

Abstract
Resuscitation from hemorrhagic shock induces profound changes in the physiologic processes of many tissues and activates inflammatory cascades that include the activation of stress transcriptional factors and upregulation of cytokine synthesis. This process is accompanied by acute organ damage (e.g., lungs and liver). We have previously demonstrated that the inducible nitric oxide synthase (iNOS) is expressed during hemorrhagic shock. We postulated that nitric oxide production from iNOS would participate in proinflammatory signaling. Using the iNOS inhibitor N6-(iminoethyl)-l-lysine or iNOS knockout mice we found that the activation of the transcriptional factors nuclear factor κB and signal transducer and activator of transcription 3 and increases in IL-6 and G-CSF messenger RNA levels in the lungs and livers measured 4 h after resuscitation from hemorrhagic shock were iNOS dependent. Furthermore, iNOS inhibition resulted in a marked reduction of lung and liver injury produced by hemorrhagic shock. Thus, induced nitric oxide is essential for the upregulation of the inflammatory response in resuscitated hemorrhagic shock and participates in end organ damage under these conditions.
Hemorrhagic shock initiates an inflammatory response characterized by the upregulation of cytokine expression (1) and accumulation of neutrophils (2) in a variety of tissues. These changes are prominent in the lungs and liver and are likely to contribute to end organ damage and resultant dysfunction after shock. The mechanisms by which hemorrhage triggers this inflammatory response remain poorly understood. Heightened adrenergic activity (3) and systemic release of proinflammatory agents from the gut (4, 5) have been hypothesized to contribute to acute lung injury after hemorrhage. In addition, reactive radicals are produced after ischemia/reperfusion and resuscitation from hemorrhagic shock, and have been implicated in a number of signal transduction pathways (6).
Among the important radicals produced during hemorrhagic shock is the bioregulatory molecule nitric oxide (NO)1 generated catalytically by three enzymes collectively termed NO synthases. We (7) and others (8) have shown that the inflammatory or inducible NO synthase (iNOS or NOS2) is upregulated in both the lungs and liver during shock. Therefore, this isoform may be capable of catalyzing the sustained production of NO after the tissue reperfusion associated with fluid resuscitation. NO can have both direct effects on cell signaling as well as indirect actions mediated by the reaction products formed when NO interacts with other molecules such as oxygen or superoxide (9). We hypothesized that enhanced NO production resulting from iNOS expression would contribute to proinflammatory signaling in hemorrhagic shock. Hemorrhagic shock experiments were therefore carried out in rats treated with the iNOS-selective inhibitor N 6-(iminoethyl)-l-lysine (L-NIL; reference 10) and mice genetically deficient in iNOS. We report here that iNOS inhibition or deficiency prevents the upregulation of the inflammatory cytokines IL-6 and granulocyte colony-stimulating factor (G-CSF), as well as the activation of key proinflammatory transcriptional factors nuclear factor (NF)-κB and signal transducer and activator of transcription (Stat)3, and reduced organ damage after hemorrhage. These data indicate that under conditions of redox stress such as hemorrhagic shock, induced NO production plays an essential role in the initiation of the inflammatory response.
Materials and Methods
Hemorrhagic Shock Protocol.
This study was approved by the University of Pittsburgh Institutional Review Board for animal experimentation and conforms to National Institutes of Health guidelines for the care and use of laboratory animals. Fasted male Sprague Dawley rats (270–290 g) were obtained from Charles River Breeding Laboratory (Cambridge, MA). iNOS-deficient mice were prepared as previously described (11). We have previously described in detail our rat model of resuscitated hemorrhagic shock (12). In brief, animals were anesthetized with methoxyfluorane, and then ventilated via endotracheal tubes using a 2.5-ml tidal volume of room air at 72 breaths/min. The right carotid artery was cannulated for continuous blood pressure monitoring, and the left jugular vein was cannulated for blood withdrawal and fluid administration. After an initial bleed of 2.25 ml/100 g body weight over 10 min, blood was withdrawn into a heparinized syringe or returned as needed to maintain a mean arterial blood pressure (MAP) of 40 mm Hg. At the point in time at which 35% of the shed blood had been returned (total shed blood volume 7.8 ± 0.5 ml), the animals were resuscitated to a MAP of 80 mm Hg by administration of the remaining shed blood plus two times the shed blood volume in lactated Ringers solution. Total shock time averaged 157 ± 2.3 min. Animals were killed 4 h after the initiation of resuscitation. Control animals underwent cannulation and anesthesia for an identical period of time as shock animals but were not bled. One group of animals (n = 6) received L-NIL (Alexis Corp., Laufelfingen, Switzerland) at 50 μg/kg/h, whereas the control group (both sham and shock animals) received saline infusion. L-NIL was dissolved in 1 ml of sterile saline fluid and was infused at the initiation of resuscitation for a period of 1 h.
The hemorrhagic shock protocol was modified as follows when performed on mice (11). The animals were anesthetized with methoxyfluorane. Both femoral arteries were surgically prepared and cannulated, one for continuous blood pressure monitoring, the contralateral artery for blood withdrawal or fluid administration. Animals were subjected to hemorrhagic shock by withdrawal of blood with a MAP maintained at 30 mm Hg for 3 h with continuous monitoring of blood pressure. Animals were resuscitated by infusion of the shed blood and intraperitoneal injection of 1 ml of saline. Animals were killed by exsanguination 4 h after resuscitation.
Hepatic Injury.
The release of the hepatocellular enzyme alanine aminotransferase (ALT) into plasma was used as an index of hepatic injury. Blood samples were collected into heparinized syringes at the end of observation period. The samples were centrifuged and the plasma was frozen at −70°C for subsequent analysis. ALT release was determined by an automated procedure using an autoanalyzer (RA 500; Technitron Inc., Tarrytown, NY).
Isolation of Organs and Cells.
After flushing the carcasses with cold (4°C) isotonic saline solution via the venous catheter, the lungs and livers were removed. Samples were immediately frozen in liquid nitrogen and stored at −80°C. Total cellular RNA was extracted from the samples using the method of Chomczyinski et al. (13). Cohort groups of rats (n = 5) were used for determination of lung wet to dry ratio and lung histology. After median sternotomy and preparation of the trachea, the left pulmonary hilus was isolated and ligated. The left lung was excised and removed for wet to dry ratio. The right lung was fixed by inflating with formaldehyde solution (4%) for histopathological examination. Tissue embedding and sectioning were performed using standard procedures. For histopathological examination, the lungs of animals were sectioned and stained with hematoxylin and eosin and for myeloperoxidase (MPO) as described (12). 10 randomly chosen fields of each lung specimen were examined at ×400 and blindly scored for number of intensely staining MPO-positive PMNs as described (12).
Reverse Transcriptase PCR Amplification.
Total RNA (2.5 μg) was subjected to first-strand cDNA synthesis using oligo (dT) primer and Moloney murine leukemia virus (MMLV) reverse transcriptase (14). Primers were designed to amplify rat G-CSF, IL-6, and iNOS with the assistance of a PCR primer design program (PCR Plan; Intelligenetics, Mountain View, CA). The primers used to amplify rat G-CSF cDNA were as described (12). The sequence of the IL-6 5′ primer was ACAGCGATGATGCACTGTCAG, corresponding to nucleotide numbers 297–317 bp of the rat IL-6 cDNA sequence (15). The sequence of the IL-6 3′ primer was ATGGTCTTGGTCCTTAGCCAC, corresponding to nucleotide numbers 633–613 bp. The sequence of the iNOS 5′ primer was TTGGGTCTTGTTAGCCTAGTC, corresponding to nucleotide numbers 114–134 bp of the rat iNOS cDNA sequence (16). The sequence of the iNOS 3′ primer was TGTGCAGTCCCAGTGAGGAAC, corresponding to nucleotide numbers 375–355 bp. The primers amplified products of 560 bp for G-CSF (12), 339 bp for IL-6, and 264 bp for iNOS. The identity of the amplified cDNA fragment obtained from reverse transcriptase PCR (RT-PCR) reaction with IL-6, G-CSF, and iNOS primers was confirmed using restriction site analysis as described (12). PCR conditions were as follows: denaturation at 94°C for 1 min, annealing at 57°C for 1 min, and polymerization at 72°C for 2 min. PCR reactions were performed in a Perkin Elmer 480 thermocycler (480; Perkin-Elmer Corp., Norwalk, CT) using different numbers of cycles to detect a linear range of input RNA. The optimal cycle number was identified as 30 cycles. Rat peritoneal macrophages elicited with thyoglycolate and RAW cells (RAW 264.7 macrophage cell line) stimulated in vitro with LPS served as positive controls for G-CSF, IL-6, and iNOS messenger RNA (mRNA). The negative control for each set of PCR reactions contained water instead of DNA template. PCR product (20% of the reaction volume) of qualitative RT-PCR was electrophorectically separated on a 2% agarose gel and stained with ethidium bromide.
For semiquantitative RT-PCR (17) γ-32P–endlabeled 5′ primer was used. 15 μl of the PCR reaction was separated on a 10% polyacrylamide gel. After gel drying and exposure to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA), the relative radioactivity of the bands was determined by volume integration using laser scanning densitometry. Each gel contained the same positive control, which permitted normalization of samples and comparison between gels.
Electrophoretic Mobility Shift Assay.
Electrophoretic mobility shift assay (EMSA) was performed using whole-tissue extracts of lung or liver sections from the experimental groups as described (18). Binding reactions were performed using 20 μg of extracted protein and radiolabeled DNA-binding elements. The activation of NF-κB was determined using the duplex oligonucleotide based on the NF-κB binding site upstream of the murine iNOS promoter (19). Activation of Stat3 was assessed using the high-affinity serum-inducible element (hSIE) duplex oligonucleotide that preferentially binds Stat3 and Stat1 (20). EMSA was performed on a 4% polyacrylamide gel as described (21). The level of transcription factor activation was quantitated using PhosphorImager analysis of gel shift band intensities. Where indicated, EMSA-binding reactions were incubated with NF-κB protein (p)50 or p65 antibodies or antibodies specific for Stat3α or Stat3β. Antibodies specific for NF-κB p50, NF-κB p65, and Stat3α were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and were generated in goats against the amino acids 350–363 of human NF-κB p50, in goats against the amino acids 531–550 of human NF-κB p65, or in rabbits against the COOH-terminal 20 of 21 amino acid residues of murine Stat3, respectively. Stat3β-specific antibody was generated at Charles River Pharmaservices (Southbridge, MA) by immunizing chickens with the COOH-terminal 10 amino acid residues of human Stat3β conjugated to thyroglobulin. The IgY fraction was purified from egg yolk by the company.
Statistics.
Unless otherwise indicated, data are presented as mean ± SEM. Comparisons of means were performed using analysis of variance (ANOVA) followed by comparison of individual pairs of means using the Scheffe test. Both tests are contained within the StatView program (4.1; Abacus Concepts, Inc., Berkeley, CA).
Results
iNOS Activity Is Required for the Upregulation of IL-6 and G-CSF mRNA Levels after Hemorrhagic Shock.
Hemorrhagi c shock followed by resuscitation results in the upregulation of proinflammatory cascades in the lung as well as in other organs (1). The inflammatory response includes the expression of iNOS during the shock phase after hemorrhage (7, 8). In these experiments, we found that lung iNOS mRNA levels were increased by 8.7-fold over sham-treated animals (P = 0.001) 4 h after resuscitation from hemorrhagic shock (Fig. 1). To determine if induced NO participates in the upregulation of cytokine expression after hemorrhagic shock, rats were subjected to resuscitated hemorrhagic shock without or with treatment with the iNOS inhibitor L-NIL. IL-6 and G-CSF mRNA levels have been previously shown to be consistently elevated in the lungs after resuscitation from hemorrhagic shock (18) where they may contribute to leukocyte recruitment and activation (22). The predominant site for G-CSF mRNA production is the bronchoepithelial cells lining the luminal side of distal bronchioles (12). 4 h after resuscitation from hemorrhagic shock, IL-6 and G-CSF mRNA were increased 3.4- and 2.5-fold, respectively, compared to sham animals (P = 0.03 and P = 0.04, respectively; Fig. 2). This upregulation was reduced 80 and 53%, respectively (P = 0.004 and P = 0.04, respectively) by administration of the iNOS inhibitor L-NIL. L-NIL alone had minimal effect on IL-6 or G-CSF mRNA levels in the sham animals (Fig. 2) and did not alter iNOS mRNA levels in the shocked animals (Fig. 1).
Figure 1.

Semiquantitative RT-PCR of iNOS mRNA from lungs of rats subjected to hemorrhagic shock and killed 4 h after resuscitation. RT-PCR reactions were performed using total RNA (2.5 μg) from the lungs of shock and sham animals receiving L-NIL (n = 6, gray bars) or saline (n = 6, black bars). Reaction products were separated on polyacrylamide gels. The gels were dried and exposed to a PhosphorImaging screen and developed using a PhosphorImager (A). In B, the radioactive signal in the region corresponding to the predicted amplified fragment of rat iNOS mRNA was quantitated using scanner laser densitometry and ImageQuant software and plotted. Values shown represent mean ± SEM. The differences between each shock and sham group were significant (P = 0.001 for each group).
Figure 2.


Semiquantitative RT-PCR of IL-6 mRNA (A and B) and G-CSF mRNA (C and D) in lungs of rats subjected to hemorrhagic shock or sham procedure without (black bars) and with (gray bars) L-NIL treatment. RT-PCR reactions were performed and analyzed as described in the legend to Fig. 1. The differences between each shock and sham group were significant (P ⩽0.03 for each group). The differences between L-NIL–treated shock and untreated shock groups were significant (P = 0.004 for IL-6 and P = 0.04 for G-CSF).
Induced NO Upregulates NF-κB and STAT Activation in Hemorrhagic Shock.
Activation of stress transcriptional factors such as NF-κB and STAT proteins contributes to the upregulation of cytokines such as IL-6 and G-CSF (23) and may also be a consequence of exposure of cells and tissues to these cytokines (24). Therefore, experiments were carried out to measure the activation of these transcriptional factors with and without iNOS inhibition. NF-κB activation was determined by EMSA using the duplex oligonucleotide based on a well-characterized NF-κB–binding sequence (19). Lungs of animals 4 h after resuscitation from shock demonstrated a 2.5-fold increase in NF-κB activation (P = 0.001) compared to sham controls (Fig. 3 A). After L-NIL treatment, levels of NF-κB activation decreased 70% in shocked animals (P = 0.003). To confirm the identity of the activated protein–DNA complex, binding assays were preincubated with a specific antibody against p50 or p65. The activated NF-κB complex was partially supershifted with p50 (Fig. 3 C), and reduced by 50% with antibody against p65 (data not shown) indicating that the complex was composed, in part, of both p50 and p65. Binding of the protein to labeled NF-κB–binding element was completely inhibited by 25-fold excess of NF-κB duplex oligonucleotide, but not by up to 100-fold excess of unlabeled hSIE duplex oligonucleotide (Fig. 3 D).
Figure 3.


Activation of NF-κB in the lungs of rats subjected to hemorrhagic shock. In A, EMSA was performed using radiolabeled NF-κB–binding element and 20 μg of protein extracts of shock and sham animals without or with L-NIL treatment. In B, the radioactive signal was quantitated by PhosphorImager analysis and plotted. The values shown are mean ± SEM. Black bars, untreated animals; gray bars, L-NIL–treated animals. The NF-κB complex in the shock groups was 2.5-fold greater than in sham controls (P = 0.001) and was reduced by 70% by L-NIL treatment (P = 0.003). In C, extracts (20 μg) from untreated shock animals were incubated with (+) or without (−) a specific antibody against the p50 portion of the NF-κB heterodimer. In D, extracts (20 μg) of the lung of a representative shock animal were incubated with the indicated fold excess of unlabeled duplex oligonucleotide or an unrelated duplex oligonucleotide (hSIE).
If iNOS-induced NF-κB activation contributes to the upregulation of cytokine expression (e.g., G-CSF), then NF-κB activity capable of binding the NF-κB–containing elements within the G-CSF promoter should be present and increased within the lungs of animals subjected to hemorrhagic shock. The promoter region of G-CSF contains a functional binding site for NF-κB (23) known as consensus decanucleotide 5′GRGRTTNCYY3′ (CK-1). EMSA using a labeled oligonucleotide based on the CK-1 sequence revealed a twofold increase of CK-1–binding activity (P = 0.03) over sham control animals in the lungs of animals subjected to hemorrhagic shock (Fig. 4, A and B). This binding activity was inhibited completely by 100-fold excess cold CK-1 duplex oligonucleotide and reduced substantially by 100-fold excess cold NF-κB duplex oligonucleotide. In contrast, binding activity was unaffected by 100-fold excess cold hSIE duplex oligonucleotide (Fig. 4 C).
Figure 4.

Increased CK-1–binding activity (BA) of lung extracts of shock animals. In A, EMSA was performed using 20 μg of extract of lung from animals subjected to hemorrhagic shock, sham animals, normal control animals, and duplex oligonucleotide. CK-1 is based on an element within the promoter region of the G-CSF gene that contains a functional NF-κB binding site. In B, CK-1 BA was quantitated by PhosphorImager analysis and the mean and ± SEM plotted. CK-1 BA in shock animals was increased twofold over sham animals (P = 0.03). In C, EMSA was performed using extracts of a representative shock lung and CK-1 duplex oligonucleotide in the presence of the indicated unlabeled duplex oligonucleotides at the indicated fold excess. The position of the CK-1–binding activity (CK-1, BA) is indicated on the left.
We have previously shown that hemorrhagic shock results in activation of STAT proteins in the lungs of rats (25). G-CSF and IL-6 are among the cytokines known to activate Stat3 after binding to their specific receptors (24). Therefore, EMSA for Stat3 was performed to determine whether the reduced cytokine expression seen with iNOS inhibition was accompanied by a reduction in Stat3 activation. The activation of Stat3 was determined in EMSA using the hSIE, a DNA-binding element that preferentially binds Stat3 and Stat1. Stat3 and Stat1 proteins bind hSIE as dimers to form three distinct serum-inducible factor (SIF) complexes, SIF-A (Stat3 homodimer), SIF-B (Stat1 and Stat3 heterodimer), and SIF-C (26). SIF-A predominates in protein extracts of hemorrhagic shock tissues (Fig. 5). After resuscitation from hemorrhagic shock, Stat3 activation (SIF-A complex) increased 4.7-fold (P = 0.002) compared to sham controls; this increase was almost completely inhibited by L-NIL treatment. Although G-CSF and IL-6 both induce activation of Stat3, each activates a distinct isoform of Stat3; IL-6 activates Stat3α (27), whereas extracts of G-CSF–stimulated PMN contained only Stat3γ, a short isoform of Stat3 derived from proteolytic cleavage of Stat3α (28, and Tweardy, D.J., unpublished data). A third isoform of Stat3, known as Stat3β (p83), is derived from alternative RNA splicing (21). Animals subjected to hemorrhagic shock demonstrated activation of all three isoforms as determined by supershift assays using antibodies specific for Stat3α and Stat3β (Fig. 5 B). Incubation with the Stat3α- or Stat3β-specific antibody resulted in supershift of Stat3α and Stat3β, respectively; however, incubation with both antibodies did not completely supershift the SIF-A complex. The nonsupershifted portion demonstrated mobility similar to Stat3γ. DNA-affinity purification of whole- tissue extracts of hemorrhagic shock lungs followed by immunoblotting with Stat3-specific monoclonal antibody that recognizes all Stat3 isoforms confirmed the activation of all three Stat3 isoforms including Stat3γ (data not shown). After L-NIL treatment, a marked reduction in the activation of all three Stat3 isoforms was observed (Fig. 5 A). Reduction in the Stat3γ isoform was consistent with reduced PMN infiltration into the lung as seen in histological assessment (see below). These data provide indirect evidence for reduced cytokine action within the tissue that is subjected to downregulation of cytokine production by the inhibition of iNOS.
Figure 5.

Increased Stat3 activation in extracts of lungs of animals subjected to hemorrhagic shock and resuscitation. In A, EMSA was performed using the hSIE duplex oligonucleotide and 20 μg of extracts from shock animals (hemorrhagic shock, HS) or sham animals treated or untreated with L-NIL. The position of the SIF-A, -B, and -C complex are indicated. In B, the SIF-A band was quantitated by PhosphorImager analysis and the mean ± SEM for each group plotted. The mean of the SIF-A complexes in the shock group was 4.7-fold greater than in sham controls (P = 0.002) and was significantly reduced by L-NIL treatment (P = 0.04). In C, extracts of a representative lung were incubated with antibodies specific for Stat3α, Stat3β, or with both antibodies. The position of the SIF-A, -B, and -C complexes and the residual SIF-A complex after supershift of Stat3α and Stat3β (Stat3γ) are indicated.
iNOS Inhibition Reduces Lung Injury in Hemorrhagic Shock.
One consequence of the upregulation of inflammatory cascades after hemorrhagic shock is organ damage manifested by accumulation of PMNs and interstitial fluid. In this study we examined whether or not the reduction in cytokine production and proinflammatory signaling in L-NIL–treated rats was associated with a decrease in lung injury. Lung injury was assessed by histology as well as wet to dry ratio. After hemorrhagic shock, lung PMN infiltration increased by 4.7-fold (P = 0.001) at 4 h resuscitation, as determined by MPO staining (Fig. 6). L-NIL treatment resulted in a 44% decrease in PMN accumulation (P = 0.02). The wet to dry ratio increased by 24% in untreated shocked animals compared to sham animals (P = 0.01, Fig. 6 C). This was accompanied by the histologic markers of tissue fluid leak, including widened interstitium due to interstitial edema (Fig. 6 A). The increase in lung wet to dry ratio comparing shock and sham animals was inhibited by L-NIL treatment by 33% (P = 0.03), which was accompanied by an obvious reduction in the histologic appearance of interstitial fluid accumulation (Fig. 6 A).
Figure 6.
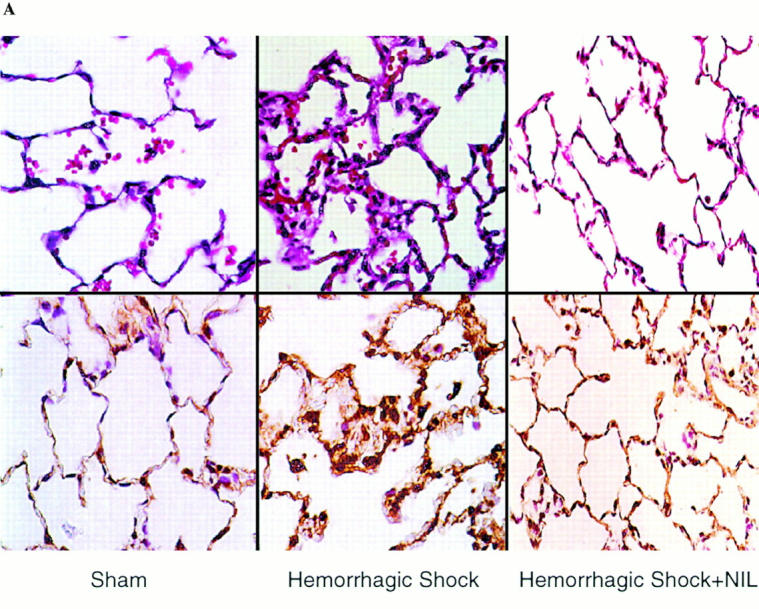


Lung injury is attenuated after L-NIL treatment of shock animals. The lungs of sham animals, untreated shock animals, and L-NIL–treated shock animals obtained 4 h after resuscitation were inflated and fixed in formaldehyde, and then sectioned, and examined at ×400. Lung injury was assessed histologically using sections stained by hematoxylin and eosin (top), and PMN accumulation was assessed by staining for MPO (bottom). In B, 10 random fields of each MPO-stained lung specimen were blindly scored for number of intensely MPO-positive PMNs. The scores were pooled for untreated animals (black bars) and L-NIL-treated animals (gray bars) and the means ± SEM were plotted. The increase in shock animals compared to sham animals was significant (P = 0.001); the decrease in shock animals treated with L-NIL compared to untreated animals was significant (P = 0.02). In C, the wet to dry ratio of each sham and shock animal was corrected by subtracting the mean value for normal animals (4.15 ± 0.3, n = 6) and the increased mean ± SEM of animals untreated (black bars) or L-NIL–treated (gray bars) was plotted. The increase in wet to dry ratio in shock animals compared to sham animals was significant (P = 0.01). The decrease in wet to dry ratio in L-NIL–treated shock animals compared to untreated shock animals was significant (P = 0.03).
iNOS Inhibition Prevents Cytokine Expression and Transcriptional Factor Activation in the Liver.
Hemorrhagic shock with resuscitation is commonly viewed as a whole body ischemia/reperfusion insult. Although the degree of ischemia may be variable in the pulmonary circulation, the splanchnic viscera experience a marked reduction in blood flow during circulatory shock and hypovolemia. To examine an organ subjected to severe ischemia/reperfusion during hemorrhagic shock and resuscitation as well as to establish whether similar proinflammatory effects of L-NIL treatment occurred outside of the lungs, we examined cytokine expression and transcriptional factor upregulation in the liver. Animals subjected to hemorrhagic shock and resuscitation demonstrated a 6.7-fold increase in IL-6 mRNA levels (P = 0.002), and a 3.3-fold increase in G-CSF mRNA levels (P = 0.003) compared to sham animals (Fig. 7). By contrast, L-NIL treatment reduced mRNA levels for IL-6 by 56% (P = 0.01) and for G-CSF by 51% (P = 0.02) when compared to untreated hemorrhaged animals. In shocked animals, the activation of NF-κB and the activation of Stat3 homodimer (SIF-A) were increased by 2.8- and 4.3-fold, respectively, in the liver compared to sham animals (P = 0.002 and P = 0.001, respectively; Fig. 8). After L-NIL treatment, shocked animals demonstrated a marked reduction in the activation of both stress transcriptional factors. NF-κB activation was inhibited by 83% (P = 0.001) compared to the untreated shock group, whereas activated Stat3 was reduced by 58% (P = 0.001). In addition, we found that liver damage measured by plasma transaminase levels was significantly reduced in the NIL-treated rats subjected to hemorrhagic shock (J. Menezes, unpublished observations). Thus, the response of the liver after hemorrhagic shock was comparable to that observed in the lung and was mediated, in part, by induced NO.
Figure 7.


Semiquantitative RT-PCR of IL-6 mRNA (A and B) and G-CSF mRNA (C and D) in livers of rats subjected to hemorrhagic shock or sham procedure without (black bars) and with (gray bars) L-NIL treatment. RT-PCR reactions were performed and analyzed as described in the legend to Fig. 1. The differences between each shock and sham group were significant (P <0.01 for each group). IL-6 mRNA levels were decreased by 56% (P = 0.01), whereas G-CSF mRNA levels decreased by 51% (P = 0.02) in L-NIL–treated shock animals compared to untreated shock animals.
Figure 8.


Activation of NF-κB and Stat3 in the liver of rats subjected to hemorrhagic shock. In A, EMSA was performed using radiolabeled NF-κB duplex oligonucleotide (A) or radiolabeled hSIE (C) and 20 μg of protein extracts of shock and sham animals without or with L-NIL treatment. The positions of NF-κB and the SIF-A, -B, and -C complexes are indicated on the left. In B and D, the radioactive signal was quantitated by PhosphorImager analysis and the mean ± SEM plotted. The values shown are mean ± SEM. Black bars, untreated animals (n = 5); gray bars, L-NIL– treated animals. In shocked animals, the activation of NF-κB and the activation of Stat3 were increased significantly compared to sham animals (P = 0.002 and P = 0.001, respectively). after L-NIL treatment NF-κB activation was reduced 83% (P = 0.001) and activation of Stat3 was reduced 58% (P = 0.001) in shocked animals compared to untreated shock animals.
Studies Using iNOS Knockout Mice.
Although L-NIL is a selective iNOS inhibitor, it may have some activity on other NO synthase isoforms (10) or may have other nonspecific effects. Therefore, to provide confirmation that the observed effects with L-NIL were due to the inhibition of the inducible form of NO synthase, we measured the activation of NF-κB and Stat3 in the lungs and the liver of iNOS-deficient and wild-type mice 4 h after hemorrhagic shock. Similar to the results seen in rats, hemorrhagic shock followed by resuscitation resulted in NF-κB and Stat3 activation in the lung (Fig. 9) and liver (Fig. 10) compared to sham-treated mice. In mice lacking the iNOS gene, the activation of NF-κB and Stat3 was reduced by 44% (P = 0.01) and 51% (P = 0.04), respectively, in the lung, and by 62% (P = 0.02) and 54% (P = 0.01), respectively, in the liver, when compared to wild-type animals. Similar to L-NIL treatment in rats, mice lacking the iNOS gene exhibited less liver damage when subjected to resuscitated shock than wild-type animals (Fig. 11).
Figure 9.


NF-κB and Stat3 activation in the lungs of wild-type (WT) and iNOS knockout (KO) mice subjected to hemorrhagic shock. EMSA was performed using radiolabeled NF-κB duplex oligonucleotide (A) or radiolabeled hSIE (C) and 20 μg of protein extracts of shock and sham wild-type or knockout mice. The position of NF-κB and the SIF-A, -B, and -C complexes are indicated on the left. In B and D, the radioactive signal was quantitated by PhosphorImager analysis and plotted. The values shown are mean ± SEM. Black bars, wild-type mice (n = 5); gray bars, knockout mice (n = 5). In shocked wild-type mice, the activation of NF-κB and the activation of Stat3 homodimer (SIFA) were increased significantly compared to sham wild-type mice (P ⩽0.01). In shocked knockout mice, NF-κB activation was reduced 44% (P = 0.01) and activation of Stat3 homodimer (SIF-A) was reduced 51% (P = 0.04) compared to shocked wild-type mice.
Figure 10.


NF-κB and Stat3 activation in the liver of wild-type (WT) and iNOS knockout (KO) mice subjected to hemorrhagic shock. EMSA was performed using radiolabeled NF-κB duplex oligonucleotide (A) or radiolabeled hSIE (C) and 20 μg of protein extracts of shock and sham wild-type or knockout mice. The position of NF-κB and the SIF-A, -B, and -C complexes are indicated on the left. In B and D, the radioactive signal was quantitated by PhosphorImager analysis and plotted. The values shown are mean ± SEM. Black bars, wild-type mice (n = 5); gray bars, knockout mice (n = 5). In shocked wild-type mice, the activation of NF-κB and the activation of Stat3 homodimer (SIFA) were increased significantly compared to sham wild-type mice (P ⩽0.01). In shocked knockout mice NF-κB activation was reduced 62% (P = 0.02) and activation of Stat3 homodimer (SIFA) was reduced 54% (P = 0.01) compared to shocked wild-type mice.
Figure 11.

Hepatic injury in wild-type (WT) and iNOS knockout (KO) mice subjected to hemorrhagic shock. The release of the hepatocellular enzyme ALT into plasma was used as an index of hepatic injury. ALT release (IU/liter) from shock and sham wild-type or knockout mice was measured using an autoanalyzer (RA 500; Technitron) and plotted. Values shown are mean ± SEM. Black bars, wild-type (WT) animals (n = 5); gray bars, knockout (KO) animals (n = 5). In shocked wild-type mice, ALT levels demonstrated a 40-fold increase compared to sham wild-type mice (P <0.01). In shocked knockout mice, ALT levels were reduced 80% (P <0.01) compared to shocked wild-type mice.
Discussion
This study was undertaken to determine to what degree induced NO contributes to the inflammatory response and subsequent end organ damage after resuscitation from hemorrhagic shock. We have extended previous studies (7, 8) by showing that iNOS is not only upregulated during shock, but that it remains elevated after resuscitation. Using either the iNOS inhibitor L-NIL or iNOS knockout mice, we demonstrate that iNOS contributes to the induced expression of the cytokines IL-6 and G-CSF in the lung and liver after hemorrhagic shock. The iNOS-dependent increase in IL-6 and G-CSF mRNA levels is associated with an iNOS-dependent increase in NF-κB and Stat3 activation in these tissues. An association between the upregulation of these proinflammatory events by NO and organ injury is shown by our experiments, demonstrating reduced PMN accumulation and edema formation in the lungs, and reduced plasma levels of liver enzymes with iNOS suppression. These data provide compelling evidence, not only that iNOS is in part responsible for lung and liver damage after hemorrhage and resuscitation, but that the induction of NO synthesis is a key event in the subsequent activation of inflammatory cascades after resuscitation.
During hemorrhagic shock, iNOS is upregulated in several sites including the lung and liver (8). The mechanism of upregulation is unclear, but could include hypoxia (6) or the action of cytokines. We have shown that iNOS expression increases in parallel with the duration of shock (7). Others have suggested that iNOS contributes to the initial vascular decompensation in hemorrhagic shock (8, 29) and we have suggested that the iNOS expression pattern is consistent with the possibility that iNOS may contribute to the progressive vascular dysfunction seen with sustained shock (7). The current data support the idea that NO can increase cytokine expression through the activation of NF-κB, and that the activation of Stat3 may be the result of local cytokine expression (24, 30). Indirect evidence for this possibility is provided by the demonstration that NF-κB binding specific for the CK-1 element in the G-CSF promoter is present in hemorrhagic shock tissues. Furthermore, activation of Stat3 isoforms characteristic of IL-6 and G-CSF stimulation were identified as part of the activated Stat3 complex. Full resuscitation of the animals requires 20 min. However, as early as 1 h after the initiation of resuscitation, we found that the levels of IL-6 and G-CSF mRNA, as well as NF-κB and Stat3 activation in lungs and livers, were elevated to levels similar to those observed at 4 h (data not shown). Thus, it is likely that the NO-mediated signaling events that are initiated in early phases of resuscitation result in the rapid activation of downstream cascades. Although NO produced by the constitutive NO synthase has well-documented signaling functions in many systems, our novel observations provide strong evidence that induced NO also participates in lung cell signaling events in inflammation. That iNOS regulation of inflammatory gene expression is perhaps a more generalized phenomenon is supported by a recent observation that the upregulation of interferon γ and the response to IL-12 after Leishmania major infection is iNOS dependent (31, and Bogdan, C., personal communication). Our results, however, do not exclude the possibility that the observed differences in both the L-NIL– treated rats and the iNOS knockout mice are due to some degree on changes in organ perfusion and oxygen delivery resulting from reduced NO availability.
NO is known to act as a signaling molecule in other circumstances either by activation of soluble guanylyl cyclase resulting in elevated cyclic guanosine 3′,5′ monophosphate (cGMP; reference 32) or through S-nitrosylation of proteins containing cysteine residues (33). Significant differences in cytokine mRNA levels (14, 18) and transcriptional factor activation (25) between the shock and sham groups was seen only after resuscitation, indicating that reperfusion was required. This suggests that redox-sensitive mechanisms were responsible for the NO-mediated signaling. Lander et al. (6, 34) have shown that NO activates p21ras through S-nitrosylation and that this occurs more efficiently in human T cells subjected to oxidative stress. Downstream events include p38 kinase activation (35) and NF-κB activation. In hemorrhagic shock, tissues are subjected to redox stress by hypoxia and oxygen radical production making S-nitrosylation of p21ras a reasonable candidate mechanism. A recent report demonstrates a role for increased phosphatidic acid in the signaling cascade involved in macrophage cytokine synthesis after hemorrhagic shock in mice (36). A relationship between phosphatidic acid and NO is not apparent at this time.
We have previously shown that nonspecific NO synthase inhibition increases organ injury in hemorrhagic shock (37), whereas here we show that the suppression of inflammation associated with selective iNOS inhibition results in a decrease in lung and liver injury. Taken together, the findings suggest that constitutive NOS is protective, but that the quantities of NO generated by iNOS cause injury. Our results suggest that organ damage in hemorrhagic shock is due, at least in part, to the proinflammatory action of NO. NO in combination with superoxide forms peroxynitrite, which could exert direct tissue toxicity. The results in hemorrhagic shock are contrasted by the observations in endotoxemia where the role of iNOS is less clear. iNOS knockout mice treated with high-dose endotoxin showed no difference in end organ damage when compared with their wild-type counterparts (11). More recently, we have shown that iNOS inhibition during endotoxemia increases apoptosis in the liver without necrosis (38). Difference between endotoxemia and hemorrhagic shock may be related to the higher levels of superoxide production and greater oxidant stress in the latter.
Approaches to remove NO in hemorrhagic shock could have therapeutic benefit. Patients suffering severe or sustained hemorrhagic shock after trauma or due to other causes of bleeding (e.g., ruptured aortic aneurysm) can develop organ injury and dysfunction. Our results indicate that selective inhibition of iNOS may be a reasonable approach. Alternatively, NO scavengers may also preserve adequate levels of NO needed to maintain perfusion while removing the excess NO which promotes inflammation and tissue injury.
Acknowledgments
Supported by National Institutes of Health grants GM-53789, GM-44100, GM-53921, and CA-72261, and by Deutsche Forschungsgemeinschaft (DFG) grant HI 614/1-1.
Abbreviations used in this paper
- ALT
alanine aminotransferase
- EMSA
electrophoretic mobility shift assay
- G-CSF
granulocyte colony-stimulating factor
- hSIE
high-affinity serum-inducible element
- iNOS
inducible NO synthase
- L-NIL
N6-(iminoethyl)-l-lysine
- MAP
mean arterial blood pressure
- MPO
myeloperoxidase
- mRNA
messenger RNA
- NF
nuclear factor
- NO
nitric oxide
- p
protein
- RT-PCR
reverse transcriptase PCR
- SIF
serum-inducible factor
- stat
signal transducer and activator of transcription
Footnotes
The present address of J. MacMicking is Laboratory of Immunology, HHMI, The Rockefeller University, 1230 York Ave., New York 10021.
References
- 1.Chaudry, I.H., W. Ertel, and A. Ayala. 1993. Alterations in inflammatory cytokine production following hemorrhage and resuscitation. In Shock, Sepsis, and Organ Failure, Third Wiggers Bernard Conference. G. Schlag, H. Redl, and D.L. Traber, editors. Springer Verlag, Berlin. 73–127.
- 2.Cotran, R.S., V. Kumar, and S.L. Robbins, editors. 1989. Robbins Pathologic Basis of Disease. 4th ed. W.B. Saunders Company, Philadelphia. 39–71.
- 3.Le Tulzo Y, Shenkar R, Kaneko D, Moine P, Fantuzzi G, Dinarello CA, Abraham E. Hemorrhage increases cytokine expression in lung mononuclear cells in mice: involvement of catecholamines in nuclear factor–κB regulation and cytokine expression. J Clin Invest. 1997;99:1516–1524. doi: 10.1172/JCI119314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deitch EA, Morrison J, Berg R, Specian RD. Effect of hemorrhagic shock on bacterial translocation, intestinal morphology, and intestinal permeability in conventional and antibiotic-decontaminated rats. Crit Care Med. 1990;18:529–536. doi: 10.1097/00003246-199005000-00014. [DOI] [PubMed] [Google Scholar]
- 5.Peitzman AB, Udekwu AO, Ochoa J, Smith S. Bacterial translocation in trauma patients. J Trauma. 1991;31:1083–1087. [PubMed] [Google Scholar]
- 6.Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- 7.Kelly E, Shah NS, Morgan NN, Watkins SC, Peitzman AB, Billiar TR. Physiologic and molecular characterization of the role of nitric oxide in hemorrhagic shock: evidence that type II nitric oxide synthase does not regulate vascular decompensation. Shock. 1997;7:157–163. doi: 10.1097/00024382-199703000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Thiemermann C, Szabo C, Mitchell JA, Vane JR. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Nat Acad Sci USA. 1993;90:267–271. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szabo C. The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock. 1996;6:79–88. doi: 10.1097/00024382-199608000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, Currie MG. l-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J Med Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- 11.MacMicking, J.D., C. Nathan, G. Hom, N. Chartrain, D.S. Fletcher, M. Trumbauer, K. Stevens, Q.W. Xie, K. Sokol, N. Hutchinson, et al. 1995. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 81:641–650. (See published erratum 81: 1170.) [DOI] [PubMed]
- 12.Hierholzer C, Kelly E, Tsukada K, Loeffert E, Watkins S, Billiar T, Tweardy D. Hemorrhagic shock induces granulocyte colony-stimulating factor (G-CSF) expression in bronchial epithelium. Am J Physiol. 1997;273:L1058–L1064. doi: 10.1152/ajplung.1997.273.5.L1058. [DOI] [PubMed] [Google Scholar]
- 13.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 14.Hierholzer C, Kelly E, Billiar TR, Tweardy DJ. Granulocyte colony-stimulating factor (G-CSF) production in hemorrhagic shock requires both the ischemic and resuscitation phase. Arch Orth Trauma Surg. 1997;116:173–176. doi: 10.1007/BF00426067. [DOI] [PubMed] [Google Scholar]
- 15.Northemann W, Braciak TA, Hattori M, Lee F, Fey GH. Structure of the rat interleukin 6 gene and its expression in macrophage-derived cells. J Biol Chem. 1989;264:16072–16082. [PubMed] [Google Scholar]
- 16.Adachi H, Iida S, Oguchi S, Ohshima H, Suzuki H, Nagasaki K, Kawasaki H, Sugimura T, Esumi H. Molecular cloning of a cDNA encoding an inducible calmodulin-dependent nitric-oxide synthase from rat liver and its expression in COS 1 cells. Eur J Biochem. 1993;217:37–43. doi: 10.1111/j.1432-1033.1993.tb18215.x. [DOI] [PubMed] [Google Scholar]
- 17.Kawasaki, E.S. 1990. Amplification of RNA. In PCR Protocols: A Guide to Methods and Applications. M.A. Innis, D.H. Gelfang, J.J. Sninsky, and T.J. White, editors. Academic Press, New York. 22–27.
- 18.Hierholzer, C., J. Kalff, Y. Kim, T. Billiar, and D. Tweardy. 1997. G-CSF and IL-6 are produced in the lung in hemorrhagic shock where they contribute to the recruitment and activation of neutrophils. In 4th International Congress on The Immune Consequences of Trauma, Shock, and Sepsis. Mechanisms and Therapeutic Approaches. E. Faist, editor. Monduzzi, Bologna. 595–598.
- 19.de Vera ME, Geller DA, Billiar TR. Hepatic inducible nitric oxide synthase: regulation and function. Biochem Soc Trans. 1995;23:1008–1013. doi: 10.1042/bst0231008. [DOI] [PubMed] [Google Scholar]
- 20.Wagner BJ, Hayes TE, Hoban CJ, Cochran BH. The SIF binding element confers sis/PDGF inducibility onto the c-fos promoter. EMBO (Eur Mol Biol Organ) J. 1990;9:4477–4484. doi: 10.1002/j.1460-2075.1990.tb07898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakraborty A, White SM, Schaefer TS, Ball ED, Dyer KF, Tweardy DJ. Granulocyte colony-stimulating factor activation of Stat3-alpha and Stat3-beta in immature normal and leukemic human myeloid cells. Blood. 1996;88:2442–2449. [PubMed] [Google Scholar]
- 22.Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 23.Dunn SM, Coles LS, Lang RK, Gerondakis S, Vadas MA, Shannon MF. Requirement for nuclear factor (NF)-kappa B p65 and NF-interleukin-6 binding elements in the tumor necrosis factor response region of the granulocyte colony-stimulating factor promoter. Blood. 1994;83:2469–2479. [PubMed] [Google Scholar]
- 24.Taniguchi T. Cytokine signaling through nonreceptor protein tyrosine kinases. Science. 1995;268:251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- 25.Hierholzer, C., J. Kalff, T. Billiar, and D. Tweardy. 1998. Activation of STAT proteins in the lung of rats following resuscitation from hemorrhagic shock. Arch. Orth. Trauma Surg. In press. [DOI] [PubMed]
- 26.Sadowski HB, Shuai K, Darnell JE, Jr, Gilman MZ. A common nuclear signal transduction pathway activated by growth factor and cytokine receptors. Science. 1993;261:1739–1744. doi: 10.1126/science.8397445. [DOI] [PubMed] [Google Scholar]
- 27.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 28.Tweardy DJ, Wright TM, Ziegler SF, Baumann H, Chakraborty A, White SM, Dyer KF, Rubin KA. Granulocyte colony-stimulating factor rapidly activates a distinct STAT-like protein in normal myeloid cells. Blood. 1995;86:4409–4416. [PubMed] [Google Scholar]
- 29.Szabo C, Thiemermann C. Invited opinion: role of nitric oxide in hemorrhagic, traumatic, and anaphylactic shock and thermal injury. Shock. 1994;2:145–155. [PubMed] [Google Scholar]
- 30.Cressman DE, Greenbaum LE, Deangelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6–deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 31.Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, Röllinghoff M, Gresser I, Bogdan C. Type 1 interferon (IFN-α/β) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity. 1998;8:77–87. doi: 10.1016/s1074-7613(00)80460-4. [DOI] [PubMed] [Google Scholar]
- 32.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 33.Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 34.Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD, Kraemer RT, Mirza UA, Chait BT, Burk SC, Quilliam LA. Redox regulation of cell signalling. Nature. 1996;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 35.Lander HM, Jacovina AT, Davis RJ, Tauras JM. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem. 1996;271:19705–19709. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- 36.Abraham E, Bursten S, Shenkar R, Allbee J, Tuder R, Woodson P, Guidot DM, Rice G, Singer JW, Repine JE. Phosphatidic acid signaling mediates lung cytokine expression and lung inflammatory injury after hemorrhage in mice. J Exp Med. 1995;181:569–575. doi: 10.1084/jem.181.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harbrecht BG, Wu B, Watkins SC, Marshall HP, Jr, Peitzman AB, Billiar TR. Inhibition of nitric oxide synthase during hemorrhagic shock increases hepatic injury. Shock. 1995;4:332–337. doi: 10.1097/00024382-199511000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Ou, J., T.M. Carlos, S.C. Watkins, J.E. Saavedra, L.K. Keefer, Y.M. Kim, B.G. Harbrecht, and T.R. Billiar. 1997. Differential effects of nonselective nitric oxide synthase (NOS) and selective inducible NOS inhibition on hepatic necrosis, apoptosis, ICAM-1 expression, and PMN accumulation during endotoxemia. NO: Biol. Chem. 1:404–416. [DOI] [PubMed]