

Abstract
Expression of the T cell antigen receptor (TCR) on the surface of thymocytes and mature T cells is dependent on the assembly of receptor subunits into TCRs in the endoplasmic reticulum (ER) and their successful traversal of the secretory pathway to the plasma membrane. TCR subunits that fail to exit the ER for the Golgi complex are degraded by nonlysosomal processes that have been referred to as “ER degradation”. The molecular basis for the loss of the TCR CD3-δ and TCR-α subunits from the ER was investigated in lymphocytes. For CD3-δ, we describe a process leading to its degradation that includes trimming of mannose residues from asparagine-linked (N-linked) oligosaccharides, generation of ubiquitinated membrane-bound intermediates, and proteasome-dependent removal from the ER membrane. When either mannosidase activity or the catalytic activity of proteasomes was inhibited, loss of CD3-δ was markedly curtailed and CD3-δ remained membrane bound in a complex with CD3-ε. TCR-α was also found to be degraded in a proteasome-dependent manner with ubiquitinated intermediates. However, no evidence of a role for mannosidases was found for TCR-α, and significant retrograde movement through the ER membrane took place even when proteasome function was inhibited. These findings provide new insights into mechanisms employed to regulate levels of TCRs, and underscore that cells use multiple mechanisms to target proteins from the ER to the cytosol for degradation.
The multi-subunit TCR is comprised of six distinct type I transmembrane polypeptides that assemble in the endoplasmic reticulum (ER)1 as an octameric complex. On most T cells, these receptor subunits consist of clonotypic TCR-α/β heterodimers in association with a set of invariant subunits including heterodimers of CD3-δ and -ε, and CD3-γ and -ε, and a TCR-ζ homodimer (1). In T cells, cell surface expression of TCRs is dependent on the proper assembly of complete TCRs in the ER, which then traverse the secretory pathway to arrive at the plasma membrane (1). Partial receptors lacking only ζ homodimers also assemble and leave the ER. However, these are largely degraded in lysosomes (2). Other partial receptors and unassembled subunits, except for ζ, are retained in the ER from where they are degraded with varying efficiencies by poorly understood mechanisms that have been referred to as “ER degradation”(3, 4). ER degradation is believed to play a particularly prominent role in immature (CD4+ CD8+) thymocytes undergoing selection in the thymus. These cells express only 10% of the number of cell surface TCRs as mature thymocytes despite adequate synthesis of all TCR subunits. This low TCR expression occurs as a consequence of as yet undefined posttranslational mechanisms that include a relative block in egress of TCRs from the ER and increased degradation of TCR components from this organelle (5, 6).
Among the TCR subunits, TCR-α and CD3-δ are particularly susceptible to degradation from the ER, whereas CD3-γ and CD3-ε generally exhibit considerably longer half-lives (3, 4, 7). The molecular basis for the selectivity of targeting for degradation among receptor subunits is largely unknown; however, it has been shown that TCR-α is uniquely unstable as a transmembrane protein due to the presence of two basic amino acids within its transmembrane domain (8, 9).
The modification of proteins with chains of ubiquitin is well-established as an important and regulated means of disposing of cytosolic and nuclear proteins by targeting for degradation in 26S proteasomes (10, 11). Recently, however, there have been several reports implicating proteasomes in the degradation of transmembrane and soluble yeast (12–14) and mammalian (15–21) proteins that were initially cotranslationally inserted into the ER. A well-studied example is that of MHC class I heavy chains, which, like TCR subunits, are type 1 transmembrane proteins with a single transmembrane domain. Newly synthesized MHC class I heavy chains undergo rapid proteasomal degradation in cells that express certain human cytomegalovirus gene products, are defective in peptide transport into the ER, or lack expression of β2 microglobulin (19–21). These MHC molecules are dislocated from the ER membrane to the cytosol with the concomitant total removal of N-linked oligosaccharides by a cytosolic N-glycanase activity. Notably, this removal from the ER membrane occurs independently of the catalytic activity of proteasomes. These dislocated proteins are then degraded in the cytosol in a proteasome-dependent manner, without evidence of ubiquitinated intermediates. Based on these observations, as well as others involving a mutant form of a soluble luminal yeast protein (13), models have been proposed for the degradation of ER proteins in which the proteins are first dislocated into the cytosol from whence they are degraded by proteasomes (20, 22–24).
ER forms of the multi-membrane spanning cystic fibrosis conductance regulator (CFTR) are also degraded by proteasomes, in this case with the generation of ubiquitinated intermediates (15, 16). It has been speculated that misfolded forms of this complex protein are recognized as such by enzymes of the ubiquitin-conjugating system and are therefore targeted for destruction. However, no information is available as to the subcellular location of ubiquitinated CFTRs, nor is it known whether CFTRs are dislocated from ER membranes before ubiquitination.
In this study, we have evaluated the degradation of murine CD3-δ and TCR-α from the ER in T lymphocytes, which continually synthesize and degrade these subunits (3, 4, 7). CD3-δ has a core molecular weight of 16 kD; the addition of three N-linked oligosaccharide chains results in its migration at ∼26 kD by SDS-PAGE (25). This protein has a luminal domain of ∼79 amino acids and an intracytoplasmic domain of 46 amino acids that includes three lysines. The TCR-α subunit has four sites of N-glycosylation and, in most T cells, forms disulfide-linked heterodimers with the TCR-β subunit after its cotranslational membrane insertion. TCR-α has a small cytoplasmic domain of five amino acids, none of which are lysines. As previously mentioned, the transmembrane domain of TCR-α includes two basic amino acids (Arg and Lys) that may destabilize this molecule in the ER, perhaps functioning as an inefficient stop-transfer signal (8, 9). We now report that both CD3-δ and TCR-α are ubiquitinated and are degraded from the ER in a proteasome-dependent manner and that for CD3-δ, removal from the ER requires the catalytic activity of proteasomes and the activity of mannosidases.
Materials and Methods
Cells and Reagents.
Thymi from C57Bl/6 ×DBA/2 mice that had been interbred for 20 generations were provided by Astrid Eder (National Cancer Institute, Bethesda, MD). 21.2.2 cells (26) and BW5147 cells (27) were maintained in complete medium as previously described (26). COS-7 cells were maintained and transfected as previously described (28). Anti-CD3-δ (R9; reference 29), affinity-purified anti-ubiquitin (anti-Ub; reference 30), 2C11 (31), #125 (7), and H28-710 (32) have all been previously described. Reagents included the following: Z-Leu-Leu-Leu-CHO (MG132; Proscript, Cambridge, MA), 50 μM; N-acetyl-Leu-Leu-norleucinal (LLnL) and N-Acetyl-Leu-Leu-methioninal (LLM; both from Boehringer Mannheim, Indianapolis, IN), 100 μM; lactacystin (LCN), 50 μM; NH4Cl, 20 mM; cycloheximide (CHX), 100 μM; deoxymannojirimycin (dMNJ; Sigma Chemical Co., St. Louis, MO), 1 mM (see Fig. 4, E only) or 2 mM; and deoxynojirimycin (dNJ; Sigma Chemical Co.), 2 mM. Cells were preincubated with DMNJ or DNJ for 2 h at 37°C before the initiation of experiments.
Figure 4.

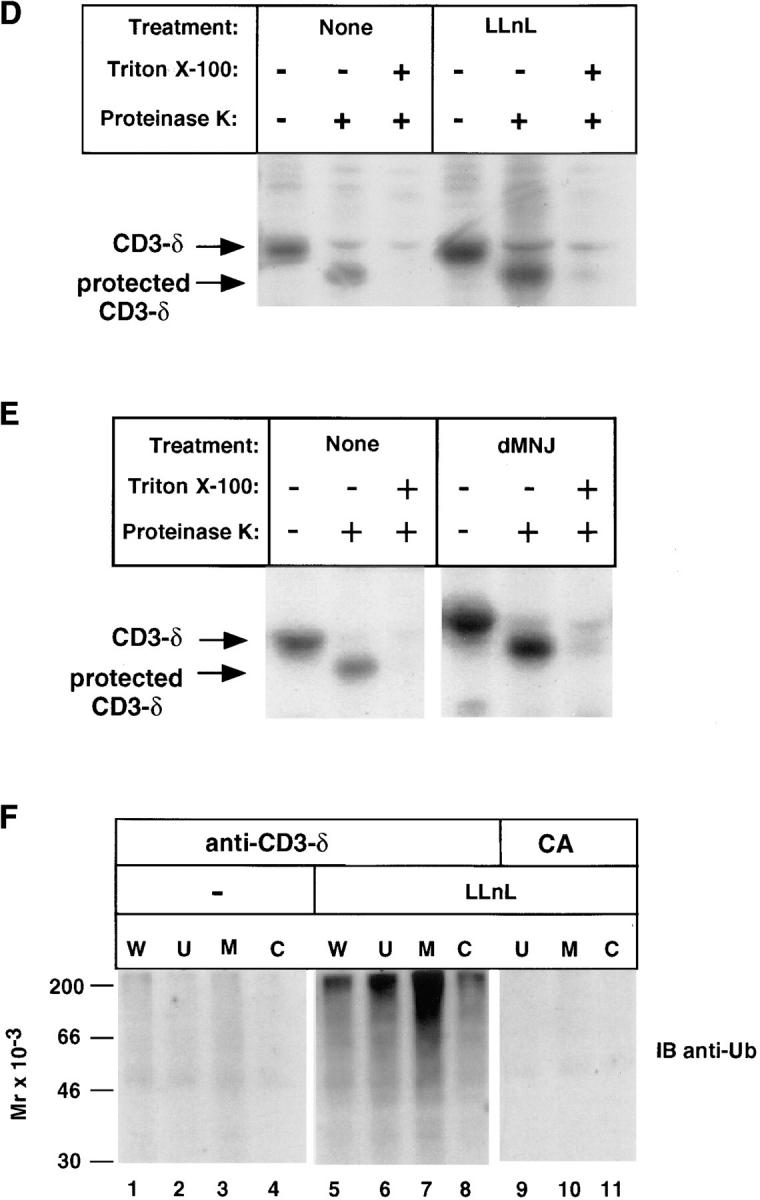
Subcellular localization of CD3-δ. (A) 21.2.2 cells were labeled with [35S]methionine for 20 min followed by a 2.5-h chase. In lanes 1–4, cells were lysed in Triton X-100 lysis buffer directly. In lanes 5–10, cells from the 2.5-h chase were broken open by mechanical shearing without detergent, as described in Materials and Methods, followed by removal of unbroken cells and nuclei (U) and separation of cytosolic (C) and membrane (M) fractions. Immunoprecipitation was with anti–CD3-δ. The amount of material used in the whole cell samples (lanes 1–4) was 1.5 × 107 cells/lane; 3 × 107 cells were subject to fractionation (lanes 5–10). (B) CD3-δ translated in rabbit reticulocytes in vitro and labeled using [35S]methionine was added to cytosol prepared as in A and immunoprecipitated with either CA or anti–CD3-δ. (C) 21.2.2 cells were labeled for 20 min and chased either in the absence or presence of LLnL for 3 h. Membrane (M) and cytosol (C) fractions were prepared as in A. Only half of the residual unbroken cells and nuclei (U) were analyzed in this experiment. Lysates were subject to sequential immunoprecipitation with 2C11(top) followed by anti–CD3-δ (bottom). In pulse-chase experiments CD3-ε labels poorly and is not well-visualized (33). (D) 21.2.2 cells were labeled for 20 min with [35S]methionine, then either treated or not with LLnL for 1 h at 4°C before a 3-h chase in the presence or absence of LLnL. Cells were then broken as in A. After removal of nuclei and unbroken cells, supernatants were divided and treated on ice either without further treatment, with the addition of proteinase K, or first treated with Triton X-100 (0.5%) followed by the addition of proteinase K as indicated. After 1 h at 4°C, PMSF was added to inactivate proteinase K, and Triton X-100 (0.5%) was then added to samples that had not been pretreated with this detergent. All samples were immunoprecipitated with 2C11. The positions of full length CD3-δ and of CD3-δ with a cleaved cytosolic tail are indicated. (E) 21.2.2 cells were subject to pulse-chase metabolic labeling as in C in the presence or absence of dMNJ. Cells were broken, and after removal of unbroken cells and nuclei supernatants were subject to proteinase K digestion and immunoprecipitation with 2C11 as in D. (F) 21.2.2 cells were incubated for 3 h either with or without LLnL. 1.5 × 107 cells were lysed directly in Triton X-100 lysis buffer (W), while the rest (5 × 107) were fractionated as in A followed by immunoprecipitation with anti–CD3-δ. Fractionated LLnL-treated samples were also immunoprecipitated with CA (lanes 9–11). Immunoblotting was with anti-Ub.
Experimental Techniques.
For “pulse-chase” metabolic labeling experiments, cells were incubated at 37°C for 20 min in methionine-free medium before labeling with Tran35S-label (ICN Radiochemicals, Costa Mesa, CA) at 1 mCi/ml (thymocytes) or 300 μCi/ml (cell lines). After labeling, cells were washed and resuspended in complete medium with the indicated additions. Cells were lysed and immunoprecipitates were washed in buffers containing Triton X-100 as previously described (33), except that 0.1% SDS was added to the wash buffer in all immunoprecipitates except those with 2C11, and 0.1% SDS was added to the lysis buffer in experiments in which samples were immunoprecipitated with anti–TCR-α. Except where noted, 1–2 × 107 cells were analyzed in each immunoprecipitation. Resolution of samples by reducing or by two-dimensional nonreducing/reducing SDS-PAGE, immunoblotting, and autoradiography were all carried out as previously described (30, 34). Except for Fig. 1 C, which was developed using enhanced chemiluminescence, all other immunoblots were developed using 125I Protein A (ICN Radiochemicals). For subcellular fractionation experiments, cells were resuspended in 0.25 M sucrose, 10 mM triethanolamine, and 1 mM EDTA, pH 7.4, for 10 min at 4°C followed by 15 passages through a 27-gauge needle. The material was subject to two sequential 5 min 1,000 g spins to remove unbroken cells and nuclei. For separation of cytosolic and membrane fractions, supernatants from the 1,000 g spin were pelleted at 100,000 g for 1 h at 4°C. Supernatants (cytosolic fractions) were directly immunoprecipitated (see Fig. 4) or immunoprecipitated after addition of lysis buffer (see Figs. 7 and 8). The buffer conditions were determined to have no effect on the ability of antibodies to immunoprecipitate TCR components. The pellet (membrane fraction) was lysed in Triton X-100 lysis buffer and immunoprecipitated. In protease protection assays the supernatant from the 1,000 g spin was treated with proteinase K at a final concentration of 100 μg/ml at 4°C, which was neutralized with 2 mM PMSF. All quantitation was carried out on gels analyzed using a Storm 820 PhosphorImager and ImageQuant software (Molecular Dynamics, Inc., Sunnyvale, CA). Coupled in vitro transcription and translation of murine CD3-δ using TNT rabbit reticulocyte lysate (Promega, Madison, WI) and 35[S]methionine (Amersham Corp., Arlington Heights, IN) was carried out as per the manufacturer's instructions. Treatment with N-glycanase (PNGase F; New England Biolabs, Inc., Beverly, MA) at 500 U/μl was carried out on washed immunoprecipitates using conditions specified by the manufacturer.
Figure 1.

Proteasomal degradation and ubiquitination of CD3-δ in thymocytes. (A and B) Thymocytes from 12- (A) or 6-wk-old (B) mice were preincubated with the indicated treatments for 1 h at 4°C before labeling for 20 (A) or 30 min (B) at 37°C with [35S]methionine followed by a chase in complete medium with the indicated additions still present. CD3-δ was immunoprecipitated (IP) from cell lysates using an anti–CD3-δ antiserum raised in rabbit (also known as R9; reference 29) and samples were resolved by SDS-PAGE under reducing conditions followed by autoradiography. The position of CD3-δ is indicated. The minor 16-kD band seen at chase points from proteasome-treated samples in B may represent a small amount of deglycosylated CD3-δ. (C) Thymocytes were incubated at 37°C as indicated, followed by immunoprecipitation of cell lysates with either anti–CD3-δ or CA. Immunoblotting (IB) was with polyclonal anti-Ub as previously described (30).
Figure 7.

Subcellular localization of TCR-α. BW5147 (A) or 21.2.2 cells (B) were labeled and chased for the indicated times followed by fractionation of cells from the chase points as described in Fig. 4 into membrane (M) and cytosol (C) fractions and immunoprecipitation with anti–TCR-α (A) or anti–TCR-α followed by anti–CD3-δ (B). Unfractionated cells from the pulse and chase are indicated by W. The number of unfractionated cell equivalents represents 15% of the number that were subject to fractionation.
Figure 8.

Protection of TCR-α in BW5147 cells. (A) Metabolically labeled cells were subject to a 2.5-h chase followed by breakage by mechanical shearing and treatment with proteinase K as described in Fig. 4. Triton X-100 lanes indicate samples that were detergent solubilized before exposure to proteinase K. Immunoprecipitates from aliquots of cells that were not subject to breakage by mechanical shearing but instead directly lysed in Triton X-100 from the pulse and chase time points are shown in B. (C) To insure that the ∼23-kD band indicated by the asterisk represented a fragment of TCR-α and not an associated protein, a proteinase K-treated sample was immunoprecipitated with anti–TCR-α and half of the sample was denatured as described in Fig. 2, followed by reimmunoprecipitation with additional anti–TCR-α. Note that the proteinase K-dependent band (asterisk) persisted, whereas other species above TCR-α were not reimmunoprecipitated. (D) Cells were labeled with [35S]methionine in the absence of LLnL and then a 2-h chase was carried out either in the presence or absence of LLnL. This was followed by proteinase K digestion.
Results
CD3-δ Is Ubiquitinated and Degraded by Proteasomes.
To determine if proteasomes play a role in degradation of CD3-δ, unfractionated thymocytes were subject to pulse-chase metabolic labeling followed by immunoprecipitation with an anti-peptide rabbit polyclonal antibody that recognizes the cytoplasmic domain of CD3-δ (anti–CD3-δ; reference 29). Newly synthesized CD3-δ was found to be rapidly degraded (Fig. 1 A); however, a peptide-aldehyde that inhibits proteasome function, LLnL (35), blocked its degradation, whereas an inhibitor of lysosome function, NH4Cl, was significantly less effective. A second potent peptide-aldehyde proteasome inhibitor, MG132 (36), as well as LCN, a chemically unrelated and highly specific proteasome inhibitor (37), both also markedly inhibited loss of CD3-δ (Fig. 1 B).
To establish whether CD3-δ undergoes ubiquitination in thymocytes, anti–CD3-δ immunoprecipitates were immunoblotted with anti-Ub antibodies capable of detecting low levels of multi-ubiquitinated proteins (reference 30, Fig. 1 C). Consistent with CD3-δ ubiquitination, in cells treated with the proteasome inhibitor LLnL discrete species as well as a higher molecular weight “smear” was easily detected in anti–CD3-δ immunoprecipitates but not in immunoprecipitates with control antiserum (CA). These were not detected in cells incubated without LLnL, indicating that these species are normally efficiently degraded by proteasomes.
To further assess proteasomal degradation of CD3-δ we turned to 21.2.2 cells (26). This cell line is a variant of the T cell hybridoma 2B4 (38) distinguished by its failure to express any form of TCR-β. This cell line is also deficient in expression of the 2B4-specific TCR-α chain, but it does express TCR-α chains derived from BW5147, the fusion partner used in the generation of 2B4 (27). Because 21.2.2 cells lack TCR-β, TCRs do not assemble, and CD3-δ remains in the ER from whence it is degraded (33). When pulse-chase metabolic labeling was carried out on these cells, LLnL and MG132 each inhibited degradation of newly synthesized CD3-δ, while a closely related peptide- aldehyde that is ineffective in blocking proteasome function, LLM (35), did not substantively affect CD3-δ loss (Fig. 2 A). As expected for this cell line, NH4Cl failed to block CD3-δ loss (Fig. 2 A), and LCN was as effective as LLnL and MG132 (data not shown).
Figure 2.

Proteasomal degradation and ubiquitination of CD3-δ in a T cell line. (A) [35S]methionine labeling of 21.2.2 cells was carried out for 20 min as in Fig. 1 followed by a 3-h chase. Immunoprecipitation was with anti–CD3-δ. (B) 21.2.2 cells were treated as in Fig. 1 C, lysates were immunoprecipitated with either anti–CD3-δ or CA, and membranes were immunoblotted with anti-Ub. (C) An anti–CD3-δ immunoprecipitate from LLnL-treated 21.2.2 cells was heated to 95°C in SDS-PAGE sample buffer containing 0.5% dithiothreitol for 5 min and then diluted into 500 μl of Triton X-100 lysis buffer. The sample was divided, reimmunoprecipitated with either anti–CD3-δ or CA, and immunoblotted with anti-Ub.
As with thymocytes, inhibition of proteasome function also resulted in an accumulation of specifically immunoprecipitated ubiquitinated species in 21.2.2 (Fig. 2 B). In Fig. 2 B immunoprecipitates were washed under conditions that minimize coimmunoprecipitation of other proteins with CD3-δ. To verify that CD3-δ itself was being ubiquitinated, anti-CD3-δ immunoprecipitates from LLnL-treated cells were denatured in SDS-PAGE sample buffer and then reimmunoprecipitated (Fig. 2 C). Only the sample specifically reimmunoprecipitated with anti–CD3-δ recovered ubiquitinated material.
Oligosaccharide Processing and CD3-δ Degradation.
Accompanying the proteasome-dependent loss of CD3-δ (Figs. 1 and 2) is a drop in molecular weight suggestive of the posttranslational action of mannosidases, which are present both in the ER and cis-Golgi. Mannosidases are responsible for the trimming of N-linked oligosaccharides after the rapid cleavage of the three glucose residues from the cotranslationally added Glc3Man9GlcNAc2 structure (Glc, glucose; Man, mannose; GlcNAc, N-acetyl glucosamine; reference 39). The resultant Man5GlcNAc2 may be further processed to generate complex carbohydrates in the medial- and trans- Golgi. To ascertain whether the change in migration is due to mannose trimming, cells were treated with dMNJ, a specific inhibitor of ER and Golgi mannosidases. As is evident, the change in migration of CD3-δ during the chase was largely abrogated by this treatment (Fig. 3 A). Strikingly, however, dMNJ also markedly inhibited the loss of CD3-δ during the chase. This effect was specific for inhibition of mannosidases, as a closely related compound, dNJ, which inhibits glucosidases but not mannosidases, affected the migration of CD3-δ but was ineffective in altering its fate (Fig. 3, B and C). Results similar to those obtained with dNJ were obtained with another glucosidase inhibitor, castanospermine (data not shown). As is evident (Fig. 3, B and C), the ability of dMNJ to inhibit loss of CD3-δ was similar in magnitude to effects seen with LLnL; and exposure of cells to the two together was not additive (data not shown). This suggests that the proteasome inhibitor and mannosidase inhibitor are functioning on the same population of CD3-δ molecules. Moreover, the half-life CD3-δ was prolonged by either LLnL or dMNJ when this TCR subunit was expressed transiently in COS-7 cells (data not shown), indicating that the effects seen do not require other T cell–specific proteins.
Figure 3.

Oligosaccharide processing and degradation of CD3-δ. (A and B) 21.2.2 cells were labeled with [35S]methionine for 20 min at 37°C as in Fig. 2 A and were chased as indicated. Immunoprecipitation was with anti–CD3-δ. (C) Quantitation of data from B.
Subcellular Localization of CD3-δ.
Subcellular fractionation was carried out to determine whether the CD3-δ that accumulates when proteasome function is inhibited is membrane-bound or cytosolic. After a 20 min labeling with [35S]methionine and a chase in medium without [35S]methionine (Fig. 4 A, lanes 1–4), cells were broken by mechanical shearing without detergent and fractionated into residual unbroken cells and nuclei (U), membranes (M), and cytosol (C) (Fig. 4 A, lanes 5–10), and immunoprecipitated with anti–CD3-δ. CD3-δ was not found in the cytosolic fractions of cells. Instead, CD3-δ localized to the membrane fraction, with LLnL-treated cells exhibiting greater levels of membrane-associated CD3-δ, commensurate with the increased amount of CD3-δ present at the end of the chase (Fig. 4 A, lanes 1–4). To verify that CD3-δ, if present, could be immunoprecipitated from the cytosolic fraction by anti–CD3-δ, CD3-δ that had been translated in vitro in rabbit reticulocyte lysate without microsomes was mixed with the cytosolic fraction. As shown (Fig. 4 B), this exogenously added material could be specifically immunoprecipitated from the cytosolic fraction with anti–CD3-δ.
CD3-ε is the only nonglycoprotein of the three CD3 subunits (1). At an early stage in the assembly of the octomeric TCR complex, CD3-δ and CD3-γ each dimerize with CD3-ε through their luminal domains. Dimerization with CD3-δ or CD3-γ allows heterodimers containing CD3-ε to be immunoprecipitated by 2C11, a monoclonal antibody that recognizes a conformation-dependent luminal epitope on CD3-ε (7, 31). The association of CD3-ε with CD3-δ and their coimmunoprecipitation with 2C11 was used to further assess the localization of CD3-δ. As is evident in both LLnL-treated and untreated samples (Fig. 4 C), the large majority of membrane-bound CD3-δ is immunoprecipitated with 2C11. This demonstrates that CD3-δ salvaged from degradation is largely bound to CD3-ε.
The results presented in Fig. 4, A and C strongly suggest that, when its degradation is inhibited, CD3-δ accumulates in ER membranes in a native configuration. To establish the topological orientation of CD3-δ with certainty, protease-protection studies were carried out. In these experiments 21.2.2 cells were labeled with [35S]methionine, and after a 3-h chase cells were broken by mechanical shearing and nuclei and residual unbroken cells were removed. The resultant postnuclear supernatant, consisting of cytosol and membrane-bound organelles, was subject to proteinase K digestion. Such treatment will degrade portions of proteins that are exposed to the cytosol, but leave protected transmembrane and luminal domains intact. Since the epitope recognized by anti–CD3-δ is cytosolic and, as expected, lost on treatment with proteinase K (data not shown), we took advantage of the luminal epitope recognized by 2C11 to immunoprecipitate residual CD3-δ after proteinase K digestion (Fig. 4, D and E). As predicted for CD3-δ in its native topological membrane orientation, its ∼4-kD cytosolic domain was degraded by proteinase K, while the luminal and transmembrane domains of CD3-δ were largely protected. As expected, solubilization of membranes with Triton X-100 before exposure to proteinase K abrogated this protection. Importantly, increased levels of CD3-δ were protected in cells in which CD3-δ degradation was prevented by inhibition of either proteasome function (Fig. 4 D) or mannosidase activity (Fig. 4 E). Also, the amount of material remaining after protease treatment (as compared with nondigested samples) was similar in cells in which proteasome or mannosidase activity was inhibited and in untreated samples. This establishes that when either proteasome function or mannosidase activity is inhibited, the CD3-δ that remains is still membrane-embedded in its native topological orientation.
To determine the location of ubiquitinated CD3-δ, anti-Ub immunoblotting of immunoprecipitates from fractionated cells was carried out (Fig. 4 F). Although some ubiquitinated CD3-δ was found in the cytosolic (C) fraction from LLnL-treated cells, the majority was membrane-bound (M; Fig. 4 F). Thus, it is apparent that ubiquitination of CD3-δ takes place while still in the ER membrane.
Degradation of TCR-α: Involvement of Proteasomes but not Mannosidases.
TCR-α chains are products of genes that undergo somatic rearrangement. In most cells TCR-α forms disulfide-linked dimers with TCR-β, also a product of a rearranged gene. The membrane proximal portion of the intraluminal domain (extracellular domain) and the entire transmembrane and cytoplasmic segments of mouse TCR-α are derived from a constant region that is invariant among TCR-α's in different cells (40). The TCR-α transmembrane segment is remarkable in having two charged amino acids (Lys and Arg), and its short cytoplasmic domain has no Lys residues (41). Recent studies in non-T cells have provided evidence for proteasome-independent dislocation of TCR-α from the ER to the cytosol accompanied by removal of its N-linked sugars (42, 43), similar to that observed for MHC class I (19–21), without evidence of accompanying ubiquitination. To assess the degradation of TCR-α we evaluated BW5147, the fusion partner used in the generation of T cell hybridomas such as 2B4 (38). This well-characterized thymoma expresses TCR-α and TCR-β but not CD3-δ (44). Using a monoclonal anti– TCR-α antibody that recognizes a constant region intraluminal epitope, newly synthesized TCR-α was found to be rapidly degraded (Fig. 5, A and B). LLnL markedly inhibited TCR-α loss, and this was accompanied by increased density both above and below TCR-α (Fig. 5 A), perhaps representing ubiquitination and partial degradation, respectively (similar results were obtained using LCN; data not shown). However, dMNJ had minimal effects on loss of this receptor subunit (Fig. 5, A and B). To ascertain the extent to which TCR-α is disulfide-linked to TCR-β, samples were resolved by two-dimensional nonreducing/reducing SDS-PAGE (Fig. 5 C). The large majority of TCR-α, including most that was salvaged by LLnL, was found not to be disulfide-linked to TCR-β. The findings obtained in BW5147 cells were verified in 21.2.2 cells (Fig. 6 A), where dMNJ inhibited degradation of CD3-δ but not TCR-α. In 21.2.2 and BW5147 a minor band above the predominant form of TCR-α is often distinguished, which may reflect expression from a second BW-specific TCR-α allele in these cells (45). Inhibition of TCR-α degradation was accompanied by the appearance of lower molecular species of 14–30 kD (Fig. 6, A and B), which unlike full-length TCR-α, were unaffected by treatment with N-glycanase (Fig. 6 B) and which were immunoreactive with anti–TCR-α by immunoblotting (data not shown). These therefore likely represent incompletely proteolyzed deglycosylated TCR-α.
Figure 5.

Degradation of TCR-α in BW5147. 35S-labeling of BW5147 cells and chase was carried out as described in Figure 2. Samples were lysed in Triton X-100 lysis buffer with 0.1% SDS to decrease nonspecific binding. Anti–TCR-α (A and C) immunoprecipitates were washed in 0.1% Triton X-100, 0.1% SDS and resolved by SDS-PAGE under reducing conditions (A) or on two-dimensional nonreducing/reducing gels (C) followed by autoradiography. The longer exposure of the left upper panel of C is to demonstrate the position of disulfide-linked TCR-α/β dimers. B shows a quantitation of the experiment in A. Quantitation was by Storm 820 PhosphorImager using ImageQuant software.
Figure 6.

Degradation of TCR-α in 21.2.2 cells. (A) Cells were labeled and chased in cold medium for the indicated times followed by sequential immunoprecipitation with anti–TCR-α (upper panel) followed by anti–CD3-δ (lower panel). (B) Cells were labeled and chased as indicated and immunoprecipitates were treated with or without N-glycanase. Positions of TCR-α, partially and fully deglycosylated TCR-α, and immunoreactive degradation products are indicated.
Proteasome-independent Retrograde Movement of TCR-α.
The location of TCR-α in T cells was assessed by separation of membrane and cytosolic fractions. In BW5147 as well as 21.2.2 cells, the large majority of TCR-α that accumulated was found in the membrane fraction (Fig. 7, A and B). In some samples treated with LLnL, more rapidly migrating forms, consistent with deglycosylation, are disproportionately represented in the cytosolic fraction. The topological orientation of TCR-α was assessed by proteinase K digestion of cells that had been broken open by mechanical shearing (Fig. 8). In contrast to CD3-δ, where a similar proportion of material was protected in samples treated with or without proteasome inhibitors, a substantially smaller percentage of the TCR-α was protected in samples in which proteasome function was inhibited as compared with samples not exposed to proteasome inhibitors (Fig. 8 A). Strikingly, loss of full length TCR-α was accompanied by the appearance of a discrete lower molecular weight immunoreactive band of ∼23 kD (Fig. 8 A). That this is a fragment of TCR-α and not an associated protein was established by heating of immunoprecipitates to 95°C in SDS sample buffer under reducing conditions and reimmunoprecipitation with anti-TCR-α (Fig. 8 C). As is evident, this 23-kD band was recovered with the same efficiency as full length TCR-α. A similar fragment was visualized when broken cells were digested with trypsin instead of proteinase K (data not shown). Since the 23-kD fragment drops from the predicted molecular weight of glycosylated TCR-α, it is likely to also be glycosylated. In fact, treatment with N-glycanase resulted in a drop in molecular weight consistent with at least one N-linked oligosaccharide (data not shown).
To assess whether this partially protected form of TCR-α represents material that never fully traversed the ER membrane, or if this reflects ongoing removal of TCR-α that had been initially fully translocated into the ER, TCR-α was labeled in the absence of LLnL and chased in its presence or absence. As can be seen in Fig. 8 D, a small amount of this partially protected TCR-α was present at the end of the pulse (lane 2); however, the amount detected clearly increased when LLnL was present during the chase. This indicates that TCR-α that had fully translocated across the ER membrane undergoes partial retrotranslocation, resulting in the retention of a discrete form of TCR-α within the ER lumen/membrane. This partial retrotranslocation occurs independent of the activity of proteasomes. However, to complete the removal/degradation of TCR-α from the ER membrane, as is the case with CD3-δ, proteasome activity is required.
Ubiquitination of TCR-α.
The smearing seen above TCR-α in lanes from LLnL-treated cells in Figs. 5 and 6 is suggestive of ubiquitination. However, recently published studies in which TCR-α was expressed in non-T cells failed to reveal evidence for this modification (42, 43). To assess whether ubiquitinated forms of TCR-α were being generated in T cells, anti-Ub immunoblotting was carried out on 21.2.2 cells. Similar to CD3-δ, specifically immunoprecipitated ubiquitinated species were, in fact, detected in the presence of the proteasome inhibitor LLnL (Fig. 9 A). These species were detected both in the cytosolic and membrane compartments. Confirmation that these represent ubiquitinated TCR-α was obtained by dissociation of samples in SDS and reimmunoprecipitation with anti– TCR-α (data not shown). Since TCR-α, unlike CD3-δ, has no lysine residues that are normally exposed to the cytosol (25, 41), ubiquitination of this subunit is either occurring on residues that have become exposed to the cytosol after the initiation of retrograde movement or is taking place on luminal residues. However, as there is no evidence for luminal ubiquitin-conjugating enzymes, the latter possibility seems unlikely. To evaluate this, anti-Ub immunoblotting was carried out on samples that had been digested with proteinase K. As is evident in Fig. 9 B, ubiquitin attached to TCR-α was largely removed with proteinase K, indicating that the lysines to which ubiquitin has been attached on TCR-α are in the cytosol. It is therefore apparent that, for TCR-α, initiation of retrograde movement out of the ER precedes ubiquitination.
Figure 9.

Ubiquitination of TCR-α in BW5147. (A) Cells were treated with or without LLnL as indicated and fractionated as described for Fig. 4. 106 cells were used in the whole cell lysate lanes (W ), and 5 × 107 cells were fractionated into cytosol and membrane fractions. Lysis and washing of immunoprecipitates was in buffer supplemented with 0.1% SDS. Immunoblotting was with anti-Ub. (B) LLnL-treated cells were broken and treated with or without proteinase K followed by immunoprecipitation and anti-Ub immunoblotting.
Discussion
Pathways to Proteasomal Degradation.
Recent findings have led to the emergence of models for degradation of transmembrane and soluble luminal proteins from the ER where fates are largely decided by proteasome-independent retrotranslocation from the ER to the cytosol through protein conducting channels followed by proteasomal degradation (20, 22–24). For MHC class I heavy chains (20) and for a mutant form of a yeast luminal protein, carboxypeptidase y (46), it has been proposed that newly synthesized proteins associate with the Sec61 translocon complex which then facilitates their movement back into the cytosol. In the case of retrotranslocation of MHC class I heavy chains, this is accompanied by complete removal of N-linked oligosaccharides, without evidence of ubiquitinated intermediates (19–21).
This study reveals that, in T cells, components of the TCR are also degraded from the ER as a consequence of pathways that terminate in proteasomes. Our evaluation of CD3-δ and TCR-α illuminates novel aspects of these degradative pathways and underscores that, although cytosolic proteasomes are likely a final destination for a large number of proteins, the mechanisms involved in targeting for proteasomal degradation vary significantly. Strikingly, even between two normal components of the TCR, the route and the determining factors leading to degradation differ. TCR-α undergoes partial retrograde movement regardless of proteasome activity, with a 23-kD glycosylated fragment remaining sequestered within the ER when proteasome function is inhibited. The partial retrotranslocation of TCR-α in the absence of proteasome function should be compared with results obtained with MHC class I and more recent studies in which TCR-α was overexpressed ectopically in nonlymphoid cells (42, 43). In these instances complete removal from the ER membrane occurred even when proteasome function was inhibited. Further distinguishing our findings from the aforementioned examples is the existence of ubiquitinated membrane-associated TCR-α. In contrast to TCR-α, the large majority of CD3-δ retains its native transmembrane location when proteasome function is inhibited. Thus, proteasome function is required both for detectable retrograde movement and destruction of this TCR subunit. Moreover, it is evident that there is a requirement for cellular mannosidase activity in proteasome-dependent CD3-δ degradation. As with TCR-α, CD3-δ also undergoes ubiquitination while still associated with the ER membrane.
When assessing differences between TCR-α and CD3-δ that might account for their relative susceptibilities to proteasome-independent retrotranslocation, the nature of their transmembrane segments must be considered. CD3-δ has a single acidic residue within its transmembrane domain, a feature common to all of the invariant TCR components (1). TCR-α, on the other hand, has two bulky basic amino acids that have been postulated to contribute to its inherent instability as a transmembrane protein (8, 9). Because of this, it is reasonable to speculate that the relatively hydrophilic nature of TCR-α's transmembrane domain has the potential to facilitate movement into and through protein-conducting channels that mediate retrograde movement. Whether or not this is a major factor in determining susceptibility to retrograde movement will require further evaluation.
Mannosidase Activity and Degradation from the ER.
Branch chain N-linked oligosaccharides are cotranslationally added to luminal asparagine residues of proteins as Glc3Man9GlcNAc2 (39). The three terminal Glc residues are generally rapidly cleaved in the ER, and ER and cis-Golgi mannosidases carry out trimming of these “high mannose” chains. This trimming involves removal of four of the mannose residues generating Man5GlcNAc2 before further processing in the Golgi complex to “hybrid” and “complex” chains (39). For a number of proteins, oligosaccharide processing plays important roles in ER “quality control”. This has been analyzed predominantly with regard to removal of glucose residues in calnexin- and calreticulin-mediated folding in the ER (47). Previous evidence of a role for mannosidase activity in determining the fate of proteins in the ER was limited to a study in which yeast pre-pro-α factor was expressed ectopically in mammalian cells (48), and more recent work on mutant α1-antitrypsin (49). Both pre-pro-α factor and α1-antitrypsin are luminal proteins without membrane anchors. Our results establish a previously unappreciated role for mannosidase activity in targeting a normal transmembrane protein, CD3-δ, for proteasomal degradation. They also demonstrate that mannosidases and proteasomes function along the same pathway leading to destruction of this receptor subunit. When 21.2.2 cells were treated with tunicamycin, which blocks the addition of N-linked oligosaccharides to proteins, nonglycosylated CD3-δ was rapidly degraded in a proteasome-dependent manner, and, in COS-7 cells, CD3-δ in which the three sites of N-glycosylation were mutated was degraded rapidly (our unpublished observations). This suggests that N-linked oligosaccharides in which mannose residues have not been trimmed stabilize CD3-δ. This could occur by enhancing the association of CD3-δ with the more stable CD3-ε. Arguing against this possibility are the findings that trimmed and untrimmed CD3-δ both coimmunoprecipitate with anti-CD3-ε, and that degradation of CD3-δ in COS-7 cells is also inhibited by dMNJ.
Man9GlcNAc2 species undergo cycles of reversible monoglucosylation (47). Since the carbohydrate-binding luminal chaperones calnexin and calreticulin preferentially associate with monoglucosylated oligosaccharides, interactions with these proteins could stabilize CD3-δ in which mannose residues have not been trimmed. However, CD3-δ does not associate with calreticulin (50) and we and others have found that the glycosylation state of CD3-δ does not substantially affect its association with calnexin (51 and our unpublished observations). Although we can not exclude with certainty that differences in the nature of calnexin interactions explain the protective effects of mannosidase inhibitors, we are left to consider that other, as yet to be characterized, carbohydrate-binding “chaperones” play a role in stabilization of untrimmed CD3-δ.
Ubiquitination and Proteasomes in Degradation from the ER.
Ubiquitinated CD3-δ and TCR-α are easily detected when proteasome function is inhibited. Ubiquitinated CD3-δ is primarily membrane-associated, whereas ubiquitinated TCR-α is found to a similar extent in both membranes and cytosol. A model integrating ubiquitination with retrograde movement is one in which ubiquitination is a requisite event preceding the initiation of retrograde movement, with this covalent modification serving to facilitate association with proteasomes and subsequent entry into protein conducting channels. However, there are observations with both TCR-α and CD3-δ that make such a simple model untenable. First, TCR-α has no lysines within its short cytoplasmic tail (41), necessitating cytoplasmic exposure of transmembrane and luminal lysines to serve as sites of ubiquitination. Second, when all of the cytoplasmic lysines of CD3-δ were mutated and this protein was transiently expressed in non-T cells, TCR-α was still subject to ubiquitination while associated with the ER (Yang, Y., J.S. Bonifacino, and A.M. Weissman, unpublished observations). These findings strongly suggest that for both TCR-α and for CD3-δ initiation of retrograde transport can precede ubiquitination.
Since the steady state level of ubiquitinated forms are low relative to the total number of molecules subject to degradation, and initiation of retrograde movement can precede ubiquitination, what then is the significance of ubiquitination in degradation from the ER? First, we point out that in most instances, when proteasome function is inhibited, proteins that are known to be ubiquitinated accumulate primarily in non-ubiquitinated forms. This is due, at least in part, to the ongoing activity of cellular deubiquitinating enzymes (11). As there are no cell permeant selective inhibitors of deubiquitinating enzymes, the percent of TCR subunits that exist transiently in a ubiquitinated form can not be determined. Since multiubiquitin chains are potent proteasome targeting signals (11), ubiquitination is required for the destruction of some ER membrane proteins (12), and proteasomes are known to associate with the ER (18, 52–54), we favor a model in which transmembrane proteins are predisposed for degradation from the ER by either having never left, or reversibly entering, protein-conducting channels, such as the translocon. In this hydrophilic milieu they have the opportunity to undergo variable degrees of movement across the membrane, enhancing their susceptibility to ubiqutination. Ubiquitination leads to association with 26S proteasomes, which facilitates complete retrograde movement and degradation. Among proteins, the relative requirements for ubiquitination and proteasomes in completing retrograde movement likely varies, with human cytomegalovirus-enhanced loss of MHC class I heavy chains at one end of the spectrum, and CD3-δ at the other. An alternative model that warrants consideration is that proteasomes are drawn to translocons by ubiquitinated chaperone-like translocon-associated proteins. This facilitates the removal/degradation of target proteins that are susceptible to retrotranslocation. For TCR subunits there is no need to postulate such an indirect mechanism, since TCR-α and CD3-δ are both ubiquitinated. However, there is at least one in vitro situation where ubiquitination of calnexin has been suggested to facilitate the degradation of a mutant form of a secreted protein (α1-antitrypsin; reference 17).
When considering the enzymes responsible for the ubiquitination of ER membrane proteins it is of note that two yeast ubiquitin conjugating enzymes (E2s), UBC6 and UBC7, have been implicated in the ubiquitination/degradation of proteins from the ER (12, 55). One of these, UBC6, has its catalytic domain localized to the cytoplasmic face of the ER membrane through a hydrophobic COOH-terminal anchor (28, 55). Interestingly, both of these E2s are implicated in the ubiquitin-mediated degradation of mutant forms of the Sec61 translocon complex (12), and we now have evidence for the existence of an enzymatically active mammalian UBC6 homolog (Tiwari, S., and A.M. Weissman, unpublished observations).
Conclusion.
Eukaryotic cells have evolved sophisticated mechanisms to degrade both transmembrane and soluble proteins of the secretory pathway that are either made in excess or that are abnormally folded. It is apparent that even among subunits of the TCR, multiple pathways exist that ultimately lead to their breakdown by cytosolic proteasomes. Degradation of CD3-δ from the ER entails trimming of mannose residues from N-linked oligosaccharides, and recognition and proteolysis by proteasomes with concomitant removal from ER membranes. For TCR-α there is no evidence that mannosidase activity plays a role, and a significant degree of exposure of this protein to the cytosol occurs regardless of proteasome function. Although light is now being cast on what has been, until recently, a mysterious process, much remains to be learned regarding the molecular details of the multiple pathways leading to degradation from the ER.
Acknowledgments
We thank Kelly Kearse, Jennifer Lippincott-Schwartz, and Jocelyn Weissman for their comments on this manuscript and Jonathan Ashwell and Richard Klausner for helpful discussions. We thank Proscript for MG132, and Astrid Eder for mouse thymi.
Footnotes
1 Abbreviations used in this paper: anti-Ub, anti-ubiquitin; CA, control antiserum; CFTR, cystic fibrosis conductance regulator; CHX, cycloheximide; dMNJ, deoxymannojirimycin; dMJ, deoxynojirimycin; ER, endoplasmic reticulum; Glc, glucose; GlcNAc, N-acetyl glucosamine; LCN, lactacystin; LLM, LLnL, N-acetyl-Leu-Leu-methioninal; N-acetyl-Leu-Leu-norleucinal; Man, mannose; Mg132, Z-Leu-Leu-Leu CHO.
References
- 1.Weissman AM. The T-cell antigen receptor: a multisubunit signaling complex. Chem Immunol. 1994;59:1–18. [PubMed] [Google Scholar]
- 2.Sussman JJ, Bonifacino JS, Lippincott-Schwartz J, Weissman AM, Saito T, Klausner RD, Ashwell JD. Failure to synthesize the T cell CD3-ζ chain: structure and function of a partial T cell receptor complex. Cell. 1988;52:85–95. doi: 10.1016/0092-8674(88)90533-8. [DOI] [PubMed] [Google Scholar]
- 3.Klausner RD, Lippincott-Schwartz J, Bonifacino JS. The T cell antigen receptor: insights into organelle biology. Annu Rev Cell Biol. 1990;6:403–431. doi: 10.1146/annurev.cb.06.110190.002155. [DOI] [PubMed] [Google Scholar]
- 4.Bonifacino JS, Lippincott-Schwartz J. Degradation of proteins within the endoplasmic reticulum. Curr Opin Cell Biol. 1991;3:592–600. doi: 10.1016/0955-0674(91)90028-w. [DOI] [PubMed] [Google Scholar]
- 5.Bonifacino JS, McCarthy SA, Maguire JE, Nakayama T, Singer DS, Klausner RD, Singer A. Novel post-translational regulation of TCR expression in CD4+ CD8+thymocytes influenced by CD4. Nature. 1990;344:247–251. doi: 10.1038/344247a0. [DOI] [PubMed] [Google Scholar]
- 6.Kearse KP, Roberts JL, Munitz TI, Wiest DL, Nakayama T, Singer A. Developmental regulation of αβ T cell antigen receptor expression results from differential stability of nascent TCR-α proteins within the endoplasmic reticulum of immature and mature T cells. EMBO (Eur Mol Biol Organ) J. 1994;13:4504–4514. doi: 10.1002/j.1460-2075.1994.tb06772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonifacino JS, Suzuki CK, Klausner RD. A peptide sequence confers retention and degradation in the endoplasmic reticulum. Science. 1990;247:79–82. doi: 10.1126/science.2294595. [DOI] [PubMed] [Google Scholar]
- 9.Shin J, Lee S, Strominger JL. Translocation of TCRα chains into the lumen of the endoplasmic reticulum and their degradation. Science. 1993;259:1901–1904. doi: 10.1126/science.8456316. [DOI] [PubMed] [Google Scholar]
- 10.Coux O, Tanaka K, Goldberg AL. Structure and function of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 11.Weissman AM. Regulating protein degradation by ubiquitination. Immunol Today. 1997;18:189–198. doi: 10.1016/s0167-5699(97)84666-x. [DOI] [PubMed] [Google Scholar]
- 12.Biederer T, Volkwein C, Sommer T. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO (Eur Mol Biol Organ) J. 1996;15:2069–2076. [PMC free article] [PubMed] [Google Scholar]
- 13.Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- 14.Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci USA. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward CL, Ōmura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 16.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 17.Qu D, Teckman JH, Ōmura S, Perlmutter DH. Degradation of a mutant secretory protein, α1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem. 1996;271:22791–22795. doi: 10.1074/jbc.271.37.22791. [DOI] [PubMed] [Google Scholar]
- 18.McGee TP, Cheng HH, Kumagai H, Ōmura S, Simoni RD. Degradation of 3-hydroxy-3-methylglutaryl-CoA reductase in endoplasmic reticulum membranes is accelerated as a result of increased susceptibility to proteolysis. J Biol Chem. 1996;271:25630–25638. doi: 10.1074/jbc.271.41.25630. [DOI] [PubMed] [Google Scholar]
- 19.Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 20.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 21.Hughes EA, Hammond C, Cresswell P. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc Natl Acad Sci USA. 1997;94:1896–1901. doi: 10.1073/pnas.94.5.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lord MJ. Protein degradation: go outside and see the proteasome. Curr Biol. 1996;6:1067–1069. doi: 10.1016/s0960-9822(02)70666-0. [DOI] [PubMed] [Google Scholar]
- 23.Bonifacino JS. Reversal of fortune for nascent proteins. Nature. 1996;384:405–406. doi: 10.1038/384405a0. [DOI] [PubMed] [Google Scholar]
- 24.Kopito RH. ER quality control: the cytoplasmic connection. Cell. 1997;88:427–430. doi: 10.1016/s0092-8674(00)81881-4. [DOI] [PubMed] [Google Scholar]
- 25.van den Elsen P, Shepley B-A, Cho M, Terhorst C. Isolation and characterization of a cDNA clone encoding the murine homolgue of the human 20K T3/T-cell receptor glycoprotein. Nature. 1985;314:542–544. doi: 10.1038/314542a0. [DOI] [PubMed] [Google Scholar]
- 26.Sussman JJ, Saito T, Shevach EM, Germain RN, Ashwell JD. Thy-1– and Ly-6–mediated lymphokine production and growth inhibition of a T cell hybridoma require co-expression of the T cell antigen receptor complex. J Immunol. 1988;140:2520–2526. [PubMed] [Google Scholar]
- 27.Hyman R, Stallings V. Complementary patterns of Thy-1 variants and evidence that antigen loss variants “pre-exist” in the parental population. J Natl Cancer Inst. 1974;52:429–437. doi: 10.1093/jnci/52.2.429. [DOI] [PubMed] [Google Scholar]
- 28.Yang M, Ellenberg J, Bonifacino JS, Weissman AM. The transmembrane domain of a carboxyl-terminal anchored protein determines localization to the endoplasmic reticulum. J Biol Chem. 1997;272:1970–1975. doi: 10.1074/jbc.272.3.1970. [DOI] [PubMed] [Google Scholar]
- 29.Samelson LE, Weissman AM, Robey FA, Berkower I, Klausner RD. Characterization of an anti-peptide antibody that recognizes the murine analogue of the human T cell antigen receptor–T3 δ chain. J Immunol. 1986;137:3254–3258. [PubMed] [Google Scholar]
- 30.Cenciarelli C, Wilhelm KJ, Jr, Guo A, Weissman AM. T cell antigen receptor ubiquitination is a consequence of receptor-mediated tyrosine kinase activation. J Biol Chem. 1996;271:8709–8713. doi: 10.1074/jbc.271.15.8709. [DOI] [PubMed] [Google Scholar]
- 31.Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci USA. 1987;84:1374–1378. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kubo RT, Born W, Kappler JW, Marrack P, Pigeon M. Characterization of a monoclonal antibody which detects all murine αβ T cell receptors. J Immunol. 1989;142:2736–2742. [Google Scholar]
- 33.Chen C, Bonifacino JS, Yuan LC, Klausner RD. Selective degradation of T cell antigen receptor chains retained in a pre-Golgi compartment. J Cell Biol. 1988;107:2149–2161. doi: 10.1083/jcb.107.6.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cenciarelli C, Hou D, Hsu K-C, Rellahan BL, Wiest DL, Smith HT, Fried VA, Weissman AM. Activation-induced ubiquitination of the T cell antigen receptor. Science. 1992;257:795–797. doi: 10.1126/science.1323144. [DOI] [PubMed] [Google Scholar]
- 35.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Gen Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 37.Fenteany G, Standaert RF, Reichard GA, Corey EJ, Schreiber SL. A β-lactone related to lactacystin induces neurite outgrowth in a neuroblastoma cell line and inhibits cell cycle progression in an osteosarcoma cell line. Proc Natl Acad Sci USA. 1994;91:3358–3362. doi: 10.1073/pnas.91.8.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hedrick SM, Matis LA, Hecht TT, Samelson LE, Longo DL, Heber E, Katz, Schwartz RH. The fine specificity of antigen and Ia determinant recognition by T cell hybridoma clones specific for pigeon cytochrome c. Cell. 1982;30:141–152. doi: 10.1016/0092-8674(82)90020-4. [DOI] [PubMed] [Google Scholar]
- 39.Elbein AD. Glycosidase inhibitors: inhibitors of N-linked oligosaccharide processing. FASEB (Fed Am Soc Exp Biol) J. 1991;5:3055–3063. doi: 10.1096/fasebj.5.15.1743438. [DOI] [PubMed] [Google Scholar]
- 40.Ashwell, J.D., and A.M. Weissman. 1995. T Cell Antigen Receptor Genes, Gene Products and Co-receptors. R.R. Rich, T.A. Fleisher, B.D. Schwartz, W.T. Shearer, and W. Strober, editors. Mosby-Year Book, Inc., St. Louis, MO. 69–93.
- 41.Chien Y, Becker DM, Lindsten T, Okamura M, Cohen DI, Davis MM. A third type of murine T-cell receptor gene. Nature. 1984;312:31–35. doi: 10.1038/312031a0. [DOI] [PubMed] [Google Scholar]
- 42.Huppa JB, Ploegh HL. The α chain of the T cell antigen receptor is degraded in the cytosol. Immunity. 1997;7:113–122. doi: 10.1016/s1074-7613(00)80514-2. [DOI] [PubMed] [Google Scholar]
- 43.Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor α chains by the proteasome. J Biol Chem. 1997;272:20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- 44.Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell. 1988;54:209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- 45.Kearse KP, Williams DB, Singer A. Persistence of glucose residues on core oligosaccharides prevents association of TCR-α and TCR-β proteins with calnexin and results specifically in accelerated degradation of nascent TCR-α proteins within the endoplasmic reticulum. EMBO (Eur Mol Biol Organ) J. 1994;13:3678–3686. doi: 10.1002/j.1460-2075.1994.tb06677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plemper RK, Bohmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388:891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- 47.Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- 48.Su K, Stoller T, Rocco J, Zemsky J, Green R. Pre-Golgi degradation of yeast prepro-α-factor expressed in a mammalian cell. J Biol Chem. 1993;268:14301–14309. [PubMed] [Google Scholar]
- 49.Liu Y, Choudhury P, Cabral CM, Sifers RN. Intracellular disposal of incompletely folded human α1-antitrypsin involves release from calnexin and post-translational trimming of asparagine-linked oligosaccharides. J Biol Chem. 1997;272:7946–7951. doi: 10.1074/jbc.272.12.7946. [DOI] [PubMed] [Google Scholar]
- 50.van Leeuwen JEM, Kearse KP. The related molecular chaperones calnexin and calreticulin differentially associate with nascent T cell antigen receptor proteins within the endoplasmic reticulum. J Biol Chem. 1996;271:25345–25349. doi: 10.1074/jbc.271.41.25345. [DOI] [PubMed] [Google Scholar]
- 51.van Leeuwen JEM, Kearse KP. Calnexin associates exclusively with individual CD3 delta and T cell antigen receptor (TCR) alpha proteins containing incompletely trimmed glycans that are not assembled into multisubunit TCR complexes. J Biol Chem. 1996;271:9660–9665. doi: 10.1074/jbc.271.16.9660. [DOI] [PubMed] [Google Scholar]
- 52.Palmer A, Rivett AJ, Thomson S, Hendil KB, Butcher GW, Fuertes G, Knecht E. Subpopulations of proteasomes in rat liver nuclei, microsomes and cytosol. Biochem J. 1996;316:401–407. doi: 10.1042/bj3160401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rivett AJ, Palmer A, Knecht E. Electron microscopic localization of the multicatalytic proteinase complex in rat liver and in cultured cells. J Histochem Cytochem. 1992;40:1165–1172. doi: 10.1177/40.8.1619280. [DOI] [PubMed] [Google Scholar]
- 54.Yang Y, Fruh K, Ahn K, Peterson PA. In vivo assembly of the proteasomal complexes, implications for antigen processing. J Biol Chem. 1995;270:27687–27694. doi: 10.1074/jbc.270.46.27687. [DOI] [PubMed] [Google Scholar]
- 55.Sommer T, Jentsch S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 1993;365:176–179. doi: 10.1038/365176a0. [DOI] [PubMed] [Google Scholar]