-CSF.9 Impairment of any of those mediators disrupts immunity. Persistent production of TNF-α has been shown to be important in the evolution of the protective immune response.24 Neutralization of this cytokine within the first 5 days of this infection blunts the ability of mice to clear infection. Likewise, we determined that administration of p75-Fc in established primary or secondary infection exacerbated infection even when it was initiated 4 days into infection. These results support our assertion that persistent production of TNF-α is critical for the optimal expression of protective immunity.
Analysis of the cytokine response by mice given the inhibitor demonstrated that none of the cytokines assayed was modulated. Only NOS2, which leads to NO production, was modulated in primary infection. These results are similar to the findings for mice given mAb to TNF-α. Animals receiving this mAb manifested a marked decrease in NO release; however, they differ from TNFR2−/− mice, in which IFN-γ is depressed.11,12 As the circulating antagonist binds TNF-α. thus removing it from engaging TNFR, the host response appears to be similar to that of TNF-α-neutralized mice. Although we measured NOS2 transcripiton, we have found that it correlates well with NO release.25 In secondary infection, no alterations were observed in IL-4 or IL-10 in the lungs of mice given p75-Fc. In contrast, the lungs of mice given mAb to TNF-α exhibited increased IL-4 and IL-10 in response to rechallenge with H. capsulatum. These disparate results indicate that the effects of biologicals can vary even though their net effect is similar. The precise mechanisms by which p75-Fc modulates immunity remain to be determined; it appears that cytokine modulation is not one of them. Recently, it was shown that T cells from mice deficient in TNF-α are dysfunctional in their ability to confer protection, and this effect is independent of their IFN-γ synthesis capacity.24 Thus, it is possible that the effect of p75-Fc is directed at the T cell compartment independent of modulating IFN-γ levels.
Additional studies found that the effect of p75-Fc on fungal burden in primary infection was similar to that of an mAb to TNF-α. On the other hand, there was a difference in the lungs of mice with secondary infection. Although the precise reason for this difference is not known, several possibilities may explain it. One of the simplest explanations is the disparity in protein content, but this seems unlikely given that the two compounds did not produce different results in primary infection. Another possibility is that the metabolism, the pharmacologic distribution, absorption, or the biologic half-life of the two reagents may differ in secondary but not primary infection. Virtually nothing is known about these parameters with either the mAb or the murine p75-Fc. A better understanding of the pharmacology of these reagents will likely improve our understanding of the differences in the impact of these agents.
In summary, p75-Fc reduced the ability of mice to survive infection with H. capsulatum. This effect was observed in both primary and secondary infection, thus supporting the importance of TNF-α in regulating immunity to both phases of infection. However, mice that had been exposed to the fungus did manifest a dose-dependent difference in susceptibility to the p75-Fc. The mechanisms by which this compound and mAb to TNF-α dampen immunity require further exploration.
Acknowledgments
This work was supported by a grant from Amgen Corporation and by Public Health Service grants AI-42747 and AI-34361 and a Merit Review from the Veterans Affairs. I thank Michael McGuinness for excellent technical assistance. Dr. Deepe has been a consultant for Amgen Corporation and has received a grant from Amgen Corporation.
References
- 1.Anderson PJ. Tumor necrosis factor inhibitors: clinical implications of their different immunogenicity profiles. Semin Arthritis Rheum. 2005;34:19–22. doi: 10.1016/j.semarthrit.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B, Cerami A. The biology of cachectin/TNF—a primary mediator of the host response. Annu Rev Immunol. 1989;7:625–655. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 3.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor-α-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 4.Lee JH, Slifman NR, Gershon SK, Edwards ET, Schwieterman WD, Siegel JN, Wise RP, Brown SL, Udall JN, Jr, Braun MM. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor α antagonists infliximab and etanercept. Arthritis Rheum. 2002;46:2565–2570. doi: 10.1002/art.10583. [DOI] [PubMed] [Google Scholar]
- 5.Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis. 2004;38:1261–1265. doi: 10.1086/383317. [DOI] [PubMed] [Google Scholar]
- 6.Deepe GS., Jr . Histoplasma capsulatum. In: Mandell BJ, Dolin R, editors. Principles and Practices of Infectious Diseases. Philadelphia: Elsevier Churchill Livingstone; 2004. pp. 3012–3026. [Google Scholar]
- 7.Allendoerfer R, Deepe GS., Jr Blockade of endogenous TNF-α exacerbates primary and secondary pulmonary histoplasmosis by differential mechanisms. J Immunol. 1998;160:6072–6082. [PubMed] [Google Scholar]
- 8.Deepe GS, Jr, Gibbons R, Woodward E. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum. J Immunol. 1999;163:4985–4993. [PubMed] [Google Scholar]
- 9.Smith JG, Magee DM, Williams DM, Graybill JR. Tumor necrosis factor-α plays a role in host defense against Histoplasma capsulatum. J Infect Dis. 1990;162:1349–1353. doi: 10.1093/infdis/162.6.1349. [DOI] [PubMed] [Google Scholar]
- 10.Wu-Hsieh BA, Lee GS, Franco M, Hofman FM. Early activation of splenic macrophages by tumor necrosis factor alpha is important in determining the outcome of experimental histoplasmosis in mice. Infect Immun. 1992;60:4230–4238. doi: 10.1128/iai.60.10.4230-4238.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou P, Miller G, Seder RA. Factors involved in regulating primary and secondary immunity to infection with Histoplasma capsulatum: TNF-α plays a critical role in maintaining secondary immunity in the absence of IFN-γ. J Immunol. 1998;160:1359–1368. [PubMed] [Google Scholar]
- 12.Zhou P, Sieve MC, Bennett J, Kwon-Chung KJ, Tewari RP, Gazzinelli RT, Sher A, Seder RA. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-γ. J Immunol. 1995;155:785–795. [PubMed] [Google Scholar]
- 13.Worsham PL, Goldman WE. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. J Med Vet Mycol. 1988;26:137–143. [PubMed] [Google Scholar]
- 14.Cain JA, Deepe GS., Jr Evolution of the primary immune response to Histoplasma capsulatum in murine lung. Infect Immun. 1998;66:1473–1481. doi: 10.1128/iai.66.4.1473-1481.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allendoerfer R, Boivin GP, Deepe GS., Jr Modulation of immune responses in murine pulmonary histoplasmosis. J Infect Dis. 1997;175:905–914. doi: 10.1086/513989. [DOI] [PubMed] [Google Scholar]
- 16.Wheat LJ, Slama TG, Norton JA, Kohler RB, Eitzen HB, French ML, Sathapatayavongs B. Risk factors for disseminated or fatal histoplasmosis. Analysis of a large urban outbreak. Ann Intern Med. 1982;96:159–163. doi: 10.7326/0003-4819-96-2-159. [DOI] [PubMed] [Google Scholar]
- 17.Brummer E, Kurita N, Yosihida S, Nishimura K, Miyaji M. Fungistatic activity of human neutrophils against Histoplasma capsulatum: correlation with phagocytosis. J Infect Dis. 1991;164:158–162. doi: 10.1093/infdis/164.1.158. [DOI] [PubMed] [Google Scholar]
- 18.Newman SL, Gootee L, Gabay JE. Human neutrophil-mediated fungistasis against Histoplasma capsulatum. Localization of fungistatic activity to the azurophil granules. J Clin Invest. 1993;92:624–631. doi: 10.1172/JCI116630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bullock WE, Wright SD. Role of the adherence-promoting receptors, CR3, LFA-1, and p150,95, in binding of Histoplasma capsulatum by human macrophages. J Exp Med. 1987;165:195–210. doi: 10.1084/jem.165.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman SL, Bucher C, Rhodes J, Bullock WE. Phagocytosis of Histoplasma capsulatum yeasts and microconidia by human cultured macrophages and alveolar macrophages. Cellular cytoskeleton requirement for attachment and ingestion. J Clin Invest. 1990;85:223–230. doi: 10.1172/JCI114416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu-Hsieh BA, Howard DH. Intracellular growth inhibition of Histoplasma capsulatum induced in murine macrophages by recombinant gamma interferon is not due to a limitation of the supply of methionine or cysteine to the fungus. Infect Immun. 1992;60:698–700. doi: 10.1128/iai.60.2.698-700.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu-Hsieh BA, Howard DH. Inhibition of the intracellular growth of Histoplasma capsulatum by recombinant murine gamma interferon. Infect Immun. 1987;55:1014–1016. doi: 10.1128/iai.55.4.1014-1016.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brummer E, Stevens DA. Effect of macrophage colony-stimulating factor (M-CSF) on macrophage morphology, phagocytosis, and intracellular multiplication of Histoplasma capsulatum. Int J Immunopharmacol. 1994;16:171–176. doi: 10.1016/0192-0561(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 24.Deepe GS, Jr, Gibbons RS. T cells require tumor necrosis factor-α to provide protective immunity in mice infected with Histoplasma capsulatum. J Infect Dis. 2006;193:322–330. doi: 10.1086/498981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deepe GS, Jr, McGuinness M. Interleukin-1 and host control of pulmonary histoplasmosis. J Infect Dis. 2006;194:855–864. doi: 10.1086/506946. [DOI] [PMC free article] [PubMed] [Google Scholar]