

Abstract
The current knowledge of CD4 function is limited to its role as a necessary coreceptor in TCR-initiated signaling. We have investigated whether CD4 regulates additional T cell functions. Using human primary resting CD4+ T cells, we demonstrate that CD4 activation is sufficient to induce lymphocyte death. Immediately after CD4 cross-linking, CD4+ T cells are rendered susceptible to apoptosis mediated by TNF or FasL. This, together with the concomitant induction of FasL within the same population, results in significant CD4+ T cell death in vitro. The CD4-dependent induction of susceptibility to apoptosis that is mediated by TNF or FasL is protein synthesis independent but phosphorylation dependent. After CD4 activation, PKC regulates susceptibility to apoptosis mediated by FasL but not the induction of susceptibility to TNF-dependent apoptosis. Moreover, significant differences between CD3 and CD4 activation were observed with regards to the kinetics of induction of CD4+ T cell susceptibility to FasL- and TNF-mediated apoptosis. Altogether, these results provide a model with which to study the molecular mechanisms regulating lymphocyte survival after CD4 activation, and highlight the potential role of CD4 in controlling lymphocyte apoptosis under physiological conditions or in disease states such as HIV infection.
The functional relevance of CD4 in the physiology of CD4+ T lymphocytes is partially understood. In the context of antigen presentation, CD4 is required to enhance the activation and proliferation that ensues after TCR engagement. An additional function of CD4 may be to control the fate of lymphocyte survival after the initial immune response. Interruption of the CD4–MHC class II interactions that occurs during antigen presentation inhibits the CD4+ T cell apoptosis triggered by subsequent reengagement of the TCR (1). This complementary function of CD4 may be of relevance not only to controlling the physiological immune response, but in disease states in which CD4 may become activated independently from TCR coactivation such as in the context of HIV infection.
Accumulating data suggest that depletion of CD4+ T cells in HIV-infected individuals is in part secondary to enhanced lymphocyte apoptosis due to an HIV-dependent dysregulation of the physiological mechanisms that control peripheral T cell homeostasis. The interaction between Fas and TNFRI with the corresponding ligands, Fas ligand (FasL)1 and TNF, controls the T cell expansion after an immune response (2–4). In HIV-infected individuals, a large portion of peripheral, and presumably uninfected, CD4+ T cells are found to be highly susceptible to FasL-mediated apoptosis (5, 6), suggesting that FasL–Fas interactions may play a role in CD4+ T cell depletion. How HIV infection directly or indirectly leads to an enhanced state of susceptibility to FasL-dependent apoptosis is currently unknown.
One potential mechanism leading to enhanced susceptibility to apoptosis includes the interaction of HIV glycoprotein (gp)120 with CD4. In vitro, CD4 activation of resting human CD4+ T cells by anti-CD4 antibodies or HIV gp120 is necessary in order for a second signal, such as TCR stimulation, to induce apoptosis (7, 8). Moreover, in in vivo murine models, activation of CD4 causes a significant CD4+ T lymphopenia secondary to an increased rate of cell apoptosis in lpr+/+ but not in lpr−/− mice (9, 10). Although these observations suggest that CD4 activation confers a “negative” signal that primes the lymphocyte to apoptosis presumably mediated by Fas–FasL interactions, several questions remain unresolved. Namely, whether CD4 activation alone is sufficient to confer susceptibility to apoptosis-inducing ligands, such as FasL or other ligands, and if so, what is the molecular mechanism(s) whereby CD4 activation leads a resting, and therefore apoptosis-resistant, T lymphocyte into a state of susceptibility to apoptosis. Previous in vitro studies have suggested that combination of specific cytokines, such as TNF and IFN-γ, that are secreted by PBLs after their activation by CD4 are sufficient to render the CD4+ T cell subpopulation susceptible to apoptosis (11). Although this suggests that indirect, CD4-dependent mechanisms regulate susceptibility, it is also possible that signal transduction pathways that are activated by CD4 cross-linking may directly lead to a state of susceptibility. This could be achieved by targeting molecules that control susceptibility to cell death pathways triggered by Fas or TNFR, among other receptors.
In this report, we have investigated whether CD4 confers susceptibility to cell death in primary CD4+ T cells, and the molecular mechanisms involved in this. Our results indicate that CD4 activation induces a net state of susceptibility to apoptosis that can be mediated not only by FasL but also by TNF. The induction of such a state of susceptibility is protein synthesis–independent, suggesting that it is mediated directly by the CD4-initiated signal transduction pathway. Moreover, we have identified protein kinase C (PKC) as a target of the CD4-initiated signal transduction pathway that is required for the induction of susceptibility to FasL-mediated apoptosis. Altogether, these studies demonstrate the importance of CD4 in regulating T cell survival, and its potential relevance in the control of CD4+ T cell homeostasis after an immune response and in the induction of CD4+ T cell depletion after HIV infection.
Materials and Methods
Cells and Culture Conditions.
To isolate CD4+ T cells, PBMCs from healthy donors were isolated from buffy coats by density gradient centrifugation (Ficoll-Hypaque; Pharmacia Biotech, Piscataway, NJ). The resulting PBMCs were depleted of monocytes by two cycles of plastic adherence, and CD3+ T cells were purified by neuraminidase-treated SRBC rosetting. CD3+ T cells were depleted of CD8+ T cells by negative selection using anti-CD8 antibodies (Caltag Labs., South San Francisco, CA) followed by goat anti–mouse IgG magnetic beads (Dynal, Lake Success, NY). The remaining cell population was repeatedly found to be 100% CD3+ T cells and ⩾98% CD4+ T cells as determined by flow cytometry in each experiment. CD4+ T cells used in the various experiments were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2 mM l-glutamine, and antibiotics (penicillin: 100 U/ml; streptomycin: 100 μg/ml; Whittaker Bioproducts, Walkersville, MD) at 2 × 106 cells/ml.
Antibodies and Reagents.
F(ab′)2 fragments of the anti-Fas monoclonal antibodies M3 and M33, and fusion proteins containing the Fc portion of mouse IgG1 and the extracellular domain of Fas (FasFc), TNFRI (TNFRFc), and CD30 (CD30Fc) have been previously described (12–14). The apoptosis-inducing anti-Fas cross-linking monoclonal antibodies (clone CH-11; IgM) was purchased from Upstate Biotechnology (Lake Placid, NY). Human recombinant TNF was purchased from Genzyme (Cambridge, MA). The anti–human FasL monoclonal antibody NOK1 conjugated with PE was provided by Dr. Hideo Yagita (Yuntendo University, Japan) (15). The PKC inhibitor GF109203X (GF) was purchased from Calbiochem (San Diego, CA). The protein synthesis inhibitor cycloheximide was purchased from Sigma Chemical Co. (St. Louis, MO).
CD4+ T Cell Cross-linking.
CD4+ T cells were incubated with the anti-CD4 antibody Leu-3a (Becton Dickinson, San Jose, CA), anti-CD3 (OKT3; American Type Culture Collection, Rockville, MD), or an isotype-matched control (mouse IgG1; Becton Dickinson) at a concentration of 5 μg/2 × 106 cells/ml for 1 h at 4°C. Recombinant HIV gp120 (Protein Sciences Corp., Meriden, CT) was added at a concentration of 5 μg/2 × 106 of CD4+ T of cells /ml for 1 h at 4°C. Anti-CD4 antibody or gp120-bound cells were then washed and transferred to 24-well Nunclon® plates (Sigma Chemical Co.) that had been previously coated with goat anti–mouse (GAM; Biosource, Camarillo, CA) or anti-gp120 antiserum (AIDS Research and References Reagent Program, National Institutes of Health, Bethesda, MD) (16, 17), and incubated for 1 h at 37°C. Secondary antibody precoating was performed using 20 μg of GAM in 200 μl, or using 20 μl of anti-gp120 serum in 200 μl of 0.05 M carbonate buffer/well for 2 h at 37°C and washed twice with 10% RPMI 1640. After the hour of CD4 cross-linking, CD4+ T cells were harvested by gentle pipetting, washed, counted, and transferred to 24-well plates at a concentration of 2 × 106 cells/ml/well or to 96-well plates at a concentration of 105 cells/ml/well. Each point was processed in triplicate.
Cell Death Induction and Analysis.
To determine Fas- or TNF-mediated apoptosis, CD4 cross-linked cells were treated with anti-Fas cross-linking IgM antibody (CH-11) or TNF for the different incubation times. Percentage of cell mortality was calculated using trypan blue dye exclusion as follows: (total number of blue cells) / (100 × total number of cells). Results from cultures from triplicate cells were used to calculate the mean and standard deviation.
Flow cytometry analysis of cell apoptosis was performed as previously described (18). In brief, cells are harvested, washed, and resuspended in 1 ml of PBS. 1 μg/ml of Hoeschst 33342 (Calbiochem, Corp.) was added and incubated for 10 min on ice. Cells were then washed and resuspended at 106 cells/ml of PBS with 2 μg/ml of propidium iodide (Sigma Chemical Co.). Cells were kept on ice and analyzed immediately on a FACStarPlus® flow cytometer and the results were calculated using CellQuest® software (Becton Dickinson). Percentage of specific apoptosis was calculated as: 100 × (percentage of apoptosis of CD4–cross-linked or IgG1 CD4+ T cells − percentage of apoptosis present in untreated [noncross-linked] CD4+ T cells [spontaneous apoptosis]/ [100 − percentage of spontaneous apoptosis]).
Flow Cytometry Analysis of FasL.
Flow cytometry for surface staining was performed on 106 CD4+ T cells. Cells were washed with PBS followed by a 20-min incubation at 4°C with 1 μg of FasL (NOK1)-PE (15) or mouse IgG1 PE isotype. Cells were then washed and fixed in 1% paraformaldehyde. Flow cytometry was performed in FACScan® and analysis was done using CellQuest® software.
Results
CD4 Activation Is Sufficient to Induce Cell Death by Apoptosis of Purified CD4+ T Cells.
Previous reports demonstrated that CD4 activation primes CD4+ T cells for the induction of T cell apoptosis triggered by a second signal provided by accessory cells or activation via the TCR (7, 8, 10, 11). To determine if CD4 activation confers susceptibility to apoptosis-inducing ligands such as FasL and TNF, preliminary experiments were performed using purified CD4+ T cells that had been CD4 cross-linked for different time periods in order to identify the optimal culture conditions. Surprisingly, in these preliminary experiments a significant mortality of the purified CD4+ T cell population that had undergone CD4 cross-linking (XL) was observed as early as at 18 h as compared to minimum cell mortality present in the control cultures treated with IgG isotype and secondary antibodies (IgG cross-linked) during the same time points (Fig. 1 A). To confirm these observations and exclude any nonspecific toxicity of anti-CD4 antibodies, we tested whether alternative modes of CD4 activation, such as that achieved by HIV gp120, would also result in the rapid induction of CD4+ T cell death. Purified resting CD4+ T cells were incubated with gp120 followed by cross-linking of anti-gp120 antibodies, and cell viability was analyzed at different time points thereafter. As shown in Fig. 1 B, significant cell death was observed 18 h after CD4 activation by gp120XL, as compared to that present in untreated, anti-gp120 antibody-treated or gp120-treated CD4+ T cell cultures, thus confirming the specificity of CD4 activation-mediated cell death. To further investigate whether the decrease in cell viability induced by CD4XL was mediated by apoptosis, flow cytometric techniques were used, which demonstrated a significant degree of apoptosis at 18 h after CD4XL but not IgGXL (Fig. 1 C). Altogether, these results indicated that CD4 activation was sufficient to trigger CD4+ T cell apoptosis in the absence of accessory cells or TCR engagement in a monocyte-depleted, ⩾98% pure primary CD4+ T cells.
Figure 1.



CD4 activation is sufficient to induce apoptosis of CD4+ T cells. (A) CD4XL or IgG1XL purified CD4+ T cells were washed, harvested, counted, and transferred to separate wells. Cell mortality was analyzed at 18 h after cross-linking as determined by trypan blue dye exclusion. (B) Pure CD4+ T cells were incubated with HIV gp120IIIB for 1 h and then transferred to anti-gp120–coated plates and processed and analyzed as in A. (C) CD4+ T cells were CD4 cross-linked or IgG cross-linked as in A, and the percentage of apoptosis was analyzed by flow cytometry in Hoeschst and propidium iodide–stained cells as described in Materials and Methods. The data in A and B represents the mean and the corresponding standard deviations of triplicate points within each experiment.
CD4XL Mediates Apoptosis through Fas–FasL Interactions and Induces Susceptibility to TNF-mediated Apoptosis.
To identify the mechanisms whereby CD4 activation directly leads to T cell apoptosis, we investigated the role of two separate apoptosis-inducing ligands and their corresponding receptors that had been previously shown to participate in T cell apoptosis (FasL and Fas and TNF and TNFR; 2–4). Purified CD4+ T cells were either CD4 cross-linked or IgG1-treated and were immediately cultured in the presence of either a Fas fusion protein (FasFc), a TNFRI fusion protein (TNFRFc), or a control fusion protein (CD30Fc). As shown in Fig. 2 A, FasFc but not TNFRFc or CD30Fc fusion proteins prevented CD4XL-induced T cell apoptosis, supporting the role of FasL–Fas interactions in CD4-mediated T cell death. This was further confirmed in additional experiments in which anti-Fas blocking (M3) or nonblocking antibodies (M33) were added immediately after CD4XL or IgG1XL. As shown in Fig. 2 B, M3 but not M33 interfered with the CD4XL-induced death of pure CD4+ T cells.
Figure 2.


CD4XL mediates apoptosis through FasL–Fas interaction. (A) Pure CD4+ T cells were CD4 cross-linked or IgG1 cross-linked in the presence of FasFc, TNFRFc, CD30Fc (each at 30 μg/ml). After 1 h of cross-linking, cells were processed as in Fig. 1 A and cell death was analyzed 18 h later. (B) IgG1XL or CD4XL of CD4+ T cells was performed either in the presence or absence of F(ab′)2 fragments of blocking (M3) or nonblocking (M33) anti-Fas antibodies used at 30 μg/ml for 18 h. Cell death was calculated as in Fig. 1 A. The data represents the mean and standard deviation of triplicate points within each experiment.
The above results suggest that CD4 activation of resting CD4+ T cells either induces a de novo state of susceptibility to FasL-mediated apoptosis (assuming a constitutive presence of FasL in their surface) and/or that it triggers de novo expression of FasL. To dissect these two mechanisms, we focused first on determining whether CD4 activation induces a state of susceptibility to FasL-dependent apoptosis. CD4+ T cells were either CD4 cross-linked or IgG1 cross-linked for 1 h followed by the addition of anti-Fas cross-linking antibodies. As shown in Fig. 3 A, addition of anti-Fas cross-linking antibodies further increased the level of CD4XL-induced apoptosis, which, as shown in Fig. 2, is mediated by Fas–FasL interactions, suggesting that CD4– cross-linked cells are susceptible to FasL-mediated apoptosis.
Figure 3.
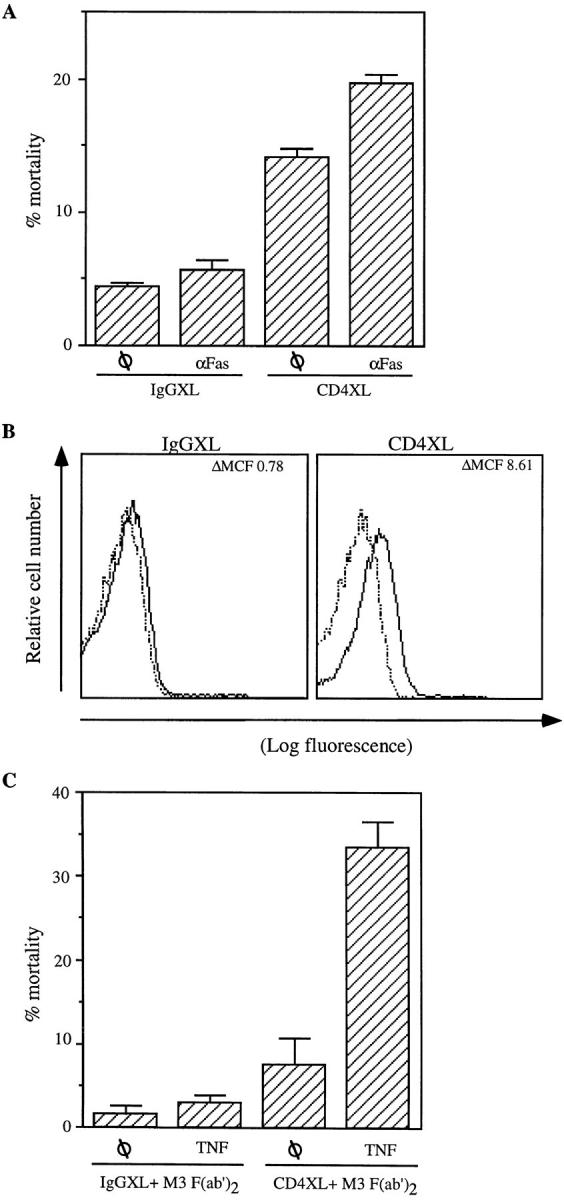
CD4XL induces susceptibility to FasL- and TNF-mediated apoptosis and FasL expression. (A) CD4+ T cells were CD4 cross-linked or IgG1 cross-linked and incubated with anti-Fas cross-linking antibody (CH-11; 250 ng/ml) for 18 h. (B) Membrane-associated FasL was measured 1 h after CD4XL or IgGXL, using isotype (dashed line) or anti-FasL (NOK-1)PE-conjugated antibodies (bold line). The mean channel fluorescence (ΔMCF) was calculated for each treatment point by subtracting the MCF for the isotype-stained sample from that of the FasL-stained sample. (C) CD4XL or IgG1XL was performed as in Fig. 1 A in the presence of M3 F(ab′)2 for 1 h, after which TNF (100 ng/ml) was added for 18 h and cell death was analyzed as in Fig. 1 A. The data represents the mean and standard deviation of triplicate points within each experiment.
We next investigated whether FasL expression is constitutive within the purified CD4+ T cell population or induced after CD4XL. CD4+ T cells were CD4 cross-linked or IgG1 cross-linked, and stained with PE-conjugated anti-FasL antibodies at different time points. FasL expression was analyzed 1, 2, and 6 h after CD4 activation. As shown in Fig. 3 B, FasL is detected in the surface of CD4+ T cells as early as 1 h after CD4XL. FasL expression was found to be rapidly induced, and in a transient fashion, since it could not be detected by 6 h after CD4 activation. From these results it is inferred that CD4 activation has a dual function that ultimately results in CD4+ T cell death within a homogeneous population. CD4 activation confers susceptibility to FasL-mediated apoptosis and induces de novo expression of FasL within the same CD4+ T cell culture.
In view of the fact that Fas and TNFRI share common intracytoplasmic proteins of the signal transduction pathways that lead to cell death by apoptosis (19, 20), we next tested whether CD4XL induced susceptibility not only to FasL-mediated apoptosis but also to TNF. To dissect the effect of FasL–Fas interactions from the possible induction of susceptibility to TNF-mediated apoptosis, CD4+ T cells that had undergone CD4XL or IgG1XL were incubated with anti-Fas blocking antibodies (M3) in the presence of TNF. As shown in Fig. 3 C, a significant degree of TNF-induced apoptosis was observed in CD4–cross-linked but not in IgG1–cross-linked T cells once FasL–Fas interactions are blocked by M3. Altogether, these results indicate that CD4 activation triggers the susceptibility to both TNF and FasL-mediated apoptosis, but that only FasL is produced after CD4 activation of a homogenous CD4+ T cell population.
Induction of Susceptibility to FasL- and TNF-mediated Apoptosis Is Protein Synthesis–independent.
To identify potential mechanisms that could explain the induction of susceptibility to apoptosis triggered by CD4 activation, we tested whether the induction of a susceptibility state to FasL- and TNF-mediated apoptosis after CD4 activation requires new protein synthesis. For this, cells were either IgG1 cross-linked or CD4 cross-linked in the presence of cycloheximide, followed by treatment with either anti-Fas cross-linking antibodies or TNF. The presence of cycloheximide caused a significant reduction in the CD4XL-induced cell death of purified CD4+ T cells (Fig. 4, compare lanes 7 and 10), suggesting that either the induction of susceptibility and/or the production of FasL is protein synthesis–dependent. To dissect these two possible mechanisms, cycloheximide-treated CD4+ T cells were CD4 cross-linked and incubated in the presence of either anti-FasXL antibodies or TNF. Results from these experiments demonstrate a significant degree of cell death mediated by either of these two ligands (Fig. 4, lanes 11 and 12). These observations indicate that the induction of a state of susceptibility to FasL- or TNF-dependent apoptosis is protein synthesis–independent, whereas the production of FasL after CD4 activation is protein synthesis–dependent. Moreover, it strengthens the observation that direct mechanisms rather than indirect ones such as those mediated by the production of cytokines participate in the induction of susceptibility to apoptosis.
Figure 4.

Induction of susceptibility to FasL- and TNF-mediated apoptosis is protein synthesis independent. CD4+ T cells were treated with CHX (10 μg/ml) for 30 min before CD4XL or IgG1XL. After 1 h of cross-linking, CD4+ T cells were transferred to separate wells and incubated either with anti-Fas crosslinking antibodies (CH-11, at a concentration of 250 ng/ml) or human recombinant TNF (100 ng/ml) for 18 h. The data represents the mean and standard deviation of triplicate points within each experiment.
PKC Mediates Susceptibility to FasL-dependent Cell Death after CD4 Activation.
To further investigate the mechanisms mediating susceptibility to FasL and TNF-dependent apoptosis after CD4 activation, we focused on protein synthesis–independent processes such as post-translational modifications of second messengers of the CD4-initiated signal transduction pathway. Although this pathway is poorly understood in primary human T cells, in transformed T cells lines, tyrosine kinases such as p56lck (21, 22), increased intracellular [Ca2+] and inositol 3,4,5-phosphate (23), p21 ras (24), and the translocation of the conventional isoforms of PKC from the cytosol to membrane (25–28) have been identified as key members of the CD4-initiated signal transduction pathway. We first analyzed if PKC is involved in mediating susceptibility to FasL-mediated cell death after CD4 activation by using GF109203X (GF). CD4+ T cells were pretreated with GF, followed by CD4XL or IgGXL, and were incubated in the presence of anti-Fas cross-linking antibodies. GF completely blocked the cell death observed after CD4XL (Fig. 5 A). Moreover, addition of anti-Fas cross-linking antibodies in the presence of GF did not induce death of CD4–cross-linked cells, suggesting that conventional PKC isoforms are involved in controlling susceptibility to FasL-mediated apoptosis in primary, CD4 activated CD4+ T cells. This result indicates that induction of susceptibility to FasL-mediated apoptosis requires PKC-dependent pathways.
Figure 5.


Inhibition of PKC reverts CD4XL-induced susceptibility to FasL- but not TNF-mediated apoptosis. (A) Pure CD4+ T cells were pretreated with 3 μM of GF for 1 h. Cells were then CD4 cross-linked or IgG1 cross-linked for 1 h and transferred to separate wells. Thereafter, anti-Fas cross-linking antibody (CH-11, at 250 ng/ml) was added for an additional 18 h. (B) CD4+ T cells were pretreated with GF as in A. Cells were CD4 cross-linked or IgG cross-linked in the presence of F(ab′)2 fragments of anti-Fas M3 antibodies. After 1 h of cross-linking, TNF (100 ng/ml) was added and incubated for an additional 18 h. (C) CD4+ T cells were pretreated or not with 3 μM GF for 1 h before PMA stimulation. Cells were then stimulated with PMA (250 ng/ml) for 1 h in the presence or absence of F(ab′)2 fragments of anti-Fas antibodies M3 or M33. Cells were washed, transferred to separate wells, and incubated for 18 h with anti-Fas cross-linking antibodies CH-11 (250 ng/ml) or TNF (100 ng/ml).
We next tested whether induction of susceptibility to TNF-mediated apoptosis triggered by CD4 activation (as shown in Fig. 3 C) requires a PKC-dependent signaling pathway. For this, CD4+ T cells were pretreated with GF and CD4XL in the presence of M3 (to prevent cell death induced by Fas–FasL interactions). TNF induced a 30% rate of CD4+ T cell death that was not reversed by pretreatment with GF (Fig. 5 B), suggesting that susceptibility to FasL- and TNF-mediated apoptosis following CD4XL is regulated by PKC-dependent and PKC-independent pathways, respectively.
To further confirm the effect of PKC in mediating susceptibility to Fas-induced cell death, we tested whether direct activation of PKC can mimic the effect of CD4 activation by using a PKC activator such as PMA. CD4+ T cells were treated with PMA for 1 h in the presence or absence of GF, and then incubated in the presence of anti-Fas cross-linking antibodies or TNF. As shown in Fig. 5 C, PMA alone induced a moderate degree of cell death, which was reversed by M3 (compare lanes 4 and 5), suggesting that PMA-induced CD4+ T cell death is mediated by FasL–Fas interactions. The reversal of the moderate degree of PMA-induced CD4+ T cell death was also blocked by GF (Fig. 5 C, lane 9), indicating that PMA induces a state of susceptibility to FasL dependent apoptosis via PKC-dependent pathways. This was further confirmed by the addition of anti-Fas cross-linking antibodies to PMA-treated cells, which further enhanced CD4+ T cell apoptosis in a GF-sensitive fashion (Fig. 5 C, lanes 7 and 10). Interestingly, TNF-mediated apoptosis was not observed in PMA-treated CD4+ T cells (lane 8), further strengthening the proof that susceptibility to TNF-mediated apoptosis of CD4-activated T cells is PKC-independent.
These results suggest that susceptibility to FasL and TNF-mediated cell death in primary CD4 T cells is regulated by at least two different pathways; PKC-dependent in the Fas signaling pathways, and PKC-independent in the TNFR signaling pathway. Both of these paths appear to be separately targeted after CD4 activation.
CD4 and CD3 Activation Shows Different Kinetics for Induction of Susceptibility to Fas-mediated Apoptosis.
T cell activation via the T cell receptor results in the activation of various PKC isoforms (28–30). Since direct activation of PKC through PMA is sufficient to induce susceptibility to Fas-mediated cell death, we asked whether CD3 activation results in susceptibility to FasL and/or TNF-mediated cell death. To compare cell death induced by CD3XL and CD4XL, cells were stimulated with anti-CD4, anti-CD3 or IgG1 for 1 h and cell viability in the absence or in the presence of an 18-h incubation with anti-Fas antibodies was analyzed at 24 and 72 h. CD4XL of resting pure CD4+ T cells induces significant cell death by 24 h, whereas CD3XL did not lead directly to cell death at this early time point (Fig. 6, lane 7) despite the increase in the expression of the activation marker CD69 (data not shown). Moreover, CD3XL CD4+ T cells were not found to be susceptible to anti-Fas antibodies added immediately after CD3XL (Fig. 6, lane 9). However, addition of anti-Fas antibodies 48 h after CD3XL resulted in a 30% cell mortality of the CD4+ T cells (Fig. 6, lane 10) In contrast to CD4 activation, CD3 activation of CD4+ T cells did not lead to susceptibility to TNF-mediated apoptosis at either the 24 or 72 h time points (Fig. 6, lanes 11 and 12). These results suggest the use of different signal transduction pathways for the induction of susceptibility to FasL and TNF-mediated apoptosis after CD4 and CD3 activation.
Figure 6.

CD4XL and CD3XL show different kinetics for induction of susceptibility to Fas-mediated apoptosis. CD4+ T cells were CD4 cross-linked, CD3 cross-linked, or IgG1 cross-linked. After 1 h of XL, CD4+ T cells were transferred to separate wells and incubated either with anti-FasXL antibodies (CH-11, at a concentration of 250 ng/ml) or human recombinant TNF (100 ng/ml) for 24 h or incubated for 72 h followed by the addition of anti-Fas or TNF as above for an additional 18 h. The data represents the mean and standard deviation of triplicate points within each experiment.
Discussion
This report demonstrates that CD4 activation of resting T cells leads directly to an aberrant state of susceptibility to apoptosis mediated by either FasL or TNF. Moreover, the simultaneous induction of FasL after CD4 activation explains the in vitro death by apoptosis of purified CD4+ T cells. The fact that protein synthesis is not required to trigger the state of susceptibility to apoptosis that ensues after CD4 activation argues for direct posttranslational modifications of second messengers of the Fas- or TNFR-dependent pathways. This argument is further supported by the fact that inhibition of PKC or tyrosine kinases reverse the CD4-mediated induction of susceptibility to FasL- but not to TNF-induced apoptosis. In addition, this study highlights potential differences in the control of lymphocyte survival by CD3- and CD4-initiated signal transduction pathways. Altogether, these observations require that the function of CD4 should be revisited and expanded to address its role as a key receptor in CD4+ T cell homeostasis as it relates to the immune response and HIV-induced immunopathogenesis.
Our observation that CD4 activation leads to a state of susceptibility to FasL-dependent apoptosis extends previous observations. CD4 activation had been shown to prime the CD4+ T cell to apoptosis if a second signal such as TCR engagement or the presence of accessory cells was provided (7, 8, 11). It is now known that such “second” signals induce the expression of FasL, as is the case for TCR activation (31–33) or constitutively express FasL, as in the case of antigen-presenting cells (14, 34). In addition, our study supports previous observations using lpr+/+ mice for which it was demonstrated that CD4 activation in vivo did not result in CD4+ T cell depletion and apoptosis as was observed in lpr−/− mice (9, 10). What was more surprising was the observation that CD4 activation could lead to the susceptibility of apoptosis triggered by TNF. The function of TNF as a key ligand in regulating peripheral T lymphocyte homeostasis is becoming more apparent (4, 6), and therefore our results may explain how CD4+ T cells may become susceptible to TNF. This may be relevant not only to the physiological control of lymphocyte survival after antigen presentation, but also to HIV pathogenesis (see below). Future studies should address whether CD4 activation leads also to a susceptibility state to additional apoptosis-inducing receptors/ligands, some of which may share common signal transduction pathways to those downstream of Fas or TNFRI.
Interfering with Fas–FasL interactions resulted in a nearly complete reversal of CD4+ T cell apoptosis after CD4 activation in in vitro cultures of purified CD4+ T cells. This implies that although CD4 triggers a significant degree of susceptibility to several apoptosis-inducing ligands, it appears to only induce the production of FasL, thus explaining the observed degree of cell death by apoptosis. Previous studies have demonstrated that CD4 activation of PBL or monocytes induces the transcription (35) and, recently, the expression of FasL (6, 36). The fact that cycloheximide blocked the CD4 mediated apoptosis by Fas–FasL interactions without interfering with the state of susceptibility supports the de novo production of FasL, which presumably occurs through transcriptional mechanisms triggered by a CD4-initiated signal transduction pathway. Therefore, the combination of the induction of susceptibility to FasL or TNF and the production of FasL by CD4 activation within primary CD4+ T cells supports the key role that CD4 may play in mediating death of this important immune cell.
When CD4 and CD3 activation were separately compared in this study, it was observed that CD3 activation alone did not directly induce cell death at earlier (24 h) or later (72 h) time points, despite the observation of CD3-mediated cell proliferation and expression of CD69. This, together with the fact that CD3 activation induced susceptibility to FasL- but not TNF-dependent apoptosis at 72 h, argues toward different signaling pathways initiated by these two lymphocyte receptors in controlling T cell survival. The role of CD4 in the context of antigen presentation of resting naive peripheral CD4+ T cells warrants further study. Although our study did not focus on the interaction between CD4- and TCR/CD3-initiated signal transduction pathways to understand how CD4 may modify lymphocyte survival in this context, a previous study showed that TCR engagement blocks the CD4+ T cell apoptosis observed in in vivo murine models after the administration of anti-CD4 antibody (37). In fact, coactivation of CD3 and CD4 resulted in the reversal of CD4-triggered cell death (Algeciras, A., and C.V. Paya, unpublished observations). It will be of interest to explore whether coactivation of TCR/CD3 and CD4 modifies FasL expression or the CD4-initiated state of susceptibility to FasL- and TNF-mediated apoptosis. Likewise, it is theoretically possible that other coactivation receptors/ligands such as CD28/B7 may also interfere with the CD4 initiated susceptibility to apoptosis by increasing the level of antiapoptotic molecules such as shown for the case of bclxL after CD28 activation (38).
In addition to the potential key role of CD4 in controlling the immune response that occurs after antigen presentation to CD4+ T cells, results from our studies have direct implications on the pathogenesis of chronic disease states, in which single activation of CD4 may take place, and which are curiously characterized by CD4+ T cell depletion. Examples include SLE and HIV infection. In SLE a significant increase in anti-CD4 antibodies has been noted, which may correlate with the lymphopenia observed in these patients (39). In the context of HIV infection, not only is there an increase in circulating levels of anti-CD4 antibodies (40–42), but more importantly, in soluble gp120 (43), anti-gp120 antibodies (44, 45), and a large number of defective HIV virions (46, 47), all of which can lead to a chronic and significant degree of CD4 activation of uninfected CD4+ T cells. As recently shown by our group, CD4+ T cells and not CD8+ T cells from HIV infected patients are in a state of susceptibility to both FasL and TNF-dependent apoptosis (6). This work further supports the potential role of the in vivo activation of CD4+ T cells by the above-mentioned HIV-related factors. In fact, ongoing studies demonstrate that rapid reductions of viral load using highly active antiretroviral therapy directly correlates with increases in peripheral CD4+ T cells counts (46, 47), suggesting that HIV-related factors may be directly involved in conferring susceptibility to apoptosis in CD4+ T cells. The steady present enhanced state of susceptibility to apoptosis mediated by FasL or TNF in these patients, together with increased levels of these two ligands (14, 47–49) may partially explain the significant degree of CD4+ T cell depletion observed in the context of HIV infection.
Results that derived from studying the mechanisms whereby CD4 activation leads to a state of susceptibility to FasL- or TNF-mediated apoptosis suggest that direct mechanisms are involved. Previous studies indicated that the production of cytokines such as TNF and IFN-γ after CD4 activation of PBMCs were sufficient to explain the susceptibility to accessory cell–mediated apoptosis (7, 8, 11); which, as recently shown by our group, constitutively express FasL (14). However, our data using purified, and therefore CD4+-monocyte depleted, CD4+ T cells indicate that protein synthesis is not required for the induction of susceptibility. These results support our previous observation that suggest that increases in TNFR and Fas membrane expression are not necessary for induction of susceptibility to FasL- and TNF-mediated apoptosis. Based on this, it is possible that CD4 directly induces a state of susceptibility of the activated CD4+ T cells, and in addition, the subsequent production of cytokines by the activated CD4+ T cell or CD4+ monocytes may result in a second wave of susceptibility to apoptosis that could affect not only these activated CD4+ T cells, but also other neighboring cells. The possibility that CD4 activation of downstream second messengers may directly influence molecules that are involved in conferring susceptibility or resistance to Fas or TNFRI-initiated apoptosis is of importance. It provides for the first time a valuable model with which to study potential modifications of members of the Fas or TNFRI signal pathways that may ensue after CD4 activation. Demonstrating that PKC is required for the induction of susceptibility to FasL-mediated apoptosis of primary CD4+ T cells is a first important step, as it implies that PKC-dependent phosphorylation is a necessary event in rendering a resistant cell into one susceptible to FasL-mediated, but interestingly not to TNFR-mediated, apoptosis. Recent studies in transformed cells support our observation, as it has been shown that failure to activate PKC is associated with resistance to FasL-mediated apoptosis (50). Because Fas and TNFRI have common intracellular molecules, such as FADD or caspase-8 (FLICE; 51–55), the selectively of PKC-dependent phosphorylation in the case of Fas-initiated signal transduction implies that either Fas itself or downstream molecules may be targeted by PKC.
In summary, our studies highlight the need to revisit the role of CD4 as an important receptor that controls T cell homeostasis. Identification of the molecular mechanism controlling this process will be of significant value in understanding the regulation of the immune response and, importantly, in determining its role in CD4+ T cell depletion in HIV-infected individuals.
Footnotes
We thank Dr. Hideo Yagita for the NOK1-PE antibody, Dr. Robert Abraham for the anti-CD3 antibody, and Teresa Hoff for outstanding secretarial assistance. We also acknowledge Dr. Paul Leibson and the rest of Carlos Paya's laboratory for helpful discussions.
Address correspondence to Carlos V. Paya, Mayo Clinic, 200 First St. SW, Guggenheim 501, Rochester, MN 55905. Phone: 507-284-3747; Fax: 507-284-3757; E-mail: paya@mayo.edu
Abbreviations used in this paper: FasL, Fas ligand; GF, PKC inhibitor GF109203X; gp, glycoprotein; PKC, protein kinase C; XL, cross-linking.
References
- 1.Boehme SA, Zheng L, Leonardo MJ. Analysis of CD4 coreceptor and activation-induced costimulatory molecules in antigen-mediated mature T lymphocyte death. J Immunol. 1995;155:1703–1712. [PubMed] [Google Scholar]
- 2.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 3.Krammer PH, Behrmann I, Daniel P, Dhein J, Debatin K-M. Regulation of apoptosis in the immune system. Curr Opin Immunol. 1994;6:279–289. doi: 10.1016/0952-7915(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 4.Sytwu H-K, Liblau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5:17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 5.Katsikis P D, Wunderlich ES, Smith CA, Herzenberg LA, Herzenberg LA. Fas antigen stimulation induces marked apoptosis of T lymphocytes in human immunodeficiency virus-infected individuals. J Exp Med. 1995;181:2029–2036. doi: 10.1084/jem.181.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badley AD, Dockrell D, Simpson M, Schut R, Lynch DH, Leibson P, Paya CV. Macrophage-dependent apoptosis of CD4+T lymphocytes from HIV-infected individuals is mediated by FasL and tumor necrosis factor. J Exp Med. 1997;185:55–64. doi: 10.1084/jem.185.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banda NK, Bernier J, Kurahara DK, Kurrle R, Haigwood N, Sekaly R-P, Finkel TH. Cross-linking CD4 by human immunodeficiency virus gp120 primers T cells for activation-induced apoptosis. J Exp Med. 1992;176:1099–1106. doi: 10.1084/jem.176.4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oyaizu N, McCloskey TW, Coronesi M, Chirmule N, Kalyanaraman VS, Pahwa S. Accelerated apoptosis in peripheral blood mononuclear cells (PBMCs) from human immunodeficiency virus type-1 infected patients and in CD4 cross-linked PBMCs from normal individuals. Blood. 1993;82:3393–3400. [PubMed] [Google Scholar]
- 9.Wang Z-Q, Dudhane A, Orlikowsky T, Clarke K, Li X, Darzynkiewicz Z, Hoffmann MK. CD4 engagement induces Fas antigen-dependent apoptosis of T cells in vivo. . Eur J Immunol. 1994;24:1549–1552. doi: 10.1002/eji.1830240714. [DOI] [PubMed] [Google Scholar]
- 10.Desbarats J, Freed JH, Campbell PA, Newell MK. Bas(CD95) expression and death-mediating function are induced by CD4 cross-linking on CD4+T cells. Proc Natl Acad Sci USA. 1996;93:11014–11018. doi: 10.1073/pnas.93.20.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oyaizu N, McCloskey TW, Than S, Hu R, Kalyanaraman VS, Pahwa S. Cross-linking of CD4 molecules upregulates Fas antigen expression in lymphocytes by inducing interferon-γ and tumor necrosis factor–α secretion. Blood. 1994;84:2622–2631. [PubMed] [Google Scholar]
- 12.Alderson MR, Tough TW, Braddy S, Davis-Smith T, Roux E, Schooley K, Miller RE, Lynch DH. Regulation of apoptosis and T cell activation by Fas-specific mAb. Int Immunol. 1994;6:1799–1806. doi: 10.1093/intimm/6.11.1799. [DOI] [PubMed] [Google Scholar]
- 13.Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, Goodwin RG, Smith CA, Ramsdell F, Lynch DH. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181:71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badley AD, McElhinny JA, Leibson PJ, Lynch DH, Alderson MR, Paya CV. Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J Virol. 1996;70:199–206. doi: 10.1128/jvi.70.1.199-206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kayagaki N, Kawasaki A, Ebata T, Ohmoto H, Ikeda S, Inoue S, Yoshino K, Okumura K, Yagita H. Metalloproteinase-mediated release of human Fas ligand. J Exp Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatch WC, Tanaka KE, Calcelli T, Rashbaum WK, Kress Y, Lyman WD. Persistent productive HIV-1 infection of a CD4-human fetal thymocyte line. J Immunol. 1992;148:3055–3061. [PubMed] [Google Scholar]
- 17.Page KA, Stearns SM, Littman DR. Analysis of mutations in the V3 domain of gp160 that affect fusion and infectivity. J Virol. 1992;66:524–533. doi: 10.1128/jvi.66.1.524-533.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ormerod MG, Sun X-M, Brown D, Snowden RT, Cohen GM. Quantification of apoptosis and necrosis by flow cytometry. Acta Oncol. 1993;32:417–424. doi: 10.3109/02841869309093620. [DOI] [PubMed] [Google Scholar]
- 19.Peter ME, Kischkel FC, Hellbardt S, Chinnaiyan AM, Krammer PH, Dixit VM. CD95 (APO-1/Fas)- associating signaling proteins. Cell Death Diff. 1996;3:161–170. [PubMed] [Google Scholar]
- 20.Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene. 1996;12:1–9. [PubMed] [Google Scholar]
- 21.Baldari CT, Di Somma MM, Milia E, Bergman M, Telford JL. Interactions between the tyrosine kinases p561ck, p59fyn and p50csk in CD4 signaling in T cells. Eur J Immunol. 1995;25:919–925. doi: 10.1002/eji.1830250409. [DOI] [PubMed] [Google Scholar]
- 22.Di Somma MM, Nuti S, Telford JL, Baldari CT. p561ck plays a key role in transducing apoptotic signals in T cells. FEBS Lett. 1995;363:101–104. doi: 10.1016/0014-5793(95)00292-h. [DOI] [PubMed] [Google Scholar]
- 23.Kornfeld H, Cruikshank WW, Pyle SW, Berman JS, Center DM. Lymphocyte activation by HIV-1 envelope glycoprotein. Nature. 1988;335:445–448. doi: 10.1038/335445a0. [DOI] [PubMed] [Google Scholar]
- 24.Baldari CT, Milia E, Di Somma MM, Baldoni F, Valitutti S, Telford JL. Distinct signaling properties identify functionally different CD4 epitopes. Eur J Immunol. 1995;25:1843–1850. doi: 10.1002/eji.1830250708. [DOI] [PubMed] [Google Scholar]
- 25.Prada NA, Cruikshank WW, Danis HL, Ryan TC, Center DM. IL-16 and other ligand-induced migration is dependent upon protein kinase C. Cell Immunol. 1996;168:100–106. doi: 10.1006/cimm.1996.0054. [DOI] [PubMed] [Google Scholar]
- 26.Ushijima H, Ando S, Kunisada T, Schröder HC, Klöcking H-P, Kijjoa A, Muller WEG. HIV-1 gp120 and NMDA induce protein kinase C translocation differentially in rat primary neuronal cultures. J Acquired Immune Defic Syndr. 1993;6:339–343. [PubMed] [Google Scholar]
- 27.Gupta S, Aggarwal S, Kim C, Gollapudi S. Human immunodeficiency virus-1 recombinant gp120 induces changes in protein kinase C isozymes—a preliminary report. Int J Immunopharmacol. 1994;16:197–204. doi: 10.1016/0192-0561(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 28.Gibellini D, Zauli G, Re MC, Lolli S, Borgatti P, Capitani S, La M, Placa CD4 engagement by HIV-1 in TF-1 hematopoietic progenitor cells increases protein kinase C activity and reduces intracellular Ca+2levels. Microbiologica (Pavia) 1994;17:85–92. [PubMed] [Google Scholar]
- 29.Berry N, Nishizuka Y. Protein kinase C and T cell activation. Euro J Biochem. 1990;189:205–214. doi: 10.1111/j.1432-1033.1990.tb15478.x. [DOI] [PubMed] [Google Scholar]
- 30.Genot EM, Parker PJ, Cantrell DA. Analysis of the role of protein kinase C-α, -ε and -ζ in T cell activation. J Biol Chem. 1995;270:9833–9839. doi: 10.1074/jbc.270.17.9833. [DOI] [PubMed] [Google Scholar]
- 31.Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, Goodwin RG, Smith CA, Ramsdell F, Lynch DH. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181:71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju S-T, Panka DJ, Cui H, Ettinger R, El-Khatib M, Sherr DH, Stanger BZ, Marshak-Rothstein A. Fas (CD95)/FasL Interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–448. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- 33.Yang Y, Merćep M, Ware CF, Ashwell JD. Fas and activation-induced Fas ligand mediate apoptosis of T cell hybridomas: inhibition of Fas ligand expression by retinoic acid and glucocorticoids. J Exp Med. 1995;181:1673–1682. doi: 10.1084/jem.181.5.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiener PA, Davis PM, Starling GC, Mehlin C, Klebanoff SJ, Ledbetter JA, Liles WC. Differential induction of apoptosis by Fas–Fas ligand interactions in human monocytes and macrophages. J Exp Med. 1997;185:1511–1516. doi: 10.1084/jem.185.8.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin K-M, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 36.Oyaizu N, Adachi Y, Hashimoto F, McCloskey TW, Hosaka N, Kayagaki N, Yagita H, Pahwa S. Monocytes express Fas ligand upon CD4 cross-linking and induce CD4+T cell apoptosis. J Immunol. 1997;158:2456–2463. [PubMed] [Google Scholar]
- 37.Wang Z, Dudhane A, Orlikowsky T, Hoffmann MK. T cell antigen receptor engagement abrogates CD4-mediated T cell deletion in vivo. . Int Immunol. 1995;7:207–211. doi: 10.1093/intimm/7.2.207. [DOI] [PubMed] [Google Scholar]
- 38.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xL . Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 39.Lenert G, Lenert P. Human CD4-reactive antibodies from SLE patients induce reversible inhibition of polyclonal T lymphocyte proliferation. Hum Immunol. 1996;49:113–121. doi: 10.1016/0198-8859(96)00054-7. [DOI] [PubMed] [Google Scholar]
- 40.Keay S, Wecksler W, Wassermann SS, Margolick J, Farzadegan H. Association between anti-CD4 antibodies and a decline in CD4+lymphocytes in human immunodefiency virus type 1 seroconverters. J Infect Dis. 1995;171:312–319. doi: 10.1093/infdis/171.2.312. [DOI] [PubMed] [Google Scholar]
- 41.Muller C, Kukel S, Bauer R. Antibodies against CD4+lymphocytes in human immunodeficiency virus type 1 seroconverters. Immunol Lett. 1994;41:163–167. doi: 10.1016/0165-2478(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 42.Burastero SE, Gaffi D, Lopalco L, Tambussi G, Borgonovo B, De Santis C, Abecasis C, Robbioni P, Gasparri A, Lazzarin A, et al. Autoantibodies to CD4 in HIV type 1–exposed seronegative individuals. AIDS Res Hum Retroviruses. 1996;12:273–280. doi: 10.1089/aid.1996.12.273. [DOI] [PubMed] [Google Scholar]
- 43.Oh SK, Cruikshank WW, Raina J, Blanchard GC, Adler WH, Walker J, Kornfeld H. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J Acquired Immune Defic Syndr. 1992;5:351–356. [PubMed] [Google Scholar]
- 44.Ellaurie M, Calvelli TA, Rubinstein A. Human immunodeficiency virus (HIV) circulating immune complexes in infected children. AIDS Res Hum Retroviruses. 1990;6:1437–1441. doi: 10.1089/aid.1990.6.1437. [DOI] [PubMed] [Google Scholar]
- 45.Feijoó E, Subirá D, Gerández-Guerrero M, Garcia R, Oritz F. Anti-CD4 activity in circulating immune complexes in HIV-infected patients. Int Arch Allergy Immunol. 1995;106:366–371. doi: 10.1159/000236868. [DOI] [PubMed] [Google Scholar]
- 46.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 47.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 48.Bilello JA, Stellrecht K, Drusano GL, Stein DS. Soluble tumor necrosis factor–α receptor type II (STNF α RII) correlates with human immunodeficiency virus (HIV) RNA copy number in HIV-infected patients. J Infect Dis. 1996;173:464–467. doi: 10.1093/infdis/173.2.464. [DOI] [PubMed] [Google Scholar]
- 49.Macchia D, Almerigogna F, Parronchi P, Ravina A, Maggie E, Romagnani S. Membrane tumour necrosis factor–α is involved in the polyclonal B cell activation induced by HIV-infected human T cells. Nature. 1993;363:464–466. doi: 10.1038/363464a0. [DOI] [PubMed] [Google Scholar]
- 50.Sawai H, Okazaki T, Takeda Y, Tashima M, Swada H, Okuma M, Kishi S, Umehara H, Domae N. Ceramide-induced translocation of protein kinase C-delta and -epsilon to the cytosol: implifications in apoptosis. J Biol Chem. 1997;272:2452–2458. doi: 10.1074/jbc.272.4.2452. [DOI] [PubMed] [Google Scholar]
- 51.Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Dixit VM. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem. 1996;9:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 52.Baker SJ, Reddy EP. Transducers of life and death: TNF receptor superfamily and associated proteins. Oncogene. 1996;12:1–9. [PubMed] [Google Scholar]
- 53.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/ FADD-interacting protease, in Fas/APO-1- and TNF receptor–induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 54.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homolgous ICE/ CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 55.Fraser A, Evan G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]