

Abstract
Heat shock proteins (hsp's) isolated from murine cancer cells can elicit protective immunity and specific cytotoxic T lymphocytes (CTLs) by channeling tumor-derived peptides bound to hsp's to the major histocompatibility class I antigen presentation pathway. Here we have investigated if hsp70 can be used in a novel peptide vaccine for the induction of protective antiviral immunity and memory CTLs. A CTL epitope from the well-defined lymphocytic choriomeningitis virus (LCMV) system was mixed with recombinant hsp70 in vitro under conditions that optimize peptide binding to hsp70. Mice were immunized with the hsp70–peptide mixture and challenged with LCMV. Virus titers were reduced 10–100-fold in these mice compared to control mice. Immunization with the hsp70–peptide mixture resulted in the development of CTL memory cells that could be reactivated during LCMV infection, and that in a 51Cr-release assay could lyse cells pulsed with the same peptide, but not cells pulsed with another LCMV peptide. These results show that hsp70 can be used with CTL epitopes to induce efficient protective antiviral immunity and the generation of peptide-specific CTLs. The results also demonstrate the usefulness of hsp70 as an alternative to adjuvants and DNA vectors for the delivery of CTL epitopes to antigen-presenting cells.
Heat shock protein 70 (hsp70)1 is a member of a family of molecular chaperones important in protein synthesis and folding. The constitutively expressed cytosolic member of this family assists the folding of newly synthesized polypeptides into their correct conformation by binding to them during protein synthesis. It also assists in the translocation of proteins across membranes into different compartments of the cell. When the cell is stressed, the inducible cytosolic member of the hsp70 family is synthesized, preventing proteins from aggregating and in this way enhancing the survival of the cell. Binding to both the constitutive and the inducible members of hsp70 is a step on the way to degradation of a protein that has become too denatured to be rescued (1, 2).
Hsp70 is among the most conserved and abundant proteins within a cell. Because of its role in protein synthesis, it is an essential molecule for the survival of the normal cell as well as for synthesis of new viral proteins in a virus-infected cell. To be able to perform its tasks, hsp70 has a broad binding specificity. In order for it to bind a peptide, the peptide needs to have a hydrophobic region of more than six amino acids with both small and large hydrophobic amino acids (3–5).
Bacterial hsp's have been long known to stimulate the host immune response, e.g., as a component in Freund's complete adjuvant. Efforts have been made to use hsp's as carrier proteins without additional adjuvants. Recently, Suzue and Young showed that antibody and cytokine production as well as lymphocyte proliferation were elicited in mice immunized with mycobacterial hsp70 covalently bound to the HIV-1 p24 protein (6). Román and Moreno showed that by immunizing mice with mycobacterial hsp70 noncovalently bound to the MHC class II influenza A peptide (pNP 206–229), proliferative T cell responses were elicited to the hsp70-binding peptide (7). In both of these studies, the bacterial hsp70 was used as a carrier protein. It is presumed that the hsp70 and its bound peptide or protein are taken up into the endosomal compartment of APCs, that they go through the exogenous processing pathway, and that the peptides get presented on MHC class II molecules.
It is well established that extracellular proteins are processed through the exogenous processing pathway and are presented on MHC class II molecules, but they do not have access to the endogenous processing pathway. This rule seems to be overcome in experiments where immunizations with hsp's isolated from murine cancer cells have been shown to elicit antitumor immunity in vivo and tumor-specific CTLs in vitro (8–10). The antitumor protection in these experiments was shown to depend on peptides binding to the hsp's purified from tumor tissue, and the results imply the use of an exogenous class I pathway. In a similar way, CTLs specific for a vesicular stomatitis virus (VSV) peptide were generated by immunizing mice with another hsp, gp96, purified from VSV-infected cells. Macrophages were shown to take up the bound peptide and present it on the MHC class I molecules of the macrophages to cytotoxic T cells (10).
These experiments showing the induction of class I–restricted CD8+ T cell responses against tumors or viruses after immunization with hsp from tumor or virus-infected cells led us to hypothesize that protective antiviral T cell responses might be induced by immunization with defined viral immunodominant T cell peptides conjugated to hsp's.
In this study, we investigated if hsp70 could be used to induce CTL-dependent protection to a viral infection in vivo. We asked if a peptide mixed in vitro with hsp70 and provided exogenously by immunization in mice would induce a protective CTL response. If so, this would show that the peptide is presented on MHC class I molecules. This ability would be of great importance in the design of new peptide vaccines, and could give us an insight into to what degree the endogenous antigen processing and presentation pathway is restricted to the origin of the antigen. We therefore mixed viral peptides from the well-defined lymphocytic choriomeningitis virus (LCMV) system (11– 13) with recombinant human hsp70 (rhsp70) in vitro and immunized mice with the rhsp70–peptide mixture. This evoked protective antiviral immunity measured in a virus plaque assay and specific CTLs measured in a 51Cr-release assay.
Materials and Methods
Mice.
C57BL/6 (H-2b) male mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and were used at 1–6 mo of age.
Viruses.
The Armstrong strain of LCMV was used in all experiments. The virus was propagated in BHK21 baby hamster kidney cells (14).
Cell lines.
MC57G (H-2b), a methylcholanthrene-induced sarcoma from C57BL/6 mice, was propagated in Eagle's MEM (Gibco Laboratories, Life Technologies Ltd., Paisley, U.K.) supplemented with 100 U/ml penicillin G, 100 μg/ml streptomycin sulfate, 2 mM l-glutamine, and 10% heat-inactivated fetal bovine serum. Vero, an African green monkey kidney cell line used in plaque assays, was propagated in MEM as above. The RMA-S cell line was derived from a Rauscher leukemia virus–induced T cell lymphoma, RBL-5, of B6 origin (H-2b; 15) and cultured in RPMI medium (Sigma Chemical Co., St. Louis, MO) supplemented as above.
Peptides, hsp70, and Binding/Mixing Procedure.
Three previously described LCMV peptides were used (12, 13, 16). Two of the peptides were from the LCMV glycoprotein (GP): peptide 33–40 (KAVYNFAT), here called 8mer, and peptide 33–45 (KAVYNFATCGIFA), here called 13mer. The third peptide was the ribonucleoprotein peptide 397–407 (QPQNGQFIHFY), here called NP. The 8mer and 13mer were synthesized at the peptide synthesis facility at Microbiology and Tumorbiology Center, Karolinska Institute (Stockholm, Sweden), by a solid-phase method using Fmoc chemistry. The NP peptide was synthesized and HPLC-purified by Dr. Robert Carroway at the University of Massachusetts peptide core facility.
The rhsp70 was produced and purified as previously described (17). The peptide binding activity of this rhsp70 has been assessed by a competition assay, in which an unfolded form of lactalbumin was complexed to rhsp70. Addition of an excess of a 7-amino acid-long peptide competed out the lactalbumin, showing that the rhsp70 used is entirely in the native form that can and does bind peptides (17). In accordance to the protocol described in Jindal et al. (17), the peptides were incubated with rhsp70 or BSA (Sigma Chemical Co.) in binding buffer (PBS with 2 mM MgCl2) at 37°C for 60 min. Then 0.5 mM ADP (Sigma Chemical Co.) was added and the incubation was continued for another 60 min at the same temperature. This protocol method was also consistent with those of others (4, 5, 18, 19). Lactoferrin (Sigma Chemical Co.) was incubated with peptide in binding buffer on ice for 60 min.
Immunizations.
Mice were immunized with 2 μg peptide (8mer or 13mer), rhsp70, BSA, or lactoferrin; with 2 μg peptide mixed with 2 μg of rhsp70, BSA, or lactoferrin; or with binding buffer only. The immunizations were done intraperitoneally, three times at 2-wk intervals (experiment 1) or two times at 3-wk intervals (experiments 2 and 3). 2 (experiment 1) or 3 (experiments 2 and 3) wk after the last immunization the mice were inoculated intraperitoneally with 5 × 104 PFU of LCMV, and on day 3 or 5 after infection the spleens were harvested. Nonimmunized mice were killed on day 8 after infection for detection of the peak CTL response.
Cytotoxicity assay.
A standard microcytotoxicity assay was used to determine CTL activity (20). RMA-S cells incubated with 8mer or NP peptides or with MC57G cells infected with LCMV were pelleted, resuspended in 100 μCi of Na-51Cr per 106 cells (Amersham Corp., Arlington Heights, IL), and incubated for 1 h at 37°C in a humidified 5% CO2 incubator. The cells were rinsed three times and resuspended to 105/ml, and 0.1 ml of the cell preparation was added to round-bottomed microtiter wells. Varying numbers of effector leukocytes were added in triplicate in 0.1 ml of medium to achieve the desired E/T ratio. For spontaneous 51Cr-release, 0.1 ml of media was added to the labeled target cells in place of effectors. For maximum 51Cr-release, 0.1 ml of 1% NP-40 (United States Biochemical Co., Cleveland, OH) was added to the labeled target cells. After 6 h at 37°C in a 5% CO2 incubator, the microtiter plates were centrifuged at 200 g for 5 min. Supernatants were removed (0.1 ml) from each well and counted on a gamma counter (model 5000; Beckman Instruments, Inc., Fullerton, CA). Data are expressed as follows: percentage of specific 51Cr-release = 100 × [(experimental cpm − spontaneous cpm) / (maximum release cpm − spontaneous release cpm)]. The killing of noninfected or nonpulsed cells was subtracted from the pulsed or infected cell data, thus giving the percentage of specific killing.
Virus Titer Assay.
Spleens from immunized mice were homogenized and centrifuged at 400 g for 15 min. Supernatants were titrated for virus plaques on Vero cell monolayers.
Results
Protective Anti-LCMV Immunity in Mice Immunized with hsp70–Mixed Peptide.
We evaluated if mixing of the 8mer or the 13mer of the GP 33 LCMV epitope with rhsp70 would enhance resistance of immunized mice to challenge with LCMV-Armstrong. For hsp70 to bind a peptide to its peptide-binding groove, the peptide needs to have a hydrophobic region of more than 6 amino acids. The 8mer, a well-defined MHC class I epitope, and the 13mer, an elongation of the 8mer with five additional amino acids, each have both small and large hydrophobic amino acids, required for binding to hsp70 (3–5). A 70:1 molar ratio of peptide/protein was chosen to facilitate the chance of most of the rhsp70 molecules binding to the LCMV peptide, according to what we had previously (17) found to be an optimal condition for peptide binding using the same source of rhsp70. After 1 h, 0.5 mM of ADP was added, as ADP greatly enhances the ability of hsp70 to bind to peptides (18).
Mice immunized with LCMV immunodominant peptides with or without prior mixing of the peptides with rhsp70 were challenged with LCMV, and viral titers in the spleens were determined by plaque assays 3 or 5 d after infection (Fig. 1).
Figure 1.

In vivo resistance to challenge with LCMV. Mice immunized with 8mer, rhsp70, or 8mer mixed with BSA have high virus titers after challenge with LCMV, whereas mice immunized with rhsp70-mixed 8mer have 8–100-fold lower virus titers. (a) Experiment 1, day 3 after infection with LCMV (n = 3). (b) Experiment 1, day 5 after infection (n = 2). (c) Experiment 2, day 3 (n = 3). (d) Experiment 2, day 5 (n = 2). (e) Experiment 3, day 5 (n = 3 or 4) N.D. = not done. Bars show the standard deviation.
In the first experiment, mice were immunized three times at 2-wk intervals before challenge with virus. At day 3 after challenge, immunization with the 8mer provided no protection compared to the buffer or rhsp70 controls, whereas immunization with the hsp70-mixed bound 8mer resulted in titers 0.8 log lower than that of rhsp70 alone. By day 5 after infection, immunization with the hsp70-mixed 8mer resulted in nearly total clearance of virus, with >100-fold clearance in comparison to rhsp70-only or 8mer-only controls. One of the two mice immunized only with buffer had cleared virus, but this was considered an anomaly, as the other mouse had a titer similar to the rhsp70-only and 8mer-only controls, and clearance by buffer-immunized mice was not seen in any other experiment.
The immunization scheme was shortened in the second and third experiments, where mice were immunized only twice before challenge with virus. Again, rhsp70 greatly enhanced the immunization capacity of the 8mer, as substantial reductions in viral titers at days 3 and 5 after infection occurred in mice immunized with the hsp70-mixed 8mer. Compared to buffer-only, 8mer-only, or rhsp70-only controls, as high as 10–100-fold reductions in viral titers were observed. In neither experiment did the 8mer alone immunize against the viral infection. As an additional control, in some experiments peptides were preincubated with BSA or human lactoferrin under conditions similar to that used with rhsp70. Neither BSA nor lactoferrin enhanced the immunization capacity of the peptides.
Similar experiments were also done with the 13mer, which was an elongated form of the 8mer. Immunization with the 13mer alone resulted in greater than a tenfold reduction in viral titers at days 3 or 5 after infection in all three experiments, and coimmunization with rhsp70 did not enhance the protective effect except in experiment 2, day 5 (Fig. 2). Thus, the hsp70 enhanced the protective immunity induced by the minimal T cell epitope but was unnecessary for immunization by the more complex epitope.
Figure 2.

Immunization with 13mer alone gives resistance to LCMV challenge. Mice immunized with the 13mer alone had lower viral titers than did buffer controls, with or without hsp70 or BSA. (a) Experiment 1, day 3 after infection with LCMV (n = 3). (b) Experiment 1, day 5 after infection (n = 2). (c) Experiment 2, day 3 (n = 3). (d) Experiment 2, day 5 (n = 2) (e) Experiment 3, day 5 (n = 3 or 4) N.D. = not done. Bars show the standard deviation.
Immunization with rhsp70-mixed Peptide Primes for LCMV-specific CTLs.
We next evaluated if the enhanced protection induced by the GP 33–derived peptides, when mixed with rhsp70, would be associated with an induction of specific CTLs. The CTL response to LCMV normally peaks at days 7–9 after infection in nonimmunized mice, but occurs on days 4 to 5 in mice previously immunized with virus (11, 20). We have found that in primary infections some CTL activity can be detected on the highly sensitive peptide-pulsed RMA-S targets 5 d after infection, whereas less killing can be detected against conventional virus-infected MC57G cell targets. For the following experiments we examined CTL killing on day 5 against both virus-infected MC57G targets and peptide-pulsed RMA-S targets.
Our results demonstrated that spleen cells from mice immunized with the rhsp70-mixed 8mer were significantly more efficient in lysing 8mer-pulsed RMA-S cells, as compared to spleen cells from mice immunized with peptide alone, with buffer, or with rhsp70 alone (Fig. 3 a). Similarly, mice immunized with the 13mer peptide mixed with rhsp70 generated significantly higher cytotoxic activity after challenge than those immunized with 13mer alone, buffer, or rhsp without peptides (Fig. 3 a). In one experiment, however, high cytotoxic activity on GP33-coated targets after challenge was obtained with spleen cells from mice immunized with the 13mer alone without rhsp70.
Figure 3.
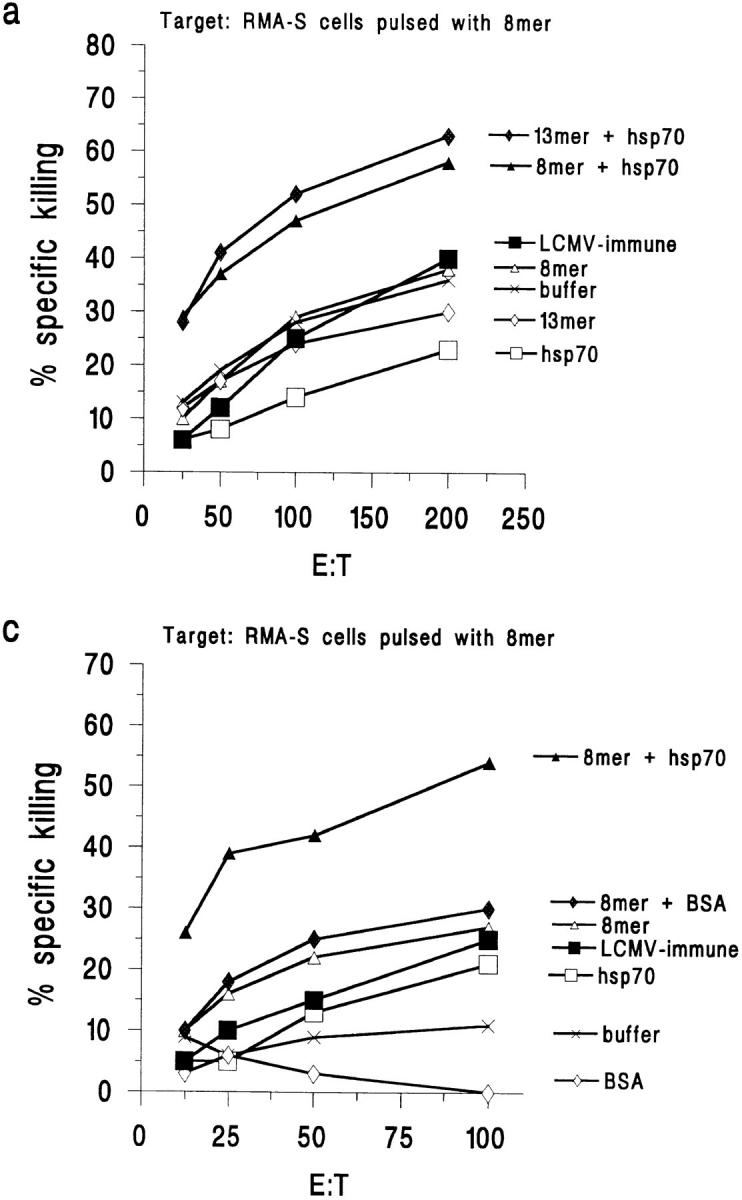
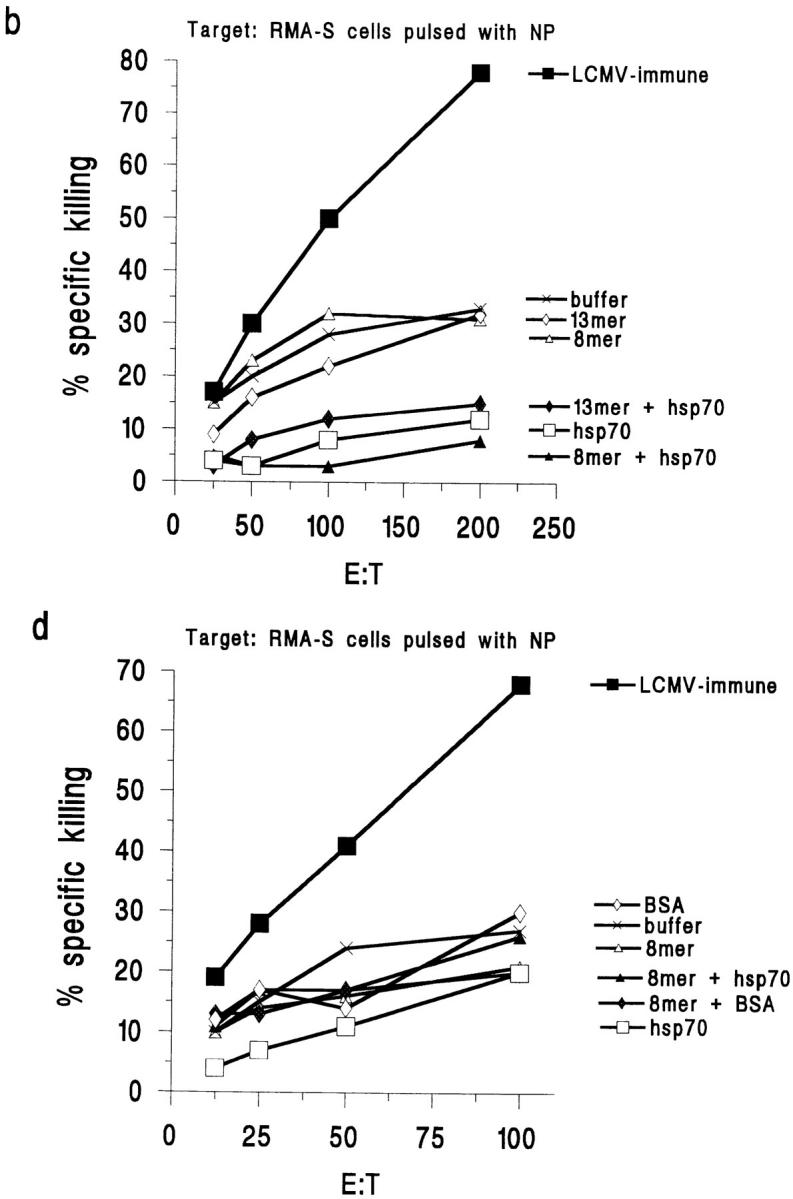
Immunization with hsp70-mixed peptide elicits peptide-specific CTLs 5 d after challenge with LCMV. (a) Immunization with rhsp70-mixed 8mer or 13mer, but not 8mer or 13mer alone or hsp70 alone, primes for 8mer-specific CTLs. Target: RMA-S pulsed with 8mer. (b) Immunization with hsp70-mixed 8mer does not prime for NP-specific CTLs. Target: RMA-S pulsed with NP. BSA cannot replace hsp70. (c) Neither BSA nor BSA with peptide prime for 8mer-specific CTLs. Target: RMA-S pulsed with 8mer. (d) Immunization with hsp70-mixed or BSA-mixed 8mer does not prime for NP-specific CTL. Target: RMA-S pulsed with NP peptide. The killing of nonpulsed RMA-S cells has been subtracted from the killing of the peptide-pulsed cells in all four panels.
The 8mer was also mixed with BSA (Fig. 3 c) or lactoferrin (data not shown) as protein carrier controls for the hsp70. Neither treatment enhanced the ability of the 8mer, when compared to the 8mer alone, to prime for a CTL response after challenge (Fig. 3 c). This is consistent with the in vivo protection results, which show that those control carrier molecules had no effect on protective immunity.
RMA-S cells pulsed with the NP peptide of the nuclear protein of LCMV were used as specificity controls in the same experiments. Although mice previously immunized with LCMV elicited a strong anti-NP response after challenge with LCMV, mice immunized with the GP33 8mer or 13mer, whether mixed with rhsp70 or not, did not elicit an enhanced anti-NP CTL response after LCMV challenge (Fig. 3, b and d). Acutely infected mice 8 d after infection were tested as positive controls, and their splenocytes mediated >80% killing of RMA-S cells pulsed with either the GP or NP peptide.
Next, we measured the cytotoxic activity of the splenocytes from the immunized mice on LCMV-infected MC57G cells, to see if the CTLs we had primed for also had the ability to lyse the less sensitive virus-infected cells. The hsp70-mixed 8mer CTLs did lyse the virus-infected cells at significant levels at day 5 in one experiment, and some low levels of lysis were detected in the other two experiments as well (data not shown). After immunizing with the hsp70-mixed 13mer, cytotoxic activity after virus challenge against LCMV-infected target cells was observed at a stronger level. Significant increases in cytotoxic activity were measured with these 13mer hsp70-primed CTLs in all experiments compared to peptide only, buffer, or hsp70 only controls (Fig. 4). Mice immunized with 13mer hsp70 developed CTL activity as high as did immune mice previously infected with LCMV on challenge. This suggests that immunizing with the hsp70-mixed 13mer would be as effective in priming for LCMV-specific CTLs as would a viral infection.
Figure 4.

Immunization with hsp70-mixed 13mer primes for LCMV-specific CTL activity 5 d after challenge with the virus. Immunization with hsp70-mixed bound 13mer, but not 13mer alone, hsp70 alone, or peptide mixed with BSA primes for CTL activity detected on LCMV-infected targets. Target: LCMV-infected cells. The killing of noninfected MC57G cells has been subtracted from the killing of the infected cells.
Discussion
We show that it is possible to induce protective immunity to LCMV with a low dose of a minimal T cell epitope if the peptide is mixed with hsp70, without the need of additional adjuvants. To our knowledge this is the first time in vivo protection to an infectious agent has been shown by immunization with a peptide–hsp complex. The protection shown here implies that the complexing of peptide to hsp70 has resulted in a more efficient CTL induction, as protection in the LCMV system is dependent on CTLs (21), particularly when immunization is carried out in the absence of neutralizing antibody epitopes. We next show that the enhanced in vivo protection was indeed associated with a more efficient induction of virus-specific CTLs on LCMV challenge. These CTLs are specific for the LCMV peptide with which we immunized and did not recognize another LCMV peptide, showing the specificity of the system. Hence, complexing of the peptide to hsp70 is likely to have resulted in a more efficient presentation of the peptide on the MHC class I complex on the cell surface of APCs. Mixing of the peptide with either of the carrier proteins BSA or lactoferrin did not result in enhanced in vivo protection or CTL generation when tested in vitro. Thus the effect of hsp70 is not a general response to any additional protein in the immunization protocol, but is specific to the properties of the hsp molecule. Such properties could involve both the known capacity of hsp70 in binding to peptides, as well as endowments of the protein we have yet to discover.
Our results indicate that exogenous antigens may play an important roles in the priming of CTLs. It has been suggested that hsp's facilitate the delivery of antigenic epitopes to MHC class I molecules (22). In preliminary experiments by Blachere et al. (23) and in a pilot experiment with a small number of animals by Heikema et al. (24), gp96 was purified from cells transfected with a gene encoding the sequence for a viral peptide from flu-NP or SV40. Splenocytes from mice immunized with this gp96 were shown to kill target cells transfected with the relevant protein or infected with the relevant virus. Experimental data with gp96 in a VSV system suggest that macrophages take up the hsp and its bound peptides, resulting in a presentation of the peptide epitope by the MHC class I molecules of the macrophage to T cells (10). It is not yet known how the hsp is taken up by APCs in vivo. For gp96 the mannose receptor has been suggested as a route of uptake by macrophages (10), but no mechanism has been suggested for the nonglycosylated hsp70. The route the peptide takes inside the cell before reaching the MHC class I molecule is also not yet known.
We show experimental support for the importance of hsp's in CTL priming. Our LCMV model shows that chaperoning of the viral peptide by hsp70 results in highly efficient CTL generation and enhanced in vivo resistance to virus replication, even when given at low doses of peptide and low doses of hsp70. During revision of this manuscript, Blachere et al. published data showing binding of a VSV peptide to gp96 and hsp70 and priming for CD8+ cells. They also show that gp96 bound to a VSV peptide can protect against challenge with a VSV-transfected tumor (25). Our data are consistent with these data, while uniquely showing that this technique can be used to induce protective antiviral immunity in vivo. Apart from the capacity of “channeling” the peptide to the MHC class I antigen presentation pathway, hsp70 may also protect the peptide from degradation. The enhanced in vivo protection was apparent with the minimal 8mer epitope of GP 33 when mixing it with hsp70, but it is notable that flanking this peptide with an additional five amino acids renders it immunogenic in vivo even when given at a quantity of only 2 μg in the absence of hsp70. The explanation for this is unclear, but it implies that the longer peptide epitope may be channeled to the MHC class I antigen presentation pathway even without hsp70, or that the 13mer is less vulnerable to degradation. The latter explanation would emphasize the protective role of hsp70 in vivo.
Efforts are being made to construct effective peptide-based vaccines against infectious agents and tumors. Peptides derived from tumor antigens have been shown to confer protection against tumor growth in murine models (26), and clinical trials have been initiated using peptides as immunotherapy in tumor patients (27). The usefulness of peptide-based vaccines is limited by the need for immunizing with a large amount of peptide and for effective and nontoxic adjuvants that also can induce specific CTL responses. Inefficient antigen presentation of the injected peptide may even lead to tolerance induction, resulting in reduced, rather than enhanced, host protection (21, 28). Using hsp's purified from infected cells or tumor cells, or as described here using an rhsp mixed in vitro with defined T cell epitopes, could considerably facilitate the efficacy of peptide-based vaccines. As hsp70 is an abundant self-protein, immunization with the whole protein is not likely to elicit immune responses to the hsp70 itself because of self tolerance (7, 29). The amount of peptide necessary to evoke a protective immune response could be greatly reduced, and an additional adjuvant is not required. These qualities, together with its efficient way of enhancing protective immunity, may render hsp70 into a good candidate for use in peptide-based vaccines.
Footnotes
Jehad Charo is thanked for critical discussions.
This study was supported by a grant for Rolf Kiessling from the Swedish Medical Research Council. Anne-Marie T. Ciupitu is supported by a fellowship from the Swedish Cancer Society. The work of Raymond Welsh is supported by U.S. Public Health Service National Institutes of Health research grants AI-17672, AR-35506, and CA-34461.
Address correspondence to Raymond M. Welsh, Department of Pathology, University of Massachusetts Medical Center, 55 Lake Ave. North, Worcester, MA 01655-0125. Phone: 508-856-5819; Fax: 508-856-5780; E-mail: raymond.welsh@banyan.ummed.edu
Abbreviations used in this paper: GP, glycoprotein; hsp, heat shock protein; LCMV, lymphocytic choriomeningitis virus; rhsp 70, recombinant human hsp70; VSV, vesicular stomatitis virus.
References
- 1.Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 2.Hartl F-U, Hlodan R, Langer T. Molecular chaperones in protein folding: the art of avoiding sticky situations. TIBS (Trends Biochem Sci) 1994;19:20–25. doi: 10.1016/0968-0004(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 3.Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething MJH. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–728. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- 4.Fourie AM, Sambrook JF, Gething MJH. Common and divergent peptide binding specificities of hsp70 molecular chaperones. J Biol Chem. 1994;269:30470–30478. [PubMed] [Google Scholar]
- 5.Craig EA, Weissman JS, Horwich AL. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell. 1994;78:365–372. doi: 10.1016/0092-8674(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 6.Suzuke K, Young RA. Adjuvant-free hsp70 fusion protein system elicits humoral and cellular immune responses to HIV-1 p24. J Immunol. 1996;156:873–879. [PubMed] [Google Scholar]
- 7.Román E, Moreno C. Synthetic peptides noncovalently bound to bacterial hsp70 elicit peptide-specific T-cell responses in vivo. . Immunology. 1996;88:487–492. doi: 10.1046/j.1365-2567.1996.d01-697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Udono H, Srivastava PK. Heat shock protein 70–associated peptides elicit specific cancer immunity. J Exp Med. 1993;178:1391–1396. doi: 10.1084/jem.178.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Udono H, Srivastava PK. Comparison of tumor-specific immunogenicities of stress-induced proteins gp96, hsp90 and hsp70. J Immunol. 1994;152:5398–5403. [PubMed] [Google Scholar]
- 10.Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein–chaperoned peptides. Science. 1995;269:1585–1588. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- 11.Zinkernagel RM, Moskophidis D, Kündig T, Oehen S, Pircher H, Hengartner H. Effector T-cell induction and T-cell memory versus peripheral deletion of T cells. Immunol Rev. 1993;131:199–223. doi: 10.1111/j.1600-065x.1993.tb01517.x. [DOI] [PubMed] [Google Scholar]
- 12.Lewicki H, Tishon A, Borrow P, Evans CF, Gairin JE, Hahn KM, Jewell DA, Wilson IA, Oldstone MBA. CTL escape viral variants. I. Generation and molecular characterization. Virology. 1995;210:29–40. doi: 10.1006/viro.1995.1314. [DOI] [PubMed] [Google Scholar]
- 13.Lewicki H, von Herrath MG, Evans CF, Whitton JL, Oldstone MBA. CTL escape viral variants. II. Biologic activity in vivo. . Virology. 1995;211:443–450. doi: 10.1006/viro.1995.1426. [DOI] [PubMed] [Google Scholar]
- 14.Welsh RM, Lampert PW, Burner PA, Oldstone MBA. Antibody-complement interactions with purified lymphocytic choriomeningitis virus. Virology. 1976;73:59–71. doi: 10.1016/0042-6822(76)90060-x. [DOI] [PubMed] [Google Scholar]
- 15.Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H-2 deficient lymphoma variants suggests alternative immune defense strategies. Nature. 1986;319:675–678. doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- 16.Selin LK, Vergilis K, Welsh RM, Nahill SR. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infection. J Exp Med. 1996;183:2489–2499. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jindal, S., S. Rosenberg, P. Murray, R.A. Young, and K.P. Williams. 1995. Human stress protein hsp70: overexpression in E. coli, purification and characterization. Bio/technology. 13:1105–1109. [DOI] [PubMed]
- 18.Palleros DR, Welch WJ, Fink AL. Interaction of hsp70 with unfolded proteins: effects of temperature and nucleotides on the kinetics of binding. Proc Natl Acad Sci USA. 1991;88:5719–5723. doi: 10.1073/pnas.88.13.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flynn GC, Chappell TG, Rothman JE. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science. 1989;245:385–390. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- 20.Welsh RM. Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J Exp Med. 1978;148:163. doi: 10.1084/jem.148.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battery M, Oehen S, Schulz M, Hengartner H, Zinkernagel RM. Vaccination with a synthetic peptide modulates lymphocytic choriomeningitis virus–mediated immunopathology. J Virol. 1992;66:1199–1201. doi: 10.1128/jvi.66.2.1199-1201.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srivastava PK, Udono H, Blanchere NE, Li Z. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics. 1994;39:93–98. doi: 10.1007/BF00188611. [DOI] [PubMed] [Google Scholar]
- 23.Blachere NE, Udono H, Janetzki S, Li Z, Heike M, Srivastava PK. Heat shock protein vaccines against cancer. J Immunother. 1993;14:352–356. doi: 10.1097/00002371-199311000-00016. [DOI] [PubMed] [Google Scholar]
- 24.Heikema A, Agsteribbe E, Wilschut J, Huckriede A. Generation of heat shock protein–based vaccines by intracellular loading of gp96 with antigenic peptides. Immunol Lett. 1997;57:69–74. doi: 10.1016/s0165-2478(97)00048-5. [DOI] [PubMed] [Google Scholar]
- 25.Blachere NE, Li Z, Chandawarkar RY, Suto R, Jaikaria NS, Basn S, Udono H, Srivastava PK. Heat shock protein–peptide complexes, reconstituted in vivo, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J Exp Med. 1997;186:1315–1322. doi: 10.1084/jem.186.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bloom MB, Perrylalley D, Robbins PF, Li Y, Elgamil M, Rosenberg SA, Yang JC. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boon T. Tumor antigens recognized by cytolytic T lymphocytes: present perspectives for specific immunotherapy. Int J Cancer. 1993;54:177–180. doi: 10.1002/ijc.2910540202. [DOI] [PubMed] [Google Scholar]
- 28.Toes RE, Blom RJ, Offringa R, Kast WM, Melief CJ. Enhanced tumor outgrowth after peptide vaccination. Functional deletion of tumor-specific CTL induced by peptide vaccination can lead to the inability to reject tumors. J Immunol. 1996;156:3911–3918. [PubMed] [Google Scholar]
- 29.Diris ML, Gralow JR, Bernhard H, Hand SL, Rubin WD, Cheever MA. Peptide-based, but not whole protein, vaccines elicit immunity to HER-2/neu, an oncogenic self-protein. J Immunol. 1996;156:3151–3158. [PubMed] [Google Scholar]