

Abstract
Experimental autoimmune encephalomyelitis (EAE) is a T cell–mediated autoimmune demyelinating disease of the central nervous system that serves as an animal model for multiple sclerosis. Antigen-specific tolerance regimens, including oral tolerance, have been used prophylactically to prevent development of acute EAE as well as a number of other autoimmune diseases. Two mechanisms have been proposed to explain the immunologic basis for disease inhibition: bystander immune suppression and clonal anergy/deletion. This report demonstrates a novel mechanism for monocyte chemotactic protein (MCP)-1 as a regulatory factor of oral tolerance. Oral administration of proteolipid protein peptide (PLP139–151) increased MCP-1 expression in the intestinal mucosa, Peyer's patch, and mesenteric lymph nodes. Increase in MCP-1 expression resulted in downregulation of mucosal interleukin (IL)-12 expression with concomitant increase in mucosal IL-4 expression. Functionally, MCP-1 upregulation was shown to regulate oral tolerance induction by the ability of antibodies to MCP-1 to inhibit tolerance induction. The anti–MCP-1 abrogation of oral tolerance induction also resulted in restoration of mucosal IL-12 expression as well as peripheral antigen-specific T helper cell 1 responses. These results demonstrate a novel and important role for MCP-1 in the regulation or oral tolerance for the prevention and treatment of autoimmune disease.
Experimental autoimmune encephalomyelitis (EAE)1 is a CD4+ T cell–mediated, demyelinating disease of the central nervous system (CNS) that serves as a model for the human disease, multiple sclerosis (MS; 1). EAE can be induced in animals by immunization with proteolipid protein (PLP), the encephalitogenic peptide sequence 139–151 (PLP139–151) emulsified in CFA (2), or alternatively by the adoptive transfer of antigen-activated PLP139–151– specific T cells to normal recipient mice (3). Characteristics of the disease include progressive ascending clinical paralysis followed by periods of remission and subsequent relapses in the SJL/J mouse (4). Analysis of mononuclear cell infiltration in the CNS has revealed that antigen-specific and nonspecific CD4+ and CD8+ T cells as well as macrophages constitute the recruited cell population (5, 6).
Peripheral, antigen-specific tolerance can be induced by oral antigen administration and has been used to prevent the induction of experimental autoimmune diseases (for review see reference 7). The mechanism of oral tolerance is not completely understood. Earlier studies demonstrated that antigen feeding induced both anergy and regulatory T cells (8–10). More recently, it was reported that high dose antigen feeding resulted in the induction of anergy/deletion (11), whereas feeding of multiple low doses of antigen induced regulatory T cells that secrete TGF-β (12, 13). Even though delivery of antigen through the intestinal mucosa appears to inhibit peripheral cell–mediated immune responses, this mode of antigen introduction can prime both mucosal and peripheral antibody responses (14, 15).
Events at the intestinal mucosa that modulate antigen uptake and processing (e.g., inhibition of oral tolerance induction by introduction of cholera toxin and antigen) also influence whether oral exposure to antigen induces peripheral tolerance or primes a peripheral immune response (16, 17). This latter information suggests that modulation of mucosal immune responses at the time of antigen feeding is a central feature in the ability to establish peripheral tolerance through oral antigen administration. Whether this involves increased lymphocyte trafficking or regulatory cytokine modulation is not well understood. Jung et al. (18) demonstrated that the chemokine MCP-1 is expressed in the mucosa after infection with bacteria, thereby suggesting that oral introduction of antigen can result in the upregulation of mucosal chemokine production.
Chemokines such as macrophage inflammatory protein 1α (MIP-1α), MCP-1, IL-8, and RANTES (regulated on activation normal T cell expressed and secreted cytokine) are molecules that induce leukocyte accumulation in tissue sites of inflammation (19). CC chemokine family members have been implicated as candidates in the immunopathology of EAE, with T cell production of MIP-1α and T cell activation gene 3 (TCA-3) shown to be required for induction of EAE (20). Additionally, MIP-1α and MCP-1 production in the CNS have been associated with acute disease symptoms in both rat (21) and murine (22–24) EAE models. These examples raise the possibility that chemokine production in the CNS of MS patients functions to drive pathogenesis of disease through the recruitment of leukocytes into the brain.
In addition to regulating leukocyte migration, chemokines appear to have additional functions. Recently, chemokine-induced signaling in human T cells has been demonstrated (25). Moreover, Taub et al. (26) have demonstrated a role for chemokines in costimulation resulting in enhanced IL-2 production as well as proliferation of T cell clones. Similarly, we have shown that chemokines can regulate T cell differentiation (27) and cytokine production (28). Chemokine expression at the intestinal mucosa as well as mucosal lymphoid tissue could affect leukocyte trafficking and/or differentiation of T helper lymphocyte effector function, thereby influencing peripheral immune responsiveness. It is well documented that immune tolerance and thus prevention of autoimmune disease can be achieved by prefeeding intact autoantigenic proteins (for review see reference 29); however, the immunologic mechanisms are not well understood. Therefore, this study was designed to explore the immune regulatory role of MCP-1 after oral antigen administration.
Materials and Methods
Animals.
Female SJL/J mice were purchased from Harlan Sprague Dawley (Indianapolis, IN). Mice were 6–7 wk old at the initiation of the experiment and were maintained on standard laboratory chow and water ad libitum. Animal care was provided in accordance with Northwestern University and National Institutes of Health (Bethesda, MD) guidelines.
Antigens.
PLP139–151 (HSLGKWLGHPDKF) was purchased from Peptides International (Louisville, KY). The amino acid composition was verified by mass spectrometry and purity (>98%) was assessed by HPLC. Theiler's virus VP2 70-86 was synthesized on an ABI Synergy machine (Perkin Elmer, Norwalk, CT) according to the manufacturer's instructions and used as the control peptide.
Antibodies.
Rabbit anti–murine MIP-1α, MCP-1, and MIP-2 antibodies were prepared by multiple site immunization of New Zealand white rabbits with recombinant proteins (R&D Systems, Minneapolis, MN) emulsified in CFA. Polyclonal antibodies were titered by direct ELISA and specificity was verified by the failure to cross-react with murine (m)IL-1α, mIL-2, mTNF, IL-6, human (h)IL-8, hRANTES, and hMIP-1. Moreover, the individual chemokine antibodies do not cross-react with other murine chemokines (e.g., anti–MCP-1 does not cross-react with MIP-1α). At the time of the experiments the titer of the antichemokine antibodies was >106. Normal rabbit serum (NRS) was used as a control immunoglobulin preparation for in vivo treatment regimens. Antichemokine antibodies were injected into individual mice from different treatment groups either intraperitoneally or intravenously with no functional differences between the two routes of administration observed.
Active EAE Induction and Clinical Disease Assessment.
EAE was induced as previously described (4). In brief, each mouse received a subcutaneous injection of 0.1 ml emulsion containing 87 μg (50 nmol) PLP139–151 and 200 μg Mycobacterium tuberculosis H37Ra in IFA (Difco, Detroit, MI) at three sites on the dorsal flank. Disease onset usually occurs within 3 wk of immunization and was assessed by observing individual animals daily, and clinical severity as follows: grade 0, no abnormality; grade 1, limp tail; grade 2, limp tail and partial hind limb weakness (waddling gait); grade 3, complete hind limb paralysis; grade 4, death. A relapse was scored when a mouse developed additional neurological deficits (⩾1 clinical grade) after a period of stabilization or improvement.
Cell Culture.
Single cell suspensions from pooled lymph nodes or spleens were cultured at a density of 2.5 × 106 cells/ml in DMEM containing 5% FCS, 1 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM nonessential amino acids and 5 × 10−5 M 2-ME (complete DMEM-5; all components from Sigma Chemical Co., St. Louis, MO) in the presence or absence of 50 μg/ml PLP peptides.
Chemokine and Cytokine ELISA.
Assessment of MCP-1 was quantitated from tissue samples using previously described ELISAs (30, 31). Mucosal samples were homogenized in 1 ml PBS and clarified by centrifugation (400 g) for 10 min. Flat-bottomed microtiter plates (Nunc, Naperville, IL) were coated with capture antibody diluted to 3.2 μg/ml in borate-buffered saline coating buffer and were incubated overnight. Nonspecific binding sites were blocked with 2% BSA in PBS for 1 h at 37°C and samples were subsequently added in triplicate for 2 h at 37°C. Biotinylated goat anti–rabbit detection antibody was added and the plates were incubated for an additional 1 h at 37°C. The wells were developed using strepavidin-peroxidase and o-phenylenediamine substrate and absorbence was read at 490 nm. Standard curves for MCP-1 were generated using a series of dilutions of purified recombinant protein (R&D Systems). Chemokine levels in mucosa were quantitated by comparison to the standard curves and expressed as pg/mg tissue. The detection limit of these assays is at least 25 pg/ml. The MCP-1 ELISA is specific and does not cross-react with any other chemokine or cytokine. The presence of IL-12 and IL-4 in tissue samples was quantitated using cytokine-specific ELISA performed as follows: microwell plates were coated with either anti–IL-12 (5 μg/ml) or anti–IL-4 (1 μg/ml) mAb (PharMingen, San Diego, CA) in Dulbecco's PBS overnight at 4°C. Nonspecific binding was reduced by blocking with PBS containing 2% BSA for 1 h at room temperature. The microwell plates were washed with 50 mM Tris 0.2% Tween 20 and dilutions of recombinant IL-12 or IL-4 (PharMingen) as well as the appropriate experimental samples were added and incubated overnight at 4°C. Either goat anti–IL-12 (20 μg/ml) or goat anti–IL-4 (10 μg/ml; R&D Systems) was added and the plates were incubated for 2 h at room temperature. The wells were developed using peroxidase-conjugated donkey anti–goat IgG (Jackson Immunoresearch, West Grove, PA) and 3,3′,5′,5′-tetramethylbenzidine dihydrochloride (TMB) substrate (DAKO Corp., Carpinteria, CA). The reaction was stopped by adding 0.18 M H2SO4 and the absorbence was read at 450 nm. Experimental samples were compared to standard curves for both IL-12 and IL-4, which were generated using a series of dilutions of recombinant protein. The data was expressed as picogram of cytokine per milligram tissue. Antigen-induced cytokine production was assayed from 48 h (for IL-4 and IFN-γ) culture supernatants after stimulation with 50 μg/ml PLP139–151. Duplicate samples were tested for the presence of IFN-γ and IL-4 (Endogen, Cambridge, MA), and the data were expressed as picogram per milliliter when compared to recombinant standards. TGF-β was analyzed from supernatants of cells grown in serum-free medium or in tissue samples using a modified ELISA protocol as previously described (12).
Results
Mucosal MCP-1 Production after Oral Antigen Administration.
We asked whether MCP-1 is expressed at mucosal tissue and lymphoid sites after introduction of oral antigen. Jung et al. (18) reported that infection of intestinal epithelium resulted in expression of an array of proinflammatory cytokine expression, including MCP-1. We have previously shown that feeding 2 mg of PLP-139–151 peptide inhibits development of EAE and induces clonal anergy as measured by a decrease in antigen-specific IL-2 and IFN-γ production in the absence of measurable TGF-β production (32). Inhibition of EAE by feeding PLP139–151 peptide is dose dependent as progressively smaller single as well as multiple doses of fed peptide failed to inhibit development of EAE (our unpublished data). Additionally, induction of oral tolerance using this single-dose peptide regimen has never resulted in an increase in TGF-β expression above that of background in antigen-specific restimulation of lymphocytes (32). In our experiments, mice were fed 2 mg of either PLP139–151 or control peptide once and 7 d later were immunized for EAE induction with PLP139–151 emulsified in CFA. Samples of proximal intestinal mucosa were harvested immediately after antigen feeding as well as at days 3, 8, 10, 14, 17, and 24 after oral administration of antigen. The tissue was homogenized and subsequently assayed for the presence of MCP-1 by ELISA. We have successfully used this approach to assay CNS tissue for the presence of chemokines (22). The results shown in Fig. 1 indicate that mucosal MCP-1 expression increased after oral administration of PLP139–151 (closed circles) and continued to increase in fed mice that were primed with PLP139–151 in CFA (arrow). These levels of MCP-1 are significantly greater (P <0.05 as indicated by asterisks) than in control peptide-fed mice primed with PLP139–151 in CFA (closed squares). Saline-fed mice did not show a level of MCP-1 expression above the detection limit of the assay (data not shown).
Figure 1.

MCP-1 production in intestinal mucosa after antigen feeding. Mice were fed either PLP139–151 or control peptide and 7 d later were primed for EAE induction by injection of PLP139–151 in CFA (arrow). Intestinal mucosa was harvested from representative mice at various timepoints after antigen feeding, homogenized, and assayed for the presence of MCP-1 protein by ELISA. The data represent the mean MCP-1 levels from duplicate assay samples. The data are representative of three identical experiments. Asterisk, P <0.05.
Peyer's Patch and Mesenteric Lymph Node Expression of MCP-1 after Oral Antigen Administration.
Antigens absorbed through the mucosal epithelium and Peyer's patches drain into the mesenteric lymph node (MLN) and portal circulation (7). We asked whether chemokine expression in the Peyer's patch and MLN could be detected after oral antigen administration. We could not detect MIP-1α expression in any mucosal tissue by ELISA, immunohistochemistry, or reverse transcriptase (RT)-PCR (data not shown), nor could we detect MCP-1 expression in Peyer's patch or MLN by ELISA or immunohistochemistry (data not shown). However, as shown in Fig. 2, we were able to use RT-PCR to detect the expression of MCP-1 at early time points in both Peyer's patch and MLN after oral antigen administration. Mice were fed either PLP139–151 or saline as a control and examined for Peyer's patch and MLN MCP-1 expression daily by RT-PCR. MCP-1 was detected in both Peyer's patch and MLN only in mice fed PLP139–151 and not in mice fed saline. Mice fed a control peptide, VP2 70-86, known to bind I-As also expressed MCP-1 in the Peyer's patch and MLN, but mice fed a control peptide, ovalbumin 323–339, that does not bind I-As with high affinity did not show mucosal MCP-1 expression (data not shown). In an effort to determine which populations of cells in the Peyer's patch and MLN expressed MCP-1, cells were separated into plastic adherent or nonadherent populations and each was subsequently analyzed for expression of MCP-1 by RT-PCR. The plastic adherent cell population was highly enriched for cells of the macrophage lineage as determined by Mac-3 immunostaining (data not shown). The results demonstrated that mucosal expression of MCP-1 was limited to macrophage-enriched adherent cells from either Peyer's patch or MLN as shown in Fig. 3 and not to the T cell–enriched nonadherent cell populations (data not shown). These results suggest that MCP-1 is expressed by mucosal immune system– adherent macrophages in response to orally administered antigen.
Figure 2.

Detection of MCP-1 mRNA expression in mucosal lymphoid tissue after antigen feeding. Mice were fed either PLP139–151 or control peptide and both Peyer's patch and MLNs were harvested and assayed for MCP-1 mRNA expression using RT-PCR. Tissue was harvested at the indicated time points (in days) after oral antigen administration. Control MCP-1 mRNA expression (C) was determined from LPS-stimulated macrophages and used as an internal control for the RT-PCR conditions. Amplified cDNA was electrophoresed through 2% agarose gels and visualized using ethidium bromide. The data are representative of three identical experiments.
Figure 3.

Detection of MCP-1 mRNA expression in the adherent cell populations of mucosal lymphoid tissue after antigen feeding. Mice were fed either PLP139–151 or control peptide and both Peyer's patch and MLNs were harvested. The adherent cells were isolated by plastic adherence (see Materials and Methods) and assayed for MCP-1 mRNA expression using RT-PCR. Adherent cells were harvested at the indicated time points (in days) after oral antigen administration. Control MCP-1 mRNA expression (C) was determined from LPS-stimulated macrophages and used as an internal control for the RT-PCR conditions. Amplified cDNA was electrophoresed through 2% agarose gels and visualized using ethidium bromide. The data are representative of three identical experiments.
Oral Administration of PLP139–151 Downregulated Mucosal-derived IL-12 and Upregualted Mucosal-derived IL-4.
Chensue et al. (33) demonstrated that increased MCP-1 production in a schistosomiasis model resulted in a decrease in IL-12 production by inflammatory macrophages. Additionally, this report together with our observation that oral administration of PLP139–151 upregulated mucosal MCP-1 expression prompted us to examine the levels of IL-12 at the mucosa after oral tolerance induction. Mice were fed either PLP139–151 or control peptide and proximal intestinal mucosa as well as spleen was harvested at days 0, 3, 8, 10, 14, 17, 20, and 24 after feeding. Tissue lysates were subsequently assayed for the presence of IL-12 and IL-4 by ELISA. The results shown in Fig. 4 demonstrate that after oral PLP139–151 administration there was a significant decrease in mucosal IL-12 expression (A, closed circles) over time when compared to control peptide–fed mice (A, closed squares). There was no detectable change in splenic IL-12 production (B). The opposite results were obtained when the tissue was analyzed for IL-4 content. As shown in Fig. 5, oral administration of PLP139–151 resulted in an increase in mucosal IL-4 production when compared to control peptide–fed tissue. We could not detect IL-4 production in whole splenic lysates after antigen feeding (data not shown). These results together with those in Fig. 1 suggest that oral administration of antigen results in an increase in mucosal MCP-1 and IL-4 expression with a concomitant decrease in IL-12 expression.
Figure 4.
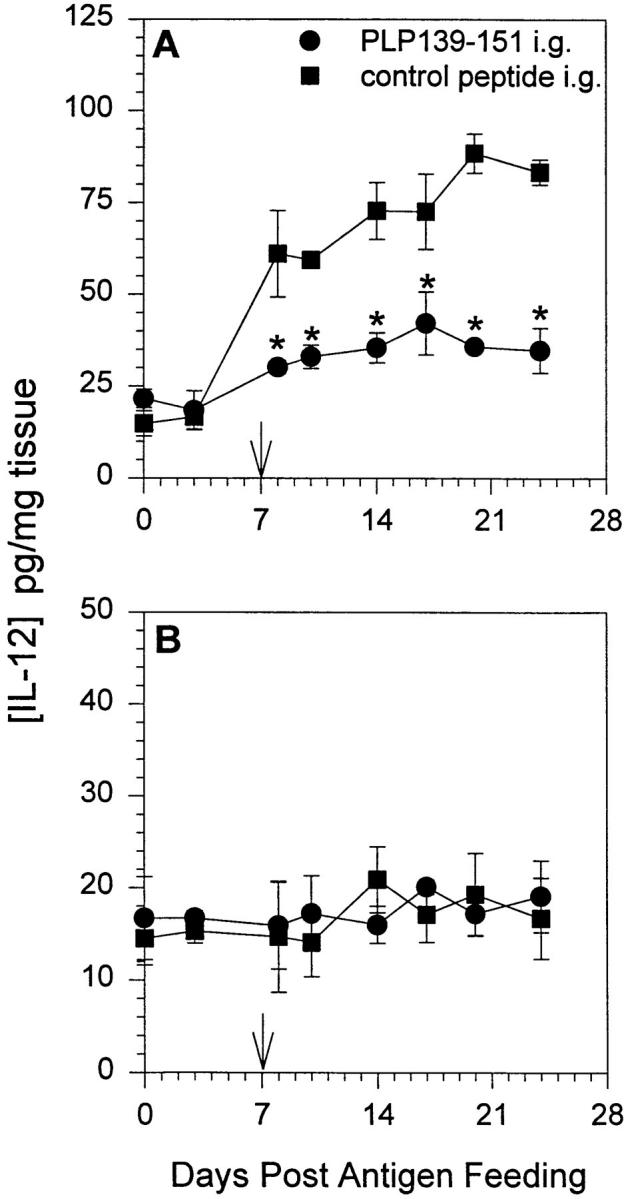
Il-12 expression in mucosal tissue and spleen after oral administration of antigen. Mice were fed either PLP139– 151 or control peptide and 7 d later were primed for EAE induction by injection of PLP139– 151 in CFA (arrow). Intestinal mucosa (A) and spleen (B) were harvested from representative mice at various time points after antigen feeding, homogenized, and assayed for the presence of IL-12 protein by ELISA. The data represent the mean IL-12 levels from duplicate assay samples. The data are representative of two identical experiments. Asterisk, P <0.05.
Figure 5.

IL-4 expression in mucosal tissue after oral administration of antigen. Mice were fed either PLP139–151 or control peptide and 7 d later were primed for EAE induction by injection of PLP139–151 in CFA (arrow). Intestinal mucosa was harvested from representative mice at various time points following antigen feeding, homogenized, and assayed for the presence of IL-4 protein by ELISA. The data represent the mean IL-4 levels from duplicate assay samples. The data are representative of two identical experiments. Asterisk, P <0.05.
Effect of In Vivo Anti–MCP-1 Treatment on Oral Tolerance Induction.
The experimental evidence to this point suggested that oral administration of antigen led to upregulation of MCP-1 and IL-4 accompanied by downregulation of IL-12. We wanted to determine whether there was a direct role for MCP-1 in the regulation of oral tolerance induction as a prevention for EAE. To test the in vivo role of MCP-1 in tolerance induction, we fed animals either PLP139–151 or control peptide and treated the recipients with either anti–MCP-1 or control NRS. The antibody treatments bracketed the oral antigen administration (i.e., 2 d before antigen feeding, the day of antigen feeding, and 2 d after antigen feeding). The results are shown in Fig. 6 A and demonstrate that mice fed control peptide developed EAE (closed squares), whereas mice fed PLP139–151 failed to develop acute EAE (closed circles). However, mice treated in vivo with anti–MCP-1 at the time of PLP139–151 feeding did not develop tolerance, rather they developed severe clinical EAE (open circles) when compared to the NRS-treated control group that were fed PLP139–151 and did not develop severe EAE (closed circles). Anti–MCP-1 treatment itself did not affect the course of EAE development as demonstrated by the failure of anti–MCP-1 to alter normal disease course (open squares). These data suggest that MCP-1 plays a functional role in oral tolerance induction.
Figure 6.

In vivo anti–MCP-1 treatment abrogates induction of oral tolerance (A) Mice were fed either PLP139–151 to induce immunologic tolerance of control peptide 7 d before disease induction with PLP139– 151 in CFA. Each of those two groups were further divided and treated with either 0.5 ml anti–MCP-1 or NRS 2 d before antigen feeding, the day of antigen feeding, and 2 d after antigen feeding. (B) Mice were fed either PLP139–151 or control peptide 7 d before disease induction with PLP139–151 in CFA. Each of those two groups was further divided and treated with either 0.5 ml anti–MCP-1 or NRS 3, 5, and 7 d after antigen feeding. All animals were followed for the development of clinical EAE according to the grading scale in the Materials and Methods. The data represent the mean clinical disease score in each group and are representative of at least two identical experiments. Animals that were fed PLP139– 151 and treated with NRS (closed circles) developed significantly decreased clinical disease compared to control peptide–fed and NRS-treated mice (closed squares), P < 0.05 days 17–25 after immunization.
Since other chemokines such as MIP-1α and RANTES have been shown to be associated with EAE (22) as well as to play a role in T cell activation and cytokine production (25–27) we tested whether in vivo treatment with antisera to these factors at the time of PLP139–151 feeding would have an effect on oral tolerance induction. Neither anti– MIP-1α nor anti-RANTES administration abrogated oral tolerance induction (data not shown). Furthermore, we investigated the temporal effect of anti–MCP-1 administration and determined whether treatment with anti–MCP-1 after feeding of PLP139–151 had the same effect as antibody treatment at the same time as peptide feeding. Groups of mice were fed one dose of 2 mg of PLP139–151 or control peptide and were treated with 0.5 ml anti–MCP-1 or NRS 3, 5, and 7 d after antigen feeding. All mice were primed with PLP139–151 emulsified in CFA and observed for the development of clinical disease signs. The results shown in Fig. 6 B demonstrate that treatment with anti– MCP-1 3 d after antigen feeding did not abrogate oral tolerance induction. These results further support the idea that MCP-1 expression at antigen feeding regulates oral tolerance induction.
Mucosal Cytokine Production after Anti–MCP-1 Treatment.
We have demonstrated that anti–MCP-1 treatment at the time of PLP139–151 feeding abrogated oral tolerance (Fig. 6 A) To directly demonstrate the effect of anti– MCP-1 treatment on mucosal cytokine expression, we performed an identical experiment to that shown in Fig. 6 A in which mice were treated with anti–MCP-1 or NRS at the time of antigen feeding. 7 d after antigen feeding, mice were immunized with PLP139–151 in CFA and 7 d after that, mice were killed and mucosal IL-12 and IL-4 expression was measured by ELISA. The results in Fig. 7 show that feeding of PLP139–151 significantly reduced mucosal IL-12 production (group B, open bar) and enhanced mucosal IL-4 production (group B, hatched bar) when compared to control-fed and control-treated animals (group A). However, mice fed PLP139–151 and treated with anti–MCP-1 at the time of oral tolerance induction (group D, open bar showed a level of mucosal-derived IL-12 similar to that of the control-fed and control-treated mice (group A) as well as control-fed and anti–MCP-1-treated mice (group C). The most important observation was that anti–MCP-1 treatment at the time of PLP139–151 antigen feeding resulted in significantly enhanced mucosal IL-12 production compared to mice fed only PLP139–151 (group B, open bar). These results suggest that MCP-1 upregulation after antigen-feeding and subsequent antigen priming result in a decrease in mucosal IL-12 expression and that neutralization of MCP-1 by in vivo antibody treatment restored mucosal IL-12 expression.
Figure 7.

Il-4 and IL-12 expression in mucosal tissue after oral administration of antigen and in vivo anti–MCP-1 treatment. Mice were fed either PLP139–151 to induce immunologic tolerance or control peptide 7 d before disease induction with PLP139–151 in CFA. Each of those two groups was treated with either 0.5 ml anti–MCP-1 or NRS 2 d before antigen feeding, the day of antigen feeding, and 2 d after antigen feeding (similar to the experiment in Fig. 2). 7 d after disease induction, intestinal mucosa was harvested and analyzed for the presence of IL-4 and IL-12. The data represent either the mean IL-4 or IL-12 levels from duplicate assay samples. The data are representative of two identical experiments.
Effect of Anti–MCP-1 Treatment on Antigen-specific T Cell Responses.
We have previously demonstrated that oral administration of PLP139–151 inhibited acute EAE development, antigen-specific proliferation, IL-2 and IFN-γ production, and increased IL-4 production (32). Since anti– MCP-1 abrogated oral tolerance induction (Fig. 6 A) and reversed the downregulation of mucosa-derived IL-12 production (Fig. 7), we asked what the effect of anti–MCP-1 treatment at the time of antigen feeding was on antigen-specific recall immune responses. Splenic lymphocytes were harvested from the experiment described in Fig. 7 and were restimulated in vitro with PLP139–151, and the culture supernatants were harvested and assayed for the presence of IL-4 and IFN-γ. The results of this experiment are shown in Fig. 8 and demonstrate that when PLP139–151 was fed to mice before EAE induction, antigen-specific IFN-γ production was significantly decreased (open bar, group B versus group A) and IL-4 production was slightly increased (hatched bar, group B versus group A). However, anti–MCP-1 treatment in vivo given at the time of antigen feeding prevented downregulation of IFN-γ production (open bar, group D versus group B), effectively returning the response to that of control-fed and -treated (open bar, group A). We could not detect a significant change in TGF-β production (data not shown). These data suggest that anti-MCP-1 treatment in vivo prevented the downregulation of IFN-γ that normally results from oral tolerance induction (reference 32 and Fig. 8), possibly by restoring IL-12 levels to that of normal control. Collectively, our data argue for a mechanism of oral tolerance induction that includes mucosal expression of MCP-1, which regulates mucosal expression of IL-12.
Figure 8.

Effect of in vivo anti–MCP-1 treatment on antigen-specific IL-4 and IFN-γ production. Splenic lymphocytes were harvested from the experimental groups shown in the figure and were cultured with PLP139–151 in vitro, and the supernatants were harvested and subsequently assayed for the presence of IL-4 and IFN-γ by ELISA. The data are representative of two identical experiments.
Discussion
Oral antigen administration has been shown to induce peripheral T cell tolerance in a number of antigen and experimental disease systems. The mechanism of oral tolerance induction is not completely understood and two hypotheses have emerged to explain the differences reported in the literature (13). The first is feeding high doses of soluble antigen, which results in clonal anergy and/or deletion (11). The second hypothesis offered to explain oral tolerance induction is the generation of an antigen-specific T cell population that has the capacity to secrete regulatory cytokines, including TGF-β, that downregulate antigen-specific immune responses in the absence of clonal anergy or deletion (34). Oral tolerance using intact neuroantigens and neuroantigen peptides has been employed primarily for the prevention of EAE by prefeeding animals followed by active induction of disease by immunization with neuroantigen in CFA (35, 36). It was only recently demonstrated that oral administration of MBP could treat ongoing chronic EAE in the B10.PL mouse (37), and that oral administration of the dominant collagen epitope could treat ongoing experimental arthritis (38); however, oral administration of the dominant PLP epitope, PLP139–151, failed to alter ongoing relapsing EAE in the SJL mouse model (39). The focus of oral tolerance mechanisms in these autoimmune disease models has been on the antigen-specific peripheral lymphocyte. In this report we described a novel mechanisms for mucosal cytokine regulation leading to the induction of oral tolerance.
MCP-1 is expressed by mucosal epithelia cells after infection with a number of bacteria (18) and can be expressed by macrophages (40). After oral administration of PLP139– 151, MCP-1 expression was upregulated in the mucosal immune tissue, including the mucosa, Peyer's patch, and MLN (Fig. 1–3). The levels of MCP-1 in the mucosa remain elevated and continue to rise in tolerized mice after immunization with PLP139–151 and CFA compared to the control-fed group (Fig. 1). We have not yet determined the mucosal cell type responsible for MCP-1 production after antigen feeding, but it appears that Peyer's patch– derived adherent Mac-3+ cells express MCP-1 after antigen feeding (Fig. 3). MCP-1 regulation of oral tolerance was shown to be biologically relevant by the ability of in vivo anti–MCP-1 treatments to abrogate oral tolerance induction at the time of antigen feeding (Fig. 6 A). The inhibition of tolerance induction by anti–MCP-1 treatment was specific as treatment regimens beginning 3 d after antigen feeding had no effect on tolerance induction (Fig. 6 A) and anti–MIP-1α and anti-RANTES treatment at the time of antigen feeding also had no effect on tolerance induction (data not shown). MCP-1 regulation of oral tolerance induction may function through two distinct mechanisms. The first is that MCP-1 directly upregulates IL-4 expression, which potentiates differentiation of Th2 cells (27). The molecular basis for MCP-1–induced Th2 differentiation is unknown, but currently under investigation. After antigen feeding and upregulation of MCP-1 expression in the mucosal immune compartment, Peyer's patch and MLN T cells may be predisposed to differentiate into Th2 cells or alternatively, MCP-1 expression could potentiate IL-4 production, which would drive peripheral Th2 differentiation (41).
The second possible mechanism postulates that MCP-1 expression could directly downregulate IL-12 production, which could result in a partial or complete block of peripheral Th1 differentiation (41) and/or enhancement of mucosal TGF-β expression (42), which in turn could result in an increase or enhancement of TGF-β–secreting regulatory cells (34). We favor the possibility that MCP-1 expression is regulating Th1 differentiation and/or TGF-β regulation based on the reports that oral tolerance can be induced in IL-4–deficient mice (43) and that treatment with anti–IL-4 does not inhibit the ability to induce oral tolerance (32). Our data suggesting that MCP-1 downregulation of mucosal IL-12 expression is central to oral tolerance induction is supported by observations from others demonstrating that orally administered IL-12 promotes a systemic antigen-specific Th1 response (44). The combination of inhibiting Th1 differentiation and potentiating Th2 differentiation may explain oral tolerance induction and why blocking these events with in vivo anti–MCP-1 administration abrogates tolerance induction (Fig. 6 A).
Another possible mechanism of MCP-1 regulation of oral tolerance induction involves T cell and/or macrophage trafficking in response to a chemotactic gradient. MCP-1 has been shown to induce T cell migration (45) and its upregulation in the mucosa after antigen feeding may promote infiltration of T cells into the mucosal immune tissues. These T cells may be preferentially activated to secrete TGF-β, migrate to the CNS, and become effector regulatory cells that downregulate EAE (46). Additionally, MCP-1 expression in the mucosa may induce migration of macrophages (47) to the site of antigen uptake and then present the fed antigen to T cells in the mucosal immune tissue. Alternatively, after migration of macrophages into the mucosal site of antigen uptake, the macrophages may migrate to peripheral immune tissue and present antigen to T cells, resulting in either induction of regulatory T cells or direct inactivation of pathogenic Th1 cells (43). Therefore, administration of neutralizing antibodies to MCP-1 in vivo would preclude the ability of mucosal-derived MCP-1 to induce T cell or macrophage migration.
The significance of MCP-1 expression in the mucosal lymphoid tissue after antigen feeding is not completely understood. MIP-1α, MIP-1β, and RANTES expression was not detected in the mucosa, Peyer's patch, or MLN after feeding of either PLP139–151, control peptide, or a non–I-As binding peptide (data not shown). It is interesting to note that although feeding a control peptide induced a quantitatively smaller mucosal MCP-1 response, it did not induce oral tolerance (Figs. 1 and 6). This suggests that there is a multilevel control of tolerance induction with a level of antigen specificity as well as a level of cytokine/chemokine regulation. The observation that mucosal MCP-1 expression is elevated after a bolus of fed antigen may reflect the constant regulation of immune responses to food antigens. This report describes an additional regulatory mechanism involved in oral tolerance induction that can be further manipulated to produce a more efficient and long-lasting immune tolerance during autoimmune disease. For example, enhancement of mucosal MCP-1 expression at the time of antigen feeding may be able to provide efficacious tolerance induction that would not be possible by antigen feeding alone. We are currently exploiting the possibilities for the development of more effective tolerogenic regimens for the treatment of ongoing autoimmune disease.
Footnotes
This work was supported by National Institutes of Health AI-35934 and NS-34510 to W.J. Karpus.
Address correspondence to William J. Karpus, Department of Pathology, Northwestern University Medical School, 303 E. Chicago Ave., W127, Chicago, IL 60611. Phone: 312-503-1005; Fax: 312-503-8240; E-mail: w-karpus@nwu.edu
Abbreviations used in this paper: CNS, central nervous system; EAE, experimental autoimmune encephalomyelitis; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; MLN, mesenteric lymph node; MS, multiple sclerosis; NRS, normal rabbit serum; PLP, proteolipid protein; RANTES, regulated on activation normal T cell expressed and secreted; RT, reverse transcriptase.
References
- 1.Martin R, McFarland HF, McFarlin DE. Immunologic aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 2.Tuohy VK, Sobel RA, Lu Z, Laursen RA, Lees MB. Myelin proteolipid protein: minimum sequence requirements for active induction of autoimmune encephalomyelitis in SWR/J and SJL/J mice. J Neuroimmunol. 1992;39:67–74. doi: 10.1016/0165-5728(92)90175-k. [DOI] [PubMed] [Google Scholar]
- 3.Satoh J, Koike F, Tabira T. Experimental allergic encephalomyelitis mediated by murine encephalitogenic T-cell lines specific for myelin proteolipid apoprotein. Ann NY Acad Sci. 1988;540:343–344. doi: 10.1111/j.1749-6632.1988.tb27093.x. [DOI] [PubMed] [Google Scholar]
- 4.McRae BL, Kennedy MK, Tan LJ, Dal M C, Canto, Miller SD. Induction of active and adoptive chronic-relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38:229–240. doi: 10.1016/0165-5728(92)90016-e. [DOI] [PubMed] [Google Scholar]
- 5.Cross AH, Cannella B, Brosnan CF, Raine CS. Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of 14C-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab Invest. 1990;63:162–170. [PubMed] [Google Scholar]
- 6.Pope JG, Karpus WJ, VanderLugt C, Miller SD. Flow cytometric and functional analyses of central nervous system–infiltrating cells in SJL/J mice with Theiler's virus–induced demyelinating disease. Evidence for a CD4+T cell-mediated pathology. J Immunol. 1996;156:4050–4058. [PubMed] [Google Scholar]
- 7.Weiner HL. Oral tolerance: immune mechanisms and treatment of autoimmune diseases. Immunol Today. 1997;18:335–343. doi: 10.1016/s0167-5699(97)01053-0. [DOI] [PubMed] [Google Scholar]
- 8.Miller SD, Hanson DG. Inhibition of specific immune responses by feeding protein antigens. IV. Evidence for tolerance and specific active suppression of cell-mediated immune responses to ovalbumin. J Immunol. 1979;123:2344–2350. [PubMed] [Google Scholar]
- 9.Asherson G L, Zembala M, Perera MA, Mayhew B, Thomas WR. Production of immunity and unresponsiveness in the mouse by feeding contact sensitizing agents and the role of suppressor cells in the Peyer's patches, mesenteric lymph nodes and other lymphoid tissues. Cell Immunol. 1977;33:145–155. doi: 10.1016/0008-8749(77)90142-3. [DOI] [PubMed] [Google Scholar]
- 10.Strobel S, Mowat AM, Drummond HE, Pickering MG, Ferguson A. Immunological responses to fed protein antigens in mice. Oral tolerance for CMI is due to activation of cyclophosphamide-sensitive cells by gut-processed antigen. Immunology. 1983;49:451–456. [PMC free article] [PubMed] [Google Scholar]
- 11.Whitacre CC, Gienapp IE, Orosz CG, Bitar DM. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J Immunol. 1991;147:2155–2163. [PubMed] [Google Scholar]
- 12.Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppresses both in vitro and in vivo immune responses by the release of transforming growth factor β after antigen-specific triggering. Proc Natl Acad Sci USA. 1992;89:421–425. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman A, Weiner HL. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc Natl Acad Sci USA. 1994;91:6688–6692. doi: 10.1073/pnas.91.14.6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Husby S, Mestecky J, Moldoveanu Z, Holland S, Elson CO. Oral tolerance in humans. T cell but not B cell tolerance after antigen feeding. J Immunol. 1994;152:4663–4670. [PubMed] [Google Scholar]
- 15.Michalek SM, McGhee JR, Kiyono H, Colwell DE, Eldridge JH, Wannemuehler MJ, Koopman WJ. The IgA response: inductive aspects, regulatory cells, and effector functions. Ann NY Acad Sci. 1983;409:48–71. doi: 10.1111/j.1749-6632.1983.tb26859.x. [DOI] [PubMed] [Google Scholar]
- 16.Elson CO, Ealding W. Cholera toxin feeding did not induce oral tolerance in mice and abrogated oral tolerance to an unrelated protein antigen. J Immunol. 1984;133:2892–2897. [PubMed] [Google Scholar]
- 17.Kay RA, Ferguson A. The immunological consequences of feeding cholera toxin. II. Mechanisms responsible for the induction of oral tolerance for DTH. Immunology. 1989;66:416–421. [PMC free article] [PubMed] [Google Scholar]
- 18.Jung HC, Eckmann L, Yang S-K, Panja A, Fierer J, Morzycka-Wroblewska E, Kagnoff MF. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. 1995;95:55–65. doi: 10.1172/JCI117676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oppenheim JJ, Zachariae CO, Mukaida N, Matsushima K. Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu Rev Immunol. 1991;9:617–648. doi: 10.1146/annurev.iy.09.040191.003153. [DOI] [PubMed] [Google Scholar]
- 20.Kuchroo VK, Martin CA, Greer JM, Ju S-T, Sobel RA, Dorf ME. Cytokines and adhesion molecules contribute to the ability of myelin proteolipid protein–specific T cell clones to mediate experimental allergic encephalomyelitis. J Immunol. 1993;151:4371–4382. [PubMed] [Google Scholar]
- 21.Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshrestha S, Guida MP, Rapoport DA, Berman JW. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol. 1993;150:2525–2533. [PubMed] [Google Scholar]
- 22.Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1α in the pathogenesis of the T cell–mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155:5003–5010. [PubMed] [Google Scholar]
- 23.Ransohoff R, Hamilton T, Tani M, Stoler M, Shick H, Major J, Estes M, Thomas D, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/ MCP-1 in experimental autoimmune encephalomyelitis. FASEB (Fed Am Soc Exp Biol) J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein–1 and other β-chemokines in murine glial cells. J Neuroimmunol. 1995;60:143–150. doi: 10.1016/0165-5728(95)00064-9. [DOI] [PubMed] [Google Scholar]
- 25.Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T cell signaling pathways by the chemokine RANTES. Science. 1995;269:1727–1730. doi: 10.1126/science.7569902. [DOI] [PubMed] [Google Scholar]
- 26.Taub DD, Turcovski-Corrales SM, Key ML, Longo DL, Murphy WJ. Chemokines and T lymphocyte activation. 1. β chemokines costimulate human T lymphocyte activation in vitro. J Immunol. 1996;156:2095–2103. [PubMed] [Google Scholar]
- 27.Karpus WJ, Jukacs NW, Kennedy KJ, Smith WS, Hurst SD, Barrett TA. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol. 1997;158:4129–4136. [PubMed] [Google Scholar]
- 28.Lukacs NW, Chensue SW, Karpus WJ, Lincoln P, Keefer C, Strieter RM, Kunkel SL. C-C chemokines differentially alter interleukin-4 production from lymphocytes. Am J Pathol. 1997;150:1861–1868. [PMC free article] [PubMed] [Google Scholar]
- 29.Weiner HL. Treatment of autoimmune diseases by oral tolerance to autoantigens. Autoimmunity. 1993;15:6–7. doi: 10.3109/08916939309008850. [DOI] [PubMed] [Google Scholar]
- 30.Lukacs NW, Kunkel SL, Strieter RM, Warmington K, Chensue SW. The role of macrophage inflammatory protein 1α in Shistosoma mansoniegg–induced granulomatous inflammation. J Exp Med. 1993;177:1551–1559. doi: 10.1084/jem.177.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lukacs NW, Chensue SW, Smith RE, Strieter RM, Warmington K, Wilke C, Kunkel SL. Production of monocyte chemoattractant protein–1 and macrophage inflammatory protein–1 alpha by inflammatory granuloma fibroblasts. Am J Pathol. 1994;144:711–718. [PMC free article] [PubMed] [Google Scholar]
- 32.Karpus WJ, Kennedy KJ, Smith WS, Miller SD. Inhibition of relapsing experimental autoimmune encephalomyelitis in SJL mice by feeding the immunodominant PLP139-151 peptide. J Neurosci Res. 1996;45:410–423. doi: 10.1002/(SICI)1097-4547(19960815)45:4<410::AID-JNR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Chensue SW, Warmington KS, Ruth JH, Sanghi PS, Lincoln P, Kunkel SL. Role of monocyte chemoattractant protein-1 (MCP-1) in Th1 (mycobacterial) and TH2 (schistosimal) antigen-induced granuloma formation: relationship to local inflammation. Th cell expression, and IL-12 production. J Immunol. 1996;157:4602–4608. [PubMed] [Google Scholar]
- 34.Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 35.Bitar DM, Whitacre CC. Suppression of experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. Cell Immunol. 1988;112:364–370. doi: 10.1016/0008-8749(88)90305-x. [DOI] [PubMed] [Google Scholar]
- 36.Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein and its fragments. J Immunol. 1988;140:440–445. [PubMed] [Google Scholar]
- 37.Meyer AL, Benson JM, Gienapp IE, Cox KL, Whitacre CC. Suppression of murine chronic relapsing experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. J Immunol. 1996;157:4230–4238. [PubMed] [Google Scholar]
- 38.Khare SD, Krco CJ, Griffiths MM, Luthra HS, David CS. Oral administration of an immunodominant human collagen peptide modulates collagen-induced arthritis. J Immunol. 1995;155:3653–3659. [PubMed] [Google Scholar]
- 39.Kennedy KJ, Smith WS, Miller SD, Karpus WJ. Induction of antigen-specific tolerance for the treatment of ongoing, relapsing autoimmune encephalomyelitis. A comparison between oral and peripheral tolerance. J Immunol. 1997;159:1036–1044. [PubMed] [Google Scholar]
- 40.Gruss H-J, Brach MA, Schumann RR, Herrmann F. Regulation of MCP-1/JE gene expression during monocytic differentiation. J Immunol. 1994;153:4907–4914. [PubMed] [Google Scholar]
- 41.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 42.Marth T, Strober W, Kelsall BL. High dose oral tolerance in ovalbumin TCR-transgenic mice: systemic neutralization of IL-12 augments TGF-β secretion and T cell apoptosis. J Immunol. 1996;157:2348–2357. [PubMed] [Google Scholar]
- 43.Garside P, Steel M, Worthey EA, Satoskar A, Alexander J, Bluethmann H, Liew FY, Mowat AM. T helper 2 cells are subject to high dose oral tolerance and are not essential for its induction. J Immunol. 1995;154:5649–5655. [PubMed] [Google Scholar]
- 44.Marinaro M, Boyaka PN, Finkelman FD, Kiyono H, Jackson RJ, Jirillo E, McGhee JR. Oral but not parenteral interleukin (IL)-12 redirects T helper 2 (Th2)-type responses to an oral vaccine without alternating mucosal IgA responses. J Exp Med. 1997;185:415–427. doi: 10.1084/jem.185.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci USA. 1994;91:3652–3656. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khoury SJ, Hancock WW, Weiner HL. Oral tolerance to myelin basic protein and natural recovery from experimental autoimmune encephalomyelitis are associated with downregulation of inflammatory cytokines and differential upregulation of transforming growth factor β, interleukin 4, and prostaglandin E expression in the brain. J Exp Med. 1992;176:1355–1364. doi: 10.1084/jem.176.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ernst CA, Zhang YJ, Hancock PR, Rutledge BJ, Corless CL, Rollins BJ. Biochemical and biologic characterization of murine monocytic chemoattractant protein-1. J Immunol. 1994;152:3541–3549. [PubMed] [Google Scholar]