

Abstract
We studied the impact of various infectious and proinflammatory agents on the induction of peripheral T cell tolerance. Adoptive transfer of CD8+ T cells from lymphocytic choriomeningitis virus (LCMV) T cell receptor transgenic mice into LCMV antigen transgenic mice expressing the LCMV glycoprotein epitope (gp) 33–41 under control of a major histocompatibility complex class I promoter led to efficient induction of peripheral tolerance after a period of transient activation. If, however, the recipient mice were challenged with viral or bacterial infections or proinflammatory agents (lipopolysaccharide or Poly:IC) early after cell transfer, tolerance induction was prevented and instead, CD8+ T cell activation leading to vigorous expansion and generation of cytolytic activity ensued. This became manifest in significant immunopathology mainly involving destruction of the splenic architecture and lysis of antigen-expressing lymphocyte and macrophage populations. Important parameters involved in the activation of host-reactive T cells by nonspecific infectious agents included the presence, localization, and quantity of the specific transgene-encoded self-antigen; in contrast, CD4+ T cells were not required. In mice surviving the acute phase, the transferred CD8+ T cells persisted at high levels in an anergic state; they were unable to generate cytolytic activity in vitro or to control LCMV infection in vivo. These results impinge on our understanding of the role of infectious agents in graft verus host reactions towards minor histocompatibility antigens.
Tcell tolerance to self-antigens expressed in the thymus primarily occurs via clonal deletion of immature thymocytes. In addition, various mechanisms of peripheral T cell tolerance have been described, which maintain unresponsiveness of clones that are potentially reactive to peripheral self-antigens, which may be underrepresented in the thymus. (a) One mechanism, ignorance, is where the antigen is not encountered by the T cell during its normal recirculation pathway in sufficient quantity to cause immune reactivity (1, 2). This state of tolerance can be broken if antigen is introduced into lymphoid tissue, i.e., into a location where T cells find appropriate qualitative and quantitative conditions to be activated (3). (b) Another is anergy, where contact with antigen induces functional unresponsiveness (4, 5). T cell anergy can be reversed if continuous contact with the tolerogen is lacking (6), and partial reversion of anergy may also be induced by infectious agents (7). (c) There is also peripheral clonal deletion where contact with antigen induces cell death (8–10). This may be the consequence of a powerful, exhaustive immune response (8, 10), but may also only be preceded by a brief period of activation, which is of limited functional relevance (11). Obviously, this form of tolerance is irreversible; it has been described, however, that proinflammatory agents such as LPS may interfere with the induction of tolerance and instead lead to T cell activation under certain conditions (12–15).
Peripheral deletion of T cells induced by MHC-presented peptide antigens has been widely studied using T cells from TCR transgenic mice adoptively transferred into mice expressing their nominal antigen. Thus, it was shown that naive H-Y–specific T cells transfused into nude male hosts largely disappeared after a period of extensive proliferation, leaving behind a small proportion of anergic cells (9). Similar observations have been made using mice expressing a transgenic TCR specific for Kb (16) or Ld (17). Evidence has also been presented that more restricted tissue-specific antigen expression may be sufficient to cause peripheral T cell deletion (18).
Surprisingly, even though large numbers of antigen-specific cytotoxic T cells were transferred and activated in the experimental systems described above, peripheral deletion usually occurred without immunopathological consequences. This is in contrast to the situation encountered during bone marrow transplantation, when transfusion of large numbers of lymphocytes into hosts ubiquitously expressing major or minor histocompatibility antigens that may be recognized by donor cells frequently leads to activation and severe immunopathology resulting in the clinical picture of graft versus host (GVH)1 disease (19). The factors important for activation versus tolerance of host-specific T cells after bone marrow transplantation have not been characterized in detail. An interesting clinical and experimental observation is, however, that infections may exacerbate and aggravate GVH disease (20).
In this study we describe a model system where naive CD8+ T cells specific for a lymphocytic choriomeningitis virus (LCMV) peptide were transferred into mice expressing this nominal LCMV epitope as a transgene under control of an ubiquitous promoter; this situation is comparable to a defined minor histocompatibility antigen difference. Similar to previous studies, we found that the specific T cells were deleted after a period of transient activation. If, however, an inflammatory process was created by viral or bacterial infections or by the injection of LPS or Poly:IC, the CD8+ T cells were activated by the transgene-encoded self-antigen, vigorously expanded, and caused significant immunopathology similar to a GVH reaction.
Materials and Methods
Gene Constructions and Transgenic Mice.
To generate mice with ubiquitous expression of an LCMV-derived CTL epitope, we constructed a hybrid LCMV/H-2Kb transgene. In this construct, the 5′ part of exon 1 of the genomic Kb gene encoding amino acids (aa) 1–12 was replaced by a cDNA fragment encoding aa 1–60 of the LCMV glycoprotein (Fig. 1 a). For this, the EcoRI fragment of the genomic Kb gene (provided by Dr. B. Arnold, Deutsches Krebsforschungszentrum, Heidelberg, Germany) was cloned into the EcoRI site of pUC 19 (pRIKb10). In this plasmid, the NruI/XmaIII fragment (encoding aa 1–12 of exon 1 of the Kb molecule) was replaced by a PCR product encoding aa 1–60 of the LCMV glycoprotein gene. The PCR product was generated from the pKOOL plasmid (1) encoding the BamHI fragment of the LCMV glycoprotein (GP) cDNA. For the PCR reaction 5′ (TCGCGAGGATTCTATCCAGTAAAAGGATGG) and 3′ (CTGTGGCATGTACGGCCGTAATG) primers were used which contained Nru1 and XmaIII restriction sites at their 5′ and 3′ ends, respectively. The LCMV GP fragment was inserted such that the following Kb aa sequence was put out of frame leading to a premature stop in exon 2 of the Kb gene. The final transgenic construct was controlled by nucleotide sequencing of the replaced region. The transgene was microinjected into fertilized C57BL/6 oocytes and founder number 8 (official nomenclature: TgN (LCMVGP33) 224 Zbz; designated H8 in this manuscript) was used in this study.
Figure 1.

Functional presentation of gp 33–41 in H8 mice. (a) The transgene construct: a fragment of the LCMV glycoprotein sequence including the gp 33–41 CTL epitope was cloned into the genomic sequence of H-2Kb. (b and c) Functional expression of gp 33–41 in peripheral lymphoid tissue. (b) Spleen cells from day 8 immune C57BL/6 mice were used as effector cells against Con A blast target cells obtained from H8 or C57BL/6 mice; as a control, C57BL/6 Con A blast target cells pulsed with gp 33–41 were used. Spontaneous release was <28%. (c) H8-, C57BL/6-, and gp 33–41-pulsed C57BL/6 spleen cells were used as stimulators for TCR318 responder spleen cells in a [3H]thymidin incorporation assay. (d) Functional gp 33 expression on fibroblasts. Embryonal fibroblasts from H8 and C57BL/6 mice were left unmanipulated or were pulsed with gp 33–41 and used as targets for effector cells obtained from LCMV-WE–infected C57BL/6 mice in a 5-h 51Cr–release assay; spontaneous release was <30%.
C57BL/6 mice were obtained from the Institut für Labortierkunde (University of Zürich, Zürich, Switzerland). The transgenic mice expressing a Vα2/Vβ8.2 T cell receptor specific for aa 33–41 of the LCMV glycoprotein 1 in association with H-2 Db (TCR327 mice) on 50% of all CD8+ T cells have been described previously (21). Perforin-deficient and recombination activating gene (RAG)1-deficient TCR318 mice (TCR318-Pfptm and TCR318-RAG1tm mice, respectively) were bred by crossing mice of line 318 to perforin-deficient (22) or RAG-1–deficient (23) animals. For preparation of bone marrow chimeras, recipient mice were exposed to 1,000 centiGrey (cGy) and 24 h later injected intravenously with 107 untreated bone marrow cells. Mice were depleted of CD4+ T cells as described (24). All mice were kept under specific pathogen-free conditions.
Infectious and Proinflammatory Agents.
LCMV strain WE (LCMV-WE) was originally obtained from F. Lehmann-Grube (Heinrich-Pette-Institut fuer Experimentelle Virologie, University of Hamburg, Hamburg, Germany) and was propagated on L 929 fibroblast cells. The LCMV-WE CTL escape variant 8.7 was generated by injection of TCR318 mice with the parental WE strain (25). Virus titers in infected organs were determined using a virus plaque assay on MC57G cells as described previously (26). Vaccinia virus WR and the recombinant vaccinia viruses expressing the LCMV nucleoprotein (NP) (obtained from Dr. H.L. Bishop, Institute of Virology, Oxford University, Oxford, UK) and aa 1–57 of the LCMV GP (obtained from Dr. Linsey Whitton, Scripps Research Institute, La Jolla, CA) were grown on BSC 40 cells and plaqued on the same cells. Listeria monocytogenes (originally obtained from R.V. Blanden, Australian National University, Canberra, Australia) was kept and prepared as described previously (27). All pathogens as well as Poly:IC (Fluka, Chemie Ab, Buchs, Switzerland) and LPS Escherichia coli 026:B6 (Chemie Brunschwig AG, Basel, Switzerland) were injected intravenously.
FACS® Analysis.
For detection of TCR318 T cells in recipient mice, spleen cells or PBLs were incubated with FITC-labeled anti-CD8, PE-labeled anti-Vα2, and biotinylated anti-TCR Vβ8 (all from PharMingen, San Diego, CA) mAb followed by Tricolor-streptavidin (CALTAG Labs., South San Francisco, CA; reference 28). For the detection of activation markers, biotinylated anti-CD62 ligand (L), CD44, and CD49d (all from PharMingen) mAb were used instead of anti-TCR Vβ8. To analyze the splenic lymphocyte subsets, spleen cells were stained with FITC anti-B220, PE anti-CD4 and biotinylated anti-CD8 (all from PharMingen), followed by Tricolor-streptavidin. Cells were analyzed on a FACScan® flow cytometer (Becton Dickinson, Mountain View, CA).
Functional CTL Assays.
For determination of ex vivo cytolytic activity, effector spleen cell suspensions were prepared. EL-4 target cells were coated with the LCMV peptides glycoprotein epitope (gp) 33–41, np 396–404 (10−6 molar for 2 h), or left untreated. 51Cr–release assays were performed according to standard protocols (29) at the indicated effector/target ratios using 5- or 15-h incubation periods. For some experiments, effector cells were restimulated for 5 d with thioglycolate-induced LCMV-WE–infected macrophages before the 51Cr–release assay. Con A blast target cells were obtained by incubating spleen cells of naive mice at 2 × 106 cells/ml in IMDM 10% FCS containing 5 μg/ml Con A. After 3 d of stimulation, the cells were harvested, purified by a Ficoll gradient, and labeled with 51Cr. Embryonal fibroblast target cells were obtained by culturing single-cell suspensions from day 14–19 embryos, which were trypsinized after decapitation and removal of the liver. They were seeded into 96-well plates at a concentration of 2 × 104 cells per well on the day before the assay. Then, 10 μl of 51Cr (10 μCi) were added for 3 h, the cells were washed three times in the plates, and effector cells were added at standard effector dilutions. For in vitro T cell proliferation assays, 5 × 105 responder spleen cells were restimulated in vitro on 2 × 105 irradiated C57BL/6 spleen cells pulsed with LCMV gp 33. Responder cell proliferation was assessed after 48 h of proliferation of culture by addition of [3H]thymidine (25 μCi/ well) for the last 12 h of incubation. T cell proliferation in vivo was assessed in the lymph nodes of recipient mice 40 h after transfer of TCR318 spleen cells labeled with the fluorescent dye 5,6 carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR). For fluorescence labeling, spleen cells were incubated at a concentration of 5 × 106 cells/ml in buffered saline solution containing 0.5 μmolar CFSE for 10 min at 37°C.
Histology and Immunohistology.
Histological procedures were performed as described previously (30) using the following primary antibodies: anti-CD8 (YTS 169; reference 24), anti-CD4 (YTS 191; reference 24), anti-B220 (PharMingen), anti–marginal metallophils (MMs; metallophilic macrophage 1; Biomedicals, Augst, Switzerland), and anti–follicular dendritic cells (FDCs; 4C11; reference 31). Primary rat mAb were revealed by a twofold sequential incubation with rabbit anti–rat Ig and rat alkaline phosphatase anti–alkaline phosphatase complex (DAKO, Glostrup, Denmark) followed by naphthol AS-B1 phosphate and New Fuchsin as substrate for the color reaction. Peripheral blood and bone marrow cytology were determined as described previously (32).
Results
Functional Expression of LCMV gp 33–41 in H8 Mice.
Functional expression of LCMV gp 33–41 in H8 mice was verified in the following experiments. (a) Con A–stimulated T cell blasts from H8 mice were effectively lysed by spleen cells from C57BL/6 mice that had been infected with 200 pfu of LCMV-WE 8 d previously (Fig. 1 b). (b) Spleen cells of H8 mice caused proliferation of LCMV-specific TCR transgenic responder cells to a similar extent as spleen cells from C57BL/6 mice exogenously loaded with the synthetic gp 33 peptide (Fig. 1 c). (c) Embryonal fibroblasts of H8 mice were specifically lysed by LCMV-specific effector cells, albeit to a lesser extent than gp 33–labeled fibroblasts obtained from C57BL/6 mice (Fig. 1 d). (d) Analysis of H8 mice crossed to mice transgenic for a gp 33–specific TCR revealed extensive thymic deletion of gp 33–specific CD8+ T cells (data not shown), leading to complete and specific tolerance of gp 33-reactive CTLs. Collectively, these data suggest that the gp 33 epitope was functionally presented by both lymphoid and nonlymphoid cells in H8 mice.
Peripheral Tolerance Induction After Transient Activation of Transferred gp 33–specific CD8+ T Cells in H8 Mice.
To study the fate of gp 33–reactive CD8+ T cells after exposure to an environment, where their nominal antigen is widely expressed both on lymphoid and nonlymphoid cells, we transfused nonirradiated H8 and littermate control mice with 5 × 107 spleen cells from RAG1-deficient TCR transgenic (TCR318-RAGtm) mice, which had been labeled with the fluorescent dye CFSE. 40 h later, CD8+ T cells of lymph nodes of recipient mice were analyzed by flow cytometry. Fig. 2 a shows that in lymph nodes of littermate control mice, one discrete peak of CFSEhi cells could be identified. In contrast, analysis of lymph node cells of H8 mice revealed several peaks of lower intensity (Fig. 2 b), suggesting that the antigen-specific donor T cells had undergone several rounds of cell division. (Fig. 2 c). Downregulation of CD62L and elevated expression of CD44 (Fig. 2 d) on CD8+ T cells further supported the observation that the TCR318-RAGtm CFSElo population had been activated after transfer into H8 mice.
Figure 2.

Peripheral tolerance induction after transient activation of transferred gp 33–specific CD8+ T cells in H8 mice. (a–d) H8 and littermate contol mice were transfused with 5 × 107 CFSE-labeled spleen cells from RAG1-deficient mice transgenic for a gp 33–41-specific T cell receptor (TCR×RAG). 40 h after transfer, mesenteric lymph node cells were analyzed by flow cytometry. CFSE staining of CD8+ cells from littermate (B6; a) or H8 recipient mice (b) is shown. CFSE+ CD8+ lymph node cells from both recipients were compared for their expression of CD62L (c) and CD44 (d). (e) Spleen cells of H8 mice transfused with 5 × 107 spleen cells from TCR318 mice were analyzed at the indicated times after transfer for gp 33–specific cytotoxicity on EL-4 target cells in a 15-h 51Cr–release assay. Spleen cells from C57BL/6 mice immunized with LCMV-WE 50 d earlier (B6 memo) were used as control. Spontaneous release was <25%. ( f ) H8 mice and littermate control (B6) mice were transfused with the indicated number of spleen cells from TCR318 mice. The percentage of CD8+ PBLs expressing the transgenic Vα and Vβ chains was monitored at the indicated time points before and after challenge infection with 200 PFU LCMV.
Although these phenotypic markers provided evidence for activation of transgenic CD8+ T cells, further analysis revealed that this was not of major functional consequence. Spleen cells of recipient mice did not show gp 33–specific cytotoxicity in a sensitive 15-h 51Cr–release assay 1, 3, 6, or 8 d after transfer of 5 × 107 TCR318 spleen cells (Fig. 2 e). Also, no signs of immunopathology could be demonstrated by clinical observation, histology (not shown), or lysis of gp 33–expressing recipient lymphocytes (Table 1, experiment A, see below). Finally, cell division appeared limited as TCR318 CD8+ T cells remained undetectable in FACS® analysis of recipient spleen cells using TCR Vα2/Vβ8–specific antibodies (Fig. 2 f, days 1–12).
Table 1.
Splenic Immunopathology Induced by Infectious and Proinflammatory Agents
| Experiment | Recipient | Total spleen cell count | Percent CD4+ | Percent CD8+ | Percent B220+ | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | TCR→ H8 | 6 × 107 | 18 | 9 | 53 | |||||
| TCR→ littermate | 5 × 107 | 17 | 10 | 56 | ||||||
| B | TCR→ H8 LCMV | 3 × 106 | 4.2 | 21 | 0.6 | |||||
| TCR→ littermate LCMV | 9 × 107 | 18 | 21 | 55 | ||||||
| C | TCR→ H8 LCMV 8.7 | 3 × 107 | 2.1 | 12.5 | 0.75 | |||||
| TCR→ H8 vacc | 8 × 107 | 9 | 22 | 15 | ||||||
| TCR→ H8 VSV | 9 × 106 | 12 | 21 | 1.2 | ||||||
| TCR→ H8 Listeria | 1 × 107 | 8.6 | 14.5 | 2.1 | ||||||
| TCR→ H8 LPS | 2 × 107 | 14.4 | 25 | 10.7 | ||||||
| TCR→ H8 Poly:IC | 5 × 107 | 0.73 | 17 | 2.4 | ||||||
| D | TCR × PKO→ H8 LCMV | 2 × 107 | 16 | 45 | 1.53 | |||||
| TCR × PKO→ B6 LCMV | 9 × 107 | 12 | 13 | 56 |
H8 and littermate control mice were transfused with 5 × 107 (A), 106 (B), or 107 (C) spleen cells from TCR318 mice or with 106 spleen cells from TCR318 mice deficient in perforin (TCR318-Pfptm; D). 1–3 d after transfer, they were injected with 500 μg Poly:IC, 10 μg LPS, 2 × 106 PFU VSV-Indiana, 2 × 106 PFU vaccinia WR, 200 PFU LCMV-WE, or WE 8.7. 20–35 d later, spleen cells of the recipient mice were counted and the relative percentage of lymphocyte subpopulations was determined by flow cytometric analysis. Data are the mean of two to six mice per group and were obtained in several separate experiments.
To evaluate the further fate of the transferred TCR318 CD8+ T cells, the recipient mice were infected with 200 PFU LCMV-WE 12 d after transfer. Fig. 2 f shows that infection of control mice transfused with as little as 106 spleen cells (containing ∼5 × 104 TCR318 CD8+ T cells) lead to vigorous expansion of the transgenic T cells, whereas in H8 mice, even after transfer of 5 × 107 spleen cells, no TCR318 T cells could be restimulated by the virus infection. Also, attempts to recover gp 33–reactive CD8+ T cells in vitro by antigen-specific restimulation in the presence of Con A supernatant were unsuccessful (data not shown). Thus, it appears likely that, as previously described in similar experimental systems, transfer of TCR318 T cells into mice widely expressing their nominal antigen leads to clonal deletion and/or induction of anergy after a period of transient activation. Experiments using bone marrow chimeras revealed that this peripheral tolerance induction was equally efficient whether the peptide was expressed only on bone marrow–derived cells or predominantly in nonlymphoid tissues (not shown).
Virus Infection Interferes with Tolerance Induction and Causes CD8+ T Cell–mediated Immunopathology.
The experiments described above confirmed the finding that high numbers of potentially host-reactive cells can be efficiently rendered tolerant in the periphery without causing damage to host tissue. To address the question of whether infectious agents may influence this tolerization process, we infected H8 and littermate control mice with 200 PFU LCMV-WE 1–3 d after transfer of 106 TCR318 spleen cells. Although infection of control mice did not lead to detectable clinical illness, H8 mice started showing signs of wasting 6–8 d after infection and between 20–40% (up to 100% if the experiments were done under non–specific pathogen-free conditions) died within 12–15 d after infection. The remaining mice continued to lose weight and all of them died 3–5 mo after infection.
Histological examination of the recipient mice 35 d after infection revealed profound alterations in splenic architecture and cellular composition in H8 mice compared to littermate controls. Macroscopically, the spleen was small and had a fibrotic appearance, the follicular structure was largely dissolved and almost all B cells, CD4 cells, FDCs, and marginal zone macrophages were absent (Fig. 3; Table 1, experiment B). Total spleen cell counts were reduced 20–50-fold and CD8+ T cells were the only cell population demonstrable at significant numbers; ∼90% of them carried the transgenic, gp 33–specific TCR. Kinetic studies showed that this splenic immunopathology was already prominent 12 d after infection and persisted for at least 60 d, which was the latest time point analyzed (Fig. 4). Interestingly, after infection of H8 mice transfused with perforin-deficient TCR318-Pfptm cells, the elimination of B cells was as pronounced (Table 1, experiment D) and the histological alterations similar (not shown), but the outcome was not lethal.
Figure 3.
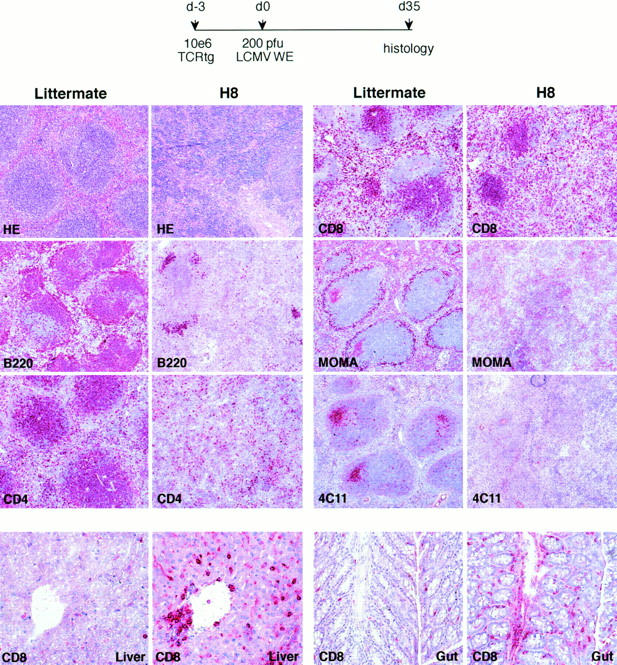
Virus-induced host-specific CD8+ T cells mediate GVH-like immunopathology. H8 and littermate control mice were transfused with 106 spleen cells from TCR318 mice and infected with 200 PFU LCMV 1 d later. At different time points after infection, tissue sections of spleen (d35), gut, and liver (d50, separate experiment) were stained with hematoxilin–eosin (HE) or with Abs of the desired specificity. B220, CD45R+ B cells; CD4, CD4+ T cells (YTS-191); CD8, CD8+ T cells (YTS-169); MOMA, marginal metallophilic macrophages; 4C11, FDCs.
Figure 4.

Kinetics of splenic immunopathology mediated by virus-induced host-specific CD8+ T cells. H8 and littermate control (B6) mice were transfused with 106 spleen cells from TCR318 mice and infected with 200 PFU LCMV 1 d later. At the indicated time points after transfer, mice were killed and the absolute number of B cells per spleen was determined by flow cytometry.
CD8+ T cell infiltrations were also observed in other target organs of GVH reactions such as liver, gut, and skin (Fig. 3). Tissue destruction was limited, however, and neither lead to prolonged elevation of liver enzymes nor to extensive eczemematous lesions of the skin. Finally, bone marrow and peripheral blood counts were significantly altered in infected H8 mice compared to negative littermate controls; bone marrow cellularity was reduced by a factor of 2–5. Peripheral white cell counts were reduced by a factor of 5, and red cell counts were similar, but reticulocyte counts were reduced by a factor of 10 and thrombocyte counts were reduced twofold. There were no obvious signs of bleeding, however, and it is unclear whether these signs of pathology alone can explain the acute deaths.
The clinical course described above showed that LCMV infection not only interfered with the induction of CD8+ T cell tolerance by host tissue, but instead caused severe immunopathology. Numeric and functional analysis of the host-specific TCR transgenic CD8+ T cells revealed that this was paralleled by their extensive proliferation (Fig. 5 a) and the generation of donor-derived (gp 33)–specific cytotoxicity (Fig. 5 b) 8 d after infection. The TCR318 T cells then returned to memory levels in transgene negative control mice (28), but remained at high levels (up to 90% of all CD8+ T cells) in H8 mice for the observation period of 60 d (Fig. 5 b).
Figure 5.

Virus infection interferes with peripheral tolerance induction and activates host-specific CD8+ T cells. H8 and littermate control (B6) mice were transfused with 106 spleen cells from TCR318 mice. 3 d later, the recipient mice were infected with 200 PFU LCMV-WE and the percentage of CD8+ PBLs expressing the transgenic Vα and Vβ chains was monitored by flow cytometric analysis (a). 8 d after challenge infection, spleen cells from some recipient mice were tested for gp 33–specific cytotoxicity on EL-4 target cells in a 5-h 51Cr–release assay; spontaneous release was <18% (b). Data from one of four similar experiments are shown.
Anergy of Persisting Host-specific CD8+ T Cells in H8 Mice.
In the following experiments, the persisting gp 33– specific CD8+ T cells were characterized phenotypically and functionally. Flow cytometric analysis of spleen cells of H8 mice 30 d after virus infection revealed an activated phenotype of the transferred TCR318 CD8+ T cells when compared to littermate controls. The transgenic TCR-α and -β chains were downregulated about four to fivefold (Fig. 6, a and b); the cells had increased forward scatter (Fig. 6 c) and expressed higher levels of CD49d (Fig. 6 d), which has been characterized as a useful marker to discriminate between acutely activated versus memory cells (28). Note that host CD8+ T cells carrying endogenous TCR chains had mostly been eliminated (Fig. 6, a and b). In strong contrast to these phenotypic signs of activation, functional analysis revealed little in vivo or in vitro activity of these cells. Restimulation in vitro with or without addition of Con A supernatant did not lead to generation of gp 33– specific cytotoxicity (Fig. 6 e). Similarly, the proliferative response to gp 33–pulsed spleen stimulator cells was strongly reduced and could not be recovered by the addition of Con A supernatant (Fig. 6 f). Finally, antiviral activity in vivo was ineffective since LCMV persisted in several organs of H8 mice 35 d after infection, whereas virus was readily controlled in littermate controls (Fig. 6 g). This was not due to the generation of CTL escape mutants as LCMV isolated from H8 mice 35 d after infection was well recognized by gp 33–stimulated transgenic effector cells in vitro as assessed by a 51Cr–release assay (data not shown).
Figure 6.

Anergy of persisting virus-induced host-specific CD8+ T cells. (a–d) Phenotypical analysis of TCR318 T cells: H8 and littermate control (B6) mice were transfused with 106 spleen cells from TCR318 mice and infected with 200 PFU LCMV-WE 3 d later. 35 d after infection, CD8+ spleen cells of recipient mice were analyzed for expression of the transgenic Vα and Vβ chains (a and b). Forward light scatter of Vα2+Vβ8+CD8+ TCR318 T cells (c) and CD49d expression of Vα2+ CD8+ T cells (d) were compared between H8 (bold lines) and littermate mice (dashed lines). (e–g) Functional analysis of TCR318 T cells. 35 d after LCMV infection, spleen cells of H8 and littermate control (B6) mice were (e) tested for gp 33–specific cytotoxicity after 5 d of restimulation on LCMV-infected macrophages in the presence of 5% Con A supernatant and ( f ) used as responder cells in a [3H]thymidine incorporation assay stimulated by C57BL/6 spleen cells pulsed with gp 33–41. In experiment g, the LCMV virus titer was analyzed in the indicated organs of the recipient mice. Data from one of two to four similar experiments are shown.
Role of Bone Marrow–derived Cells in the Induction of CTL-mediated Immunopathology.
To address the question of which type of APC is involved in the observed activation of host-specific T cells in the context of an infection, we studied activation of CD8+ T cells and immunopathology in bone marrow chimeric mice. Infection of H8–B6 chimeras 3 d after transfer of 106 TCR318 cells showed that antigen exclusively expressed on bone marrow–derived cells is sufficient for the induction of T cell proliferation (Fig. 7 a), the generation of host-specific cytolytic activity (Fig. 7 b), and subsequent lethal immunopathology. It was also sufficient for the maintenance of a high number of anergic T cells in the chronic phase. Most of these features, including acute lethal immunopathology, were also observed in the B6–H8 chimeras (Fig. 7, a and b) where the specific antigen is largely expressed on non–bone marrow– derived cells. An exception was the maintenance of anergic T cells in the peripheral circulation, and to a lesser extent, in the spleen.
Figure 7.

Antigen presentation by bone marrow–derived versus epithelial cells for activation of host-specific CTLs. 6 wk after bone marrow reconstitution, H8–B6 and B6–H8 bone marrow chimeras were transfused with 106 spleen cells from TCR318 mice. 3 d later, the recipient mice were infected with 200 PFU LCMV-WE and the percentage of CD8+ PBLs expressing the transgenic Vα and Vβ chains was monitored by flow cytometric analysis (a). 8 d after challenge infection, spleen cells from some recipient mice were tested for gp 33–specific cytotoxicity on EL-4 target cells in a 5-h 51Cr–release assay; spontaneous release was <18% (b). † Deaths.
A Variety of Infectious and Inflammatory Stimuli Can Interfere with Tolerance Induction.
Next, we investigated whether antigen-unrelated third-party infections are also able to abolish tolerance induction and lead to activation and immunopathology in this model. H8 and littermate control mice transfused with 107 TCR318 spleen cells (corresponding to 106 TCR318 CD8+ T cells) were challenged with a variety of different, completely unrelated pathogens. Infections with vesicular stomatitis virus (VSV), vaccinia virus (to a minor extent), and the intracellular bacterium Listeria monocytogenes all elicited a similar phenotype. This included expansion of TCR318 CD8+ T cells (Fig. 8 a), the generation of gp 33–specific ex vivo cytotoxic activity (Fig. 8 c), and the destruction of host lymphocytes (Fig. 8 d; Table 1). Interestingly, injection with 500 μg Poly:IC or 10 μg LPS also induced activation (Fig. 8 b) and immunopathology (albeit not acutely lethal) in H8 mice (Table 1, Fig. 8 d). Both infectious as well as inflammatory agents mediated their effect without involvement of T helper cells as depletion of CD4+ T cells before infection did not change the observed phenotype (Fig. 8 e).
Figure 8.

Various infectious and inflammatory stimuli can interfere with tolerance induction and activate host-specific CD8+ T cells. H8 (closed symbols) and littermate control mice (open symbols) were transfused with 107 spleen cells from TCR318 mice and 1 d later injected with 500 μg Poly:IC, 10 μg LPS, 2 × 106 PFU VSV-Indiana, 2 × 106 PFU vaccinia WR, 200 PFU LCMV-WE 8.7, or 200 PFU LCMV-WE. The percentage of CD8+ PBLs expressing the transgenic Vα and Vβ chains was monitored by flow cytometry (a, b, e, and f ). 8 d after challenge infection, spleen cells from some recipient mice were tested for gp 33–specific cytotoxicity on EL-4 target cells in a 5-h 51Cr–release assay (c). 20 d after infection, mice were killed and the absolute number of B cells per spleen was determined by flow cytometry (d ). In experiment e, mice were depleted of CD4+ T cells 3 and 1 d before cell transfer. In experiment f, the number of transferred TCR318 spleen cells was varied as indicated and mice were infected either with LCMV-WE (closed symbols) or with LCMV-WE 8.7 (open symbols).
Initial experiments had indicated that tenfold more (i.e., 107) TCR318 spleen cells had to be transferred to elicit the described phenotype with antigen-unrelated third-party pathogens, when compared to infection with the specific LCMV. To evaluate, whether this was due to particular properties of LCMV infection or due to the fact that LCMV-WE introduces the specific gp 33 epitope in a different context, we infected H8 mice transfused with different amounts of TCR318 spleen cells with LCMV-WE or with the LCMV-WE 8.7 escape mutant, which is not recognized by the transgenic TCR. Fig. 8 f shows that 100-fold more TCR318 CD8+ T cells were required for the mutant virus to induce measurable proliferation, indicating that introduction of the nominal antigen in the context of the LCMV infection strongly enhanced the response to the transgene encoded self-antigen.
Discussion
This study shows that antigen-unrelated viral and bacterial infections as well as proinflammatory agents such as LPS and Poly:IC can influence the induction of peripheral CD8+ T cell tolerance to a widely expressed self-antigen in vivo. Importantly, these stimuli not only prevented establishment of tolerance, but instead enhanced activation of cytolytic CD8+ effector T cells that subsequently caused significant immunopathology.
Efficient induction of peripheral T cell tolerance after a transient period of proliferation has been described previously in several systems using adoptive transfer of TCR transgenic T cells into mice widely expressing their nominal antigen (9, 16–18, 33). Tolerance was found to be a consequence of extensive proliferation and subsequent clonal deletion, leaving a small T cell population in an anergic state; T cell anergy was reversible after transfer into an antigen-free environment (6, 34). In the present experiments we found only limited activation and proliferation of antigen-specific T cells, for which antigen expression on bone marrow–derived cells was sufficient. This is similar to a recent study where antigen expression in the host was more restricted due to the use of a tissue-specific promoter (18). Differences in the extent of T cell proliferation observed in the present compared to previous studies may be explained by differences in the TCR transgenic mice and the use of nude (9) or scid (17) recipient mice to increase sensitivity for the study of transferred T cells. The finding that 12 d after transfer, TCR318 T cells could not be restimulated by infection with LCMV, even after transfer into antigen-free recipients (data not shown), suggests that clonal deletion was mainly responsible for the tolerance induction observed. A contribution of anergy can, however, not be formally excluded. Whatever the mechanism, our data confirm that peripheral tolerization of potentially autoreactive T cells involves a period of transient activation that is usually of little functional or immunopathological consequence.
A role for infectious agents in breaking peripheral T cell tolerance has been documented in previous studies. Infection of mice expressing the LCMV glycoprotein in the β cells of the endocrine pancreas with LCMV leads to activation of LCMV-specific CTLs and to the induction of diabetes (1, 2). Also, infection with the nematode Nipostrongulus brasiliensis has been reported to partially reverse staphylococcal enterotoxin β–induced anergy of CD4+ T cells as assessed by proliferation and the restoration of IL-4 production (7, 35). While in these experiments established tolerance is reversed, other experiments showed that some stimuli may also abolish the induction of tolerance. It was originally reported by Claman more than 30 yr ago, that bacterial LPS can interfere with induction of B cell unresponsiveness to γ-globulin (12). Later studies showed that LPS also prevents the induction CD4+ T cell tolerance (36), a finding that was recently extended to the induction of tolerance to superantigens (15). Earlier reports had also documented that interference with tolerance induction can lead to activation (13, 14).
This study shows that a variety of pathogens and proinflammatory agents such as LPS or Poly:IC can interfere with the induction of peripheral CD8+ T cell tolerance to widely expressed antigens. The most important aspect of our findings is that this conversion from efficient tolerance induction to immunity towards tissue antigens can lead to significant CD8+ T cell–mediated immunopathology. Our results emphasize the importance of the antigen-specific component in this antihost immune reactivity. (a) In the absence of the specific antigen, infections with VSV, Listeria, or vaccinia virus or treatment with Poly:IC or LPS did not lead to measurable LCMV-specific T cell activation (transfer into C57BL/6 mice + antigen-unrelated infection), confirming that nonspecific “bystander activation” is of low efficacy and limited biological consequence (37). (b) In the absence of an infection, the transgene-encoded self-antigen efficiently tolerized the antigen-reactive CD8+ T cells (transfer into H8 mice, no infection). (c) If, however, mice were infected in the presence of the specific antigen (transfer into H8 mice + antigen-unrelated or antigen-specific infection), these infections were able to activate CTLs specific for the transgene-encoded self-antigen that caused immunopathology. The observed pathology was only partially mediated by perforin. Although activation of perforin-deficient gp 33–specific CD8+ T cells in H8 mice did not lead to lethality (which is presumably a consequence of target cell destruction outside lymphoid tissue), the CD8+ T cell–mediated destruction of lymphoid cells was almost as pronounced as in mice transfused with perforin-competent cells. Whether this reflects cytotoxicity mediated by CD95L (38, 39) or TNF-α (40, 41) remains to be tested.
Which parameters determine the conversion from tolerance to CTL-mediated immunopathology? Several factors were evaluated. (a) The importance of localization and quantity of the specific antigen (3) was well illustrated by the comparison between the effects of infection with LCMV-WE8.7 (carrying a mutated gp 33 epitope; reference 25) and the wild-type virus. Although these viruses do not differ in their capacity to replicate and to induce costimulatory molecules or cytokines, 50–100-fold more TCR318 T cells had to be transferred to observe the described phenotype after infection with the mutant compared to the wild-type virus. Since wild-type virus introduces the specific antigen in addition the transgene encoded peptide mainly into APCs, this illustrates the role of specificity and indicates the importance of antigen localization and quantity for the outcome of antigen contact of CTLs. (b) The role of the APC type required to present the host antigen was studied in bone marrow chimera experiments, which clearly show that in the presence of an infection, antigen presentation by bone marrow–derived cells is sufficient to elicit the described phenotype. The important question of whether antigen presentation by non–bone marrow–derived epithelial cells is sufficient to induce CTL activation could not be conclusively addressed in this experimental model. It is well known that lethal irradiation, as used in our study, spares some bone marrow–derived cells, in particular dendritic cells. The reduced number of chronically persisting antigen-specific CTLs in mice expressing the antigen mostly on non–bone marrow–derived cells may be explained by the fact that the initiating inflammatory reaction is controlled faster due to the lack of significant lymphoid pathology in these mice. (c) Previous studies have postulated that under certain conditions CD4+ T cells are important for changing CD8+ T cell tolerance into activation (11). It has also been suggested that Th1s may promote CD8+ T cell activation. In our model, CD4+ T cells were completely dispensable for the induction of CTL-mediated immunopathology. (d) The fact that the proinflammatory substances Poly:IC or LPS could elicit the same effect as viral or bacterial infections indicates that a replicating pathogen is not required for antigen-specific CTL activation under the conditions studied. The effects of Poly:IC and LPS are severalfold. A previous study showed that interference with superantigen-induced deletion was not mediated by the induction of the costimulatory molecule B7 on APCs (42); this remains, however, an untested possibility. All infectious and proinflammatory agents used have in common that they induce the secretion of various cytokines. In this context, two recent studies have shown that both Poly:IC and LPS can act indirectly on CD8+ T cell proliferation, presumably by stimulation of type I interferon production by APCs (43). Several other redundant cytokine pathways are conceivable including IL-1, IL-2, IL-12, TNF, and IFN-γ (15, 44). Whatever the precise molecular mechanism, it is important to note that for a CD8+, T cell encounter of antigen is not sufficient, even if it is presented on a “professional” APC. Under in vivo conditions, the inflammatory milieu is necessary to convert this encounter into an immunogenic, activating signal.
Some of the immunopathological consequences of infection observed in this study are reminiscent of alterations characteristic of GVH reactions (GVHRs; reference 45). An impact of infectious agents on the exacerbation of GVHRs was demonstrated more than 20 yr ago (20) and enhancement of donor antihost reactivity has been documented in humans and animal models after the infection with CMV (46, 47) and, interestingly, also after the administration of endotoxin (48) or Poly:IC (49). Although the experimental setup (involving the transfusion of TCR transgenic donor spleen cells) and the fact that there was comparatively mild pathology outside lymphoid tissue renders an analogy of our model to this clinical condition rather limited, some of our findings may illustrate principles relevant for activation of host-specific T cells during GVHRs; CD8+ T cell–mediated antihost reactivity can be induced by a variety of bacterial and viral infectious agents. This does probably not involve activation of specific T cells cross-reactive to tissue antigens (50, 51) or nonspecific bystander activation (37, 43). As demonstrated in this study, activation is crucially dependent on the presence of the specific antigen. Infections may then provide an environment that interferes with the induction of tolerance and instead promotes activation of cytolytic CD8+ T cells to a sufficient extent to cause extensive GVHRs.
Footnotes
We thank Karin Riem for excellent technical assistance.
This work was supported by the Swiss National Foundation grant 31-32179.91 (to H. Hengartner), grant 31-32195.91 (to R. Zinkernagel), and the Deutsche Forschungsgemeinschaft (to S. Ehl).
Address correspondence to Dr. Stephan Ehl, Institute of Experimental Immunology, Department of Pathology, University of Zürich, Schmelzbergstr. 12, CH-8091 Zürich, Switzerland. Phone: 411-255-2328; Fax: 411-255-4420; E-mail: stephehl@usz.unizh.ch
The present address of J. Hombach is Global Program for Vaccines and Immunization, World Health Organization, CH-1211 Geneva 27, Switzerland. The present address of H. Pircher is Institute for Medical Microbiology and Hygiene, Department of Immunology, University of Freiburg, D-79104 Freiburg, Germany.
Abbreviations used in this paper: aa, amino acids; CFSE, 5,6 carboxyfluorescein diacetate succinimidyl ester; FDC, follicular dendritic cell; GP, glycoprotein; GVH, graft versus host; GVHR, GVH reaction; H8 mice, LCMV gp33-transgenic mice; L, ligand; LCMV, lymphocytic choriomeningitis virus; NP, nucleoprotein; RAG, recombination activating gene; VSV, vesicular stomatitis virus.
J. Hombach and S. Ehl contributed equally to this work.
References
- 1.Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 2.Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 3.Zinkernagel R, Ehl S, Aichele P, Oehen S, Kundig T, Hengartner H. Antigen localization regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol Rev. 1997;156:199–209. doi: 10.1111/j.1600-065x.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- 4.Schonrich G, Kalinke U, Momburg F, Malissen M, Schmitt VA, Malissen B, Hammerling GJ, Arnold B. Down-regulation of T cell receptors on self-reactive T cells as a novel mechanism for extrathymic tolerance induction. Cell. 1991;65:293–304. doi: 10.1016/0092-8674(91)90163-s. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 6.Rocha B, Tanchot C, von Boehmer BH. Clonal anergy blocks in vivo growth of mature T cells and can be reversed in the absence of antigen. J Exp Med. 1993;177:1517–1521. doi: 10.1084/jem.177.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rocken M, Urban JF, Shevach EM. Infection breaks T-cell tolerance. Nature. 1992;359:79–82. doi: 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]
- 8.Webb S, Morris C, Sprent J. Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell. 1990;63:1249–1256. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- 9.Rocha B, von Boehmer BH. Peripheral selection of the T cell repertoire. Science. 1991;251:1225–1228. doi: 10.1126/science.1900951. [DOI] [PubMed] [Google Scholar]
- 10.Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 11.Guerder S, Matzinger P. A fail-safe mechanism for maintaining self-tolerance. J Exp Med. 1992;176:553–564. doi: 10.1084/jem.176.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claman HN. Tolerance to a protein antigen in adult mice and the effect of non-specific factors. J Immunol. 1963;91:833–841. [PubMed] [Google Scholar]
- 13.Louis JA, Chiller JM, Weigle WO. The ability of bacterial lipopolysaccharide to modulate the induction of unresponsiveness to a state of immunity. Cellular parameters. J Exp Med. 1973;138:1481–1495. doi: 10.1084/jem.138.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parks DE, Walker SM, Weigle WO. Bacterial lipopolysaccharide (endotoxin) interferes with the induction of tolerance and primes thymus-derived lymphocytes. J Immunol. 1981;126:938–942. [PubMed] [Google Scholar]
- 15.Vella AT, McCormack JE, Linsley PS, Kappler JW, Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2:261–270. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 16.Bertolino P, Heath WR, Hardy CL, Morahan G, Miller JF. Peripheral deletion of autoreactive CD8+ T cells in transgenic mice expressing H-2Kb in the liver. Eur J Immunol. 1995;25:1932–1942. doi: 10.1002/eji.1830250721. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L. The fate of adoptively transferred antigen-specific T cells in vivo. Eur J Immunol. 1996;26:2208–2214. doi: 10.1002/eji.1830260937. [DOI] [PubMed] [Google Scholar]
- 18.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I–restricted exogenous presentation of self antigens in vivo. J Exp Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Billingham RE. The biology of graft-versus-host reactions. Harvey Lect. 1966;62:21–78. [PubMed] [Google Scholar]
- 20.van Bekkum D, Roodenburg J, Heidt P, van der Waaij D. Mitigation of secondary disease of allogeneic mouse radiation chimeras by modification of the intestinal microflora. J Natl Cancer Inst. 1974;52:401–404. doi: 10.1093/jnci/52.2.401. [DOI] [PubMed] [Google Scholar]
- 21.Pircher H, Mak TW, Lang R, Ballhausen W, Ruedi E, Hengartner H, Zinkernagel RM, Burki K. T cell tolerance to Mlsa encoded antigens in T cell receptor V beta 8.1 chain transgenic mice. EMBO (Eur Mol Biol Organ) J. 1989;8:719–727. doi: 10.1002/j.1460-2075.1989.tb03431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 23.Mombaerts P, Jaconini J, Johnson R, Herrup K, Tonegawa S, Papaiounou V. RAG-1 deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 24.Cobbold SP, Jayasuriya A, Nash A, Prospero TD, Waldmann H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature. 1984;312:548–551. doi: 10.1038/312548a0. [DOI] [PubMed] [Google Scholar]
- 25.Pircher H, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell–resistant virus variants in vivo. Nature. 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 26.Battegay M, Cooper S, Althage A, Banziger J, Hengartner H, Zinkernagel RM. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24- or 96-well plates. J Virol Methods. 1991;33:191–198. doi: 10.1016/0166-0934(91)90018-u. [DOI] [PubMed] [Google Scholar]
- 27.Fehr T, Schoedon G, Odermatt B, Holtschke T, Schneemann M, Bachmann M, Mak T, Horak I, Zinkernagel RM. Crucial role of interferon consensus sequence binding protein, but neither of interferon regulatory factor 1 nor of nitric oxide synthesis for protection against murine listeriosis. J Exp Med. 1997;185:921–931. doi: 10.1084/jem.185.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmerman C, Brduscha RK, Blaser C, Zinkernagel RM, Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J Exp Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zinkernagel RM, Leist T, Hengartner H, Althage A. Susceptibility to lymphocytic choriomeningitis virus isolates correlates directly with early and high cytotoxic T cell activity, as well as with footpad swelling reaction, and all three are regulated by H-2D. J Exp Med. 1985;162:2125–2141. doi: 10.1084/jem.162.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aichele P, Brduscha-Riem K, Oehen S, Odermatt B, Zinkernagel R, Hengartner H, Pircher H. Peptide-antigen treatment of virus-immune mice: antigen-specific tolerance versus immunopathology. Immunity. 1997;6:519–528. doi: 10.1016/s1074-7613(00)80340-4. [DOI] [PubMed] [Google Scholar]
- 31.Gray D, Kosco M, Stockinger B. Novel pathways of antigen presentation for the maintenance of memory. Int Immunol. 1991;3:141–148. doi: 10.1093/intimm/3.2.141. [DOI] [PubMed] [Google Scholar]
- 32.Binder D, Fehr J, Hengartner H, Zinkernagel R. Virus-induced transient bone marrow aplasia: major role of interferon-α/β during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med. 1997;185:517–530. doi: 10.1084/jem.185.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocha B, Grandien A, Freitas AA. Anergy and exhaustion are independent mechanisms of peripheral T cell tolerance. J Exp Med. 1995;181:993–1003. doi: 10.1084/jem.181.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alferink J, Schittek B, Schonrich G, Hammerling GJ, Arnold B. Long life span of tolerant T cells and the role of antigen in maintenance of peripheral tolerance. Int Immunol. 1995;7:331–336. doi: 10.1093/intimm/7.2.331. [DOI] [PubMed] [Google Scholar]
- 35.Rocken M, Urban J, Shevach EM. Antigen-specific activation, tolerization, and reactivation of the interleukin 4 pathway in vivo. J Exp Med. 1994;179:1885–1893. doi: 10.1084/jem.179.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiller JM, Weigle WO. Termination of tolerance to human gamma globulin in mice by antigen and bacterial lipopolysaccharide (endotoxin) J Exp Med. 1973;137:740–750. doi: 10.1084/jem.137.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ehl S, Hombach J, Aichele P, Hengartner H, Zinkernagel RM. Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J Exp Med. 1997;185:1241–1251. doi: 10.1084/jem.185.7.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker M, Altman N, Podack E, Levy R. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J Exp Med. 1996;183:2645–2656. doi: 10.1084/jem.183.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braun M, Lowin B, French L, Acha-Orbea H, Tschopp J. Cytotoxic T cells deficient in both functional Fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J Exp Med. 1996;183:657–661. doi: 10.1084/jem.183.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piguet P, Grau B, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions in the acute phase of graft-versus-host disease. J Exp Med. 1987;166:1280–1289. doi: 10.1084/jem.166.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Speiser D, Bachmann M, Frick T, McKall-Faienza K, Griffiths E, Pfeffer K, Mak T, Ohashi P. TNF receptor p55 controls early acute graft-versus-host disease. J Immunol. 1997;158:5185–5190. [PubMed] [Google Scholar]
- 42.Vella AT, Mitchell T, Groth B, Linsley PS, Green JM, Thompson CB, Kappler JW, Marrack P. CD28 engagement and proinflammatory cytokines contribute to T cell expansion and long-term survival in vivo. J Immunol. 1997;158:4714–4720. [PubMed] [Google Scholar]
- 43.Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 44.Weigle WO, Scheuer WV, Hobbs MV, Morgan EL, Parks DE. Modulation of the induction and circumvention of immunological tolerance to human gamma-globulin by interleukin 1. J Immunol. 1987;138:2069–2074. [PubMed] [Google Scholar]
- 45.Rappaport H, Khalil A, Halle PO, Pritchard L, Dantchev D, Mathe G. Histopathologic sequence of events in adult mice undergoing lethal graft-versus-host reaction developed across H-2 and/or non–H-2 histocompatibility barriers. Am J Pathol. 1979;96:121–142. [PMC free article] [PubMed] [Google Scholar]
- 46.Grundy JE, Shanley JD, Shearer GM. Augmentation of graft-versus-host reaction by cytomegalovirus infection resulting in interstitial pneumonitis. Transplantation. 1985;39:548–553. doi: 10.1097/00007890-198505000-00018. [DOI] [PubMed] [Google Scholar]
- 47.Lonnqvist B, Ringden O, Wahren B, Gahrton G, Lundgren G. Cytomegalovirus infection associated with and preceding chronic graft-versus-host disease. Transplantation. 1984;38:465–468. doi: 10.1097/00007890-198411000-00004. [DOI] [PubMed] [Google Scholar]
- 48.Howard J. Increased sensitivity to bacterial endotoxin of F1 hybrid mice undergoing graft-versus-host reaction. Nature. 1961;190:1122. doi: 10.1038/1901122a0. [DOI] [PubMed] [Google Scholar]
- 49.Peres A, Amlani S, Kornbluth M, Seemayer TA, Lapp WS. The effects of polyinosinic:polycytidylic acid on the graft-versus-host reaction. III. Increased severity of the reaction with delayed pI:C treatment. Transplantation. 1989;48:80–84. [PubMed] [Google Scholar]
- 50.Yang H, Welsh RM. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol. 1986;136:1186–1193. [PubMed] [Google Scholar]
- 51.Yang HY, Dundon PL, Nahill SR, Welsh RM. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. 1989;142:1710–1718. [PubMed] [Google Scholar]