

Abstract
Mutations in the tyrosine kinase, Btk, result in a mild immunodeficiency in mice (xid). While B lymphocytes from xid mice do not proliferate to anti-immunoglobulin (Ig), we show here induction of the complete complement of cell cycle regulatory molecules, though the level of induction is about half that detected in normal B cells. Cell cycle analysis reveals that anti-Ig stimulated xid B cells enter S phase, but fail to complete the cell cycle, exhibiting a high rate of apoptosis. This correlated with a decreased ability to induce the anti-apoptosis regulatory protein, Bcl-xL. Ectopic expression of Bcl-xL in xid B cells permitted anti-Ig induced cell cycle progression demonstrating dual requirements for induction of anti-apoptotic proteins plus cell cycle regulatory proteins during antigen receptor mediated proliferation. Furthermore, our results link one of the immunodeficient traits caused by mutant Btk with the failure to properly regulate Bcl-xL.
Bcell development proceeds as a series of sequential check points designed to ensure the production and coupling of functional heavy and light chains, and the integrity of the Ig signal transduction cascade (1). Mutations which lead to disruption of the Ig receptor complex or downstream signaling components can have dramatic effects on the generation and/or function of developing B cells (2–4). The X chromosome–encoded cytoplasmic Bruton's tyrosine kinase (Btk)1 is required for efficient signaling through the antigen receptor and B cell development in both mice and humans (5–8). In humans, mutations in Btk result in X-linked agammaglobulinemia (XLA), a severe immunodeficiency that manifests as a block in B cell development at the pre–B cell stage, rendering affected males virtually devoid of peripheral B cells (9–11). A spontaneous mutation in Btk in mice results in the milder X-linked immunodeficiency, xid, which is characterized by reduced numbers of splenic B cells and a failure of these B cells to enter the long-lived B cell pool (12, 13). Additionally, xid B cells have an unusual surface phenotype that is not characteristic of either immature or mature B cells (14, 15), suggesting some defect in development after their generation in the bone marrow. Furthermore, peritoneal B1 cells are absent (16) and there are specific Ig isotype deficiencies. However, despite these immunodeficient traits, the mice are robust and have a normal life span. The variability in severity of disease between xid and XLA reflects genuine species-specific differences in the requirement for Btk during B cell development rather than differences in site-specific mutations in Btk (7, 8, 11).
Although Btk is thought to be a component of several signal transduction pathways, it is almost certainly the disruption of the Ig signal transduction pathway that results in the immunodeficiencies of XLA and xid. In vitro cross-linking of surface IgM on xid B cells fails to promote cell cycle progression, consistent with the observation that XLA and xid phenotypes include a failure to expand specific B cell populations (pre–B cells in humans and B1 cells in mice) whose development is thought to require productive antigen receptor signaling (17, 18).
The molecular control of cell cycle progression involves the sequential activation of a family of serine/threonine kinases known as cyclin-dependent kinases, or cdks, and their subsequent phosphorylation of specific substrates (19, 20). cdks are activated in part by physical coupling with cyclins, a family of regulatory subunits that are induced at phase-specific stages during the cell cycle. The decision to enter S phase occurs late in G1, at the restriction point, R, after which the cells are committed to DNA replication and cellular division. A major component of the restriction point is the phosphorylation and inactivation of the protein production of the retinoblastoma gene, Rb, during G1 by the cyclin D–associated kinase activity (21). However, despite the requirement for Rb phosphorylation for S phase entry, inactivation of Rb does not guarantee cell cycle progression since Rb phosphorylation can be detected in cell lines undergoing apoptosis (22, 23). In such systems, ectopic expression of the antiapoptotic molecule, Bcl-2, permits orderly cell cycle progression. These and other experiments led to the hypothesis that cellular division can only be achieved with the engagement of the proliferative machinery in the presence of antiapoptotic proteins (22, 24, 25).
The Bcl-2–related antiapoptosis regulatory protein, Bcl-xL, is expressed at different stages during B cell ontogeny (26–29). Intriguingly, Bcl-xL is expressed at stages of B cell development arrested in XLA (pre–B cells) and xid (B1 B cells) (29) and is known to be upregulated as a consequence of antigen receptor cross-linking (27–29). The importance of Bcl-xL expression during B cell development is demonstrated by the expansion of the pro-pre-B cell compartment in two independently generated bcl-xL transgenic mouse models (26, 27) and the dramatic loss of peripheral lymphoid system in chimeric mice homozygous for a targeted deletion in bcl-x (30).
Given that the defects in XLA and xid both affect expansion of B cells at developmental stages dependent on Ig signaling, we explored the influence of defective Btk on the induction of cell cycle regulatory proteins after anti-Ig stimulation. Our results indicate that the Ig signal transduction cascade that uses mutant Btk does stimulate the induction of cyclins, cdks, and kinase activity; however, there is a specific failure to normally upregulate Bcl-xL. The induction of cell cycle regulatory machinery via mutant Btk was sufficient to promote proliferation in the presence of transgene-driven Bcl-xL expression. These results demonstrate a specific requirement for the antiapoptotic protein Bcl-xL during antigen receptor–induced B cell proliferation and links certain immunodeficient traits in xid with failure to induce this viability-enhancing protein.
Materials and Methods
Mice.
Male and female CBA/N (xid) mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and used as breeding pairs at DNAX Research Institute. CBA/Ca mice were obtained from The Jackson Laboratory and used as control mice. Males from two previously described and independently derived strains of bcl-x transgenic mice (bcl-x-87; reference 26, and bcl-x- 81; reference 25) were used in breedings with female CBA/N (xid).
Reagents.
Antibodies used in this study were monoclonal rat anti–mouse cyclin D3 and D2, rabbit anti–mouse cyclin E, cyclin A, cyclin B, cdk2, cdk4, and cdk6; these reagents were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse anti–mouse cell division cycle (cdc)2, mouse anti–human Rb, and anti–Bcl-2 antibodies used for Western blotting were purchased from PharMingen (San Diego, CA). The anti-CD43 FITC, anti-B220 PE, anti-CD5 PE, and anti-B220 FITC used in flow cytometric analysis were purchased from PharMingen. Anti-Bcl-xL and anti-Bad were purchased from Transduction Laboratories (Lexington, KY). Horseradish peroxidase–conjugated anti-rat, rabbit, and mouse secondary reagents were purchased from Amersham Corp. (Arlington Heights, IL). The anti-cdc2 antibody used for immunoprecipitations was purchased from GIBCO BRL (Gaithersburg, MD). The substrate for the kinase assay, histone H1, was purchased from Boehringer Mannheim (Indianapolis, IN). The affinity purified F(ab′)2 goat anti–mouse IgM used to stimulate B cells was purchased from Cappel Labs. (Malvern, PA). The rat anti–mouse monoclonal anti-CD40 antibody has been described previously (31).
Purification of Splenic B Cells.
To isolate splenic B cells, single cell suspensions were prepared from spleens of unprimed mice. Red blood cells were lysed using red blood cell lysis buffer (Sigma Chemical Co., St. Louis, MO) according to the manufacturer's instructions. To deplete T cells, splenocytes were stained using anti–Thy-1 (Dupont-NEN, Boston, MA) and anti-CD4 (RL172), followed by incubation in rabbit complement (Cedarlane Labs. Ltd., Hornby, Ontario, Canada) at 37°C for 45 min. These cells were consistently >90% B2201/IgM1 and included both the low and high density B cells.
In Vitro Stimulations and Whole Cell Lysate Preparation.
The ex- pression of cell cycle regulatory proteins in anti-Ig–stimulated B cell populations was evaluated using procedures previously described (32). In brief, 50-ml cultures were set up at 106 B cells/ml in 75 cm2 flasks (Becton Dickinson Labware, Lincoln Park, NJ) and stimulated with 25 μg/ml F(ab′)2 anti-IgM. Cells were collected at 6, 12, 24, 36, 48, and 72 h after stimulation. All stimulations were carried out in supplemented RPMI containing 10% FCS (JR Scientific, Woodland, CA), 5 × 105 M 2-ME (Polysciences, Inc., Warrington, PA), 2 mM glutamate (JR Scientific), 10 mM Hepes buffer (Irvine Scientific, Santa Ana, CA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Irvine Scientific). After stimulation, cells were recovered, washed three times in cold PBS, divided into three pellets, and stored at −80°C for later use in Western blotting and in vitro kinase assays. When all time points were collected, one pellet from each time point was lysed in NP-40 lysis buffer (1% NP-40, 250 mM NaCl, 1 mM Hepes, pH 7.5, and 1 mM dithiothreotol [DTT; United States Biochemical Corp., Cleveland, OH] with protease inhibitors added; final concentration: 5 μg/ml aprotinin [Sigma Chemical Co.], 125 μg/ml Pefabloc [Boehringer Mannheim], 5 μg/ml pepstatin [Sigma Chemical Co.]). Protein concentrations were calculated using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA) according to the manufacturer's procedures. An equal volume of 2× SDS sample buffer (Novex, San Diego, CA) with 2-ME was then added to the total cell lysates. 125 μg of lysate was run per time point on 12% Novex reducing gels according to manufacturer's suggested protocol (6% gels were used to detect the Rb protein), and transferred onto Immobilon PVDF membranes (Millipore, Bedford, MA) at 30 V overnight in 4°C. The mouse plasmacytoma line P3X was obtained from the American Type Culture Collection (Rockville, MD) and used as a positive control on all gels.
In Vitro Kinase Assay.
cdk2- and cdc2-dependent kinase assays were performed using the histone substrate according to procedures described elsewhere (33). In brief, frozen pellets were lysed in 1 ml 0.1% NP-40 lysis buffer containing 50 mM Hepes, pH 7.0, and 250 mM NaCl in the presence of the above protease inhibitors. Protein concentration was calculated as described above, and 200 μg of total protein was used for each kinase assay. In brief, lysates were precleared with normal rabbit serum followed by Zysorbin (Zymed, South San Francisco, CA). 1 μg of anti-cdk2, anti-cdc2, or normal rabbit sera was added to the lysate and incubated at 4°C for 4 h with rocking. Protein G beads (Pharmacia Biotech, Inc., Piscataway, NJ) were added and the lysates were rotated in the cold for another hour. Immunoprecipitates were washed 4 times in lysis buffer followed by two washes in kinase buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 1 mM DTT). The kinase reactions were carried out in a 50 μl volume containing 2.5 μg histone H1 and 5 μCi [32P]γATP (Amersham Corp.) per reaction. Reactions proceeded for 30 min at room temperature with rocking. Samples were run on 12% Novex precast gels, fixed, dried, and exposed to phosphor screen (Molecular Dynamics, Sunnyvale, CA). The results were read on a Storm 860 phosphorimager (Molecular Dynamics).
Cell Cycle Analysis.
Cell cycle analysis was performed using a modified method of Carayon and Bord (34). Purified B cells were cultured at 106/ml in 200-μl aliquots in the presence of 50 μM bromodeoxyuridine (BrdU; Sigma Chemical Co.) and an equimolar concentration of cytidine (Sigma Chemical Co.) plus 25 μg/ml anti-IgM for times indicated. Cells were harvested and stored in PBS with 0.5% paraformaldehyde overnight. PBS with 5% Tween 20 was added to permeabilize the nuclear membrane. The cells were then washed and resuspended in a solution of 10 mg/ml DNAse I (Sigma Chemical Co.) plus anti-BrdU (Becton Dickinson, Mountain View, CA). Finally propidium iodide (PI) is added and then analysis is performed on FACScalibur® using Cellquest software (Becton Dickinson).
Thymidine Incorporation.
200-μl aliquots (2 × 105 total cells) of stimulated cells were placed in 96-well flat-bottomed plates at times indicated and pulsed for 45 min in 1 μCi [3H]thymidine (Amersham Corp.). Cells were harvested and incorporated cpms were counted using a PHD cell harvester (Cambridge Technology Corp., Cambridge, MA).
Testing of Serum IgM and IgG3 Levels by ELISA.
Sera was col- lected from the experimental mice. Samples were serially diluted and compared with known standards of purified IgM and IgG3 as previously described (35).
Results
Induction of Complete Complement of Cell Cycle Regulatory Proteins in Anti-Ig–stimulated xid B Cells.
Consistent with a previous report (36), purified B cells from xid mice showed little [3H]thymidine incorporation in response to stimulation with 25 μg/ml anti-IgM (Fig. 1 A). In contrast, the level of [3H]thymidine incorporation in xid B cells after stimulation with 25 μg/ml anti-CD40 was comparable to normal B cells (Fig. 1 B). Since B cells from xid mice have a selective inability to proliferate in response to anti-IgM stimulation, it was possible that the xid mutation in Btk affected induction of proteins that control cell cycle progression in response to antigen receptor cross-linking. To test this, lysates were prepared from anti-IgM stimulated xid and control B cells at different times after stimulation through 72 h and Western blot analysis of cell cycle regulatory proteins was performed. Fig. 2 shows the induction of regulatory proteins that control the entry into G1 (i.e., cyclin D2, cyclin D3, cdk4, cdk6, and Rb). Cyclins D2 and D3 were induced after anti-Ig stimulation of xid B cells with the same kinetics as in the control CBA/Ca B cells, the only difference being the slightly greater induction of the faster migrating form of cyclin D3 in the control B cells, the significance of which is not known (Fig. 2, A and B). Similarly, both cdk4 and cdk6 were present to comparable extents in both the anti-Ig–stimulated control and xid B cells (Fig. 2, C and D). Most notable was the comparable induction and hyperphosphorylation of Rb in both xid and control B cells after anti-Ig stimulation (Fig. 2 E). Surprisingly, despite the inability of xid B cells to proliferate, the induction of cyclins and cdks involved in S phase progression were also detected. Specifically, both cyclins E and A (Fig. 3, A and B), along with their kinase partners cdk2 and cdc2 (Fig. 3, C and D) were detected at about half the levels seen in control B cells. The only significant difference in these two populations was found at the level of expression of cyclin B, the regulatory protein that governs the progression through mitosis. Although cyclin B was only weakly induced in anti-Ig–stimulated xid B cells, it was easily detected in control B cells (Fig. 3 C).
Figure 1.

Defective anti-IgM–triggered proliferation in xid B cells. B cells from xid (black bar) and CBA/Ca (gray bar) were stimulated with mAbs against CD40 (A) or F(ab′)2 anti-IgM (B) and pulsed with [3H]thymidine for 1 h at the time indicated. Standard errors are shown and results are representative of three independent experiments.
Since both the cyclins and cdk partners required for cell cycle progression were induced in anti-Ig–stimulated xid B cells despite the inability of these cells to proliferate, we performed cdk2- and cdc2-associated kinase assays to determine if cyclin/cdk complexes were assembled correctly and that activating posttranslational modifications had occurred. Fig. 4 shows that both cdk2- and cdc2-associated kinase activities are clearly present in anti-IgM–stimulated xid B cells at 48 and 72 h after stimulation, albeit at approximately half the levels detected in anti-Ig–treated control B cells. Collectively, these data indicate that anti-Ig–stimulated xid B cells induce substantial levels of active cell cycle regulatory proteins despite their inability to complete the cell cycle in response to this stimulus.
Figure 4.

In vitro kinase assays from anti-IgM–stimulated B cells. Stimulated B cells were collected at the time points indicated and used to measure (A) cdk2- and (B) cdc2-associated specific kinase activity using Histone H1 as the substrate in the presence of [32P]gATP. Kinase activity was quantitated by comparing the amount of 32P incorporated into the substrate by phosphorimager analysis. Kinase activity detected in xid (black bars) and CBA/Ca (gray bars) are shown as arbitrary units. Similar results were obtained in three independent experiments.
Anti-Ig–stimulated xid B Cells Successfully Pass the G1 Restriction Point, then Enter an Aborted S Phase.
Since cell cycle regulatory protein induction and kinase activity were detectable in anti-Ig–stimulated xid B cells, we carefully examined the various stages of cell cycle progression using BrdU and PI labeling to distinguish the discrete steps of cell cycle progression. As previously described (34, 37), the combination of long-term BrdU incorporation plus PI staining allows the discrimination of cells that are in (a) the original G0/G1 phase; (b) the S phase; (c) the G0/G1 phase of the second cell cycle; and finally (d) apoptotic cells (as illustrated diagrammatically in Fig. 5, top). Anti-IgM–stimulated control B cells clearly revealed a characteristic S phase entry that began as early as 24 h after stimulation (Fig. 5, center, arrow) (for review see reference 38). By 36 h, cells were beginning to successfully accumulate in the second round of the cell cycle and could still be detected in S phase at 48 and 72 h after stimulation. Such detailed analyses revealed that anti-IgM–stimulated xid B cells also successfully passed the restriction point and entered S phase at 36 h (Fig. 5, bottom, arrow), albeit some 12 h later than in the control B cells. These data presumably explain the small amounts of thymidine incorporation that can be detected at 48 and 72 h after stimulation of xid B cells with anti-Ig (Fig. 1). However, BrdU and PI costaining convincingly established that there was no accumulation of anti-Ig–stimulated cells in the second round of the cell cycle (Fig. 5, bottom), indicating that the cells do not successfully complete S phase. Indeed, by 48 and 72 h after stimulation, >85% of the anti-Ig–stimulated xid B cells are apoptotic (Fig. 5, bottom).
Figure 5.

BrdU incorporation and PI analysis of anti-IgM–stimulated B cells from CBA/Ca and xid B mice. The diagram at the top indicates the cell cycle position of cells at the time the cells were harvested. This analysis identifies cells in G0 (G0), S phase (S), and those which have completed S phase and are now in the second cell cycle G0 phase (2nd G0), and apoptotic cells (Apo). B cells from CBA/Ca (top row) and xid (bottom row) mice were stimulated with 25 μg/ml of anti-IgM in the presence of BrdU. Cells were harvested at time points indicated and fixed to permit intranuclear staining of DNA with PI. The initial entry of B cells from xid and CBA/Ca is indicated with an arrow. Analysis was performed on FACSCalibur® using CellQuest software.
These analyses are also consistent with previous reports showing that xid B cells are more prone to undergo apoptosis than control B cells (29, 39). The increased susceptibility of xid B cells to apoptosis was evident as early as 12 h after stimulation with anti-Ig, when 20% of the cells were found to be apoptotic by PI staining compared with 5% of the normal B cells (Fig. 5). The increased susceptibility to apoptosis was unrelated to anti-Ig stimulation, since xid B cells cultured in media alone died at a significantly faster rate than did their non-xid counterparts (data not shown). Even in the presence of the strong mitogen, anti-CD40, that clearly stimulates xid B cells to proliferate (see Fig. 1), there is a higher proportion of apoptotic cells in the cultures than in the anti–CD40-stimulated control cells (data not shown). Thus, in addition to the selective inability to proliferate to anti-IgM stimulation, xid B cells are more apoptosis-susceptible than are their normal B cell counterparts.
Diminished Induction of Bcl-xL in Anti-Ig–stimulated xid B Cells.
To explore the mechanism underlying the increased apoptosis susceptibility of xid B cells, we evaluated expression of the apoptotic regulatory proteins Bcl-2, Bax, and Bcl-xL in this population. As shown in Fig. 6, A and B, the levels of Bcl-2 and Bax are comparable in anti-Ig–stimulated xid and control B cells. In contrast, there was a striking and consistent decrease in Bcl-xL induction in anti-Ig– stimulated xid B cells compared with normal B cells (Fig. 6 C). The induction of Bcl-xL in anti-Ig–stimulated normal B cells was found to begin as early as 12 h after stimulation, presumably exerting its antiapoptotic effects long before the cells enter S phase.
Figure 6.

Analysis of Bcl-2 and related proteins. B cells were stimulated as described in the legend to Fig. 2 and were collected at the time points indicated. Western blots were prepared and screened with antibodies specific for Bcl-2 (A), Bax (B) or Bcl-xL (C). Horseradish peroxidase conjugates were used as secondary reagents followed by visualization with enhanced chemiluminescence.
Bcl-xL Transgene Permits Anti-Ig–induced xid B Cell Proliferation.
To determine whether the diminished induction of Bcl-xL in anti-Ig–stimulated xid B cells contributed to their failure to proliferate, we evaluated the consequence of introducing two independently generated bcl-x transgenes into these animals. CBA/N (xid) female mice, homozygous for the mutation in the X-linked gene Btk, were bred to male mice that were heterozygous for a bcl-x transgene (bcl-x-87 or bcl-x-81). These crosses provided offspring in which all of the males were xid while the females were non-xid. Additionally, the offspring were either heterozygous for the bcl-x transgene or were wild-type. The presence of the bcl-x transgene permitted B cells from F1 male (xid.bcl-x-87) mice to proliferate to anti-IgM stimulation; these cells successfully completed the cell cycle and could be detected in the G0/G1 stage of second cell cycle (Fig. 7), confirming our observations that mutations in Btk do not block cell cycle machinery induction. However, it should be noted that there was still a delay in entry into S phase that was not detected in the female F1 (xid.bcl-x-87) mice, suggesting that mutant Btk continued to effect the rate of entry into cell cycle. Indeed, although splenic B cells from transgene positive male F1 (xid.bcl-x-87) mice did proliferate to anti-Ig, the proportion of responding cells was only about half of that detected in the female F1 (xid. bcl-x-87) mice. Consistent with this, we detected only about half the amount of [3H]thymidine incorporation in the male F1 (xid.bcl-x-87) mice compared to that in the female F1 (xid.bcl-x-87) mice (Table 1).
Figure 7.

BrdU and PI analysis of stimulated B cells from xid and F1 (xid.bcl-x-87) offspring. B cells from xid, female F1 (xid.bcl-x-87), and male F1 (xid.bcl-x-87) mice were stimulated with 25 μg/ml anti-IgM in the presence of BrdU and collected at the indicated time points. Cells were prepared and analyzed as described in the legend to Fig. 5. Arrows indicate the initial entry of cells into S phase.
Table 1.
Comparison of [3H]thymidine Incorporation in F1 (xid.bcl-x) and xid Mice
| Mouse | [3H]thymidine* incorporation | Transgene | Btk‡ | Phenotype | ||||
|---|---|---|---|---|---|---|---|---|
| Female F1 (xid.bcl-x-87) | 15,489 | yes | +/− | non-xid | ||||
| Female F1 (xid.bcl-x-87) | 12,300 | yes | +/− | non-xid | ||||
| Female F1 (xid.bcl-x-81) | 14,032 | yes | +/− | non-xid | ||||
| Female F1 (xid.bcl-x-81) | 11,089 | yes | +/− | non-xid | ||||
| Male F1 (xid.bcl-x-87) | 7,050 | yes | − | xid | ||||
| Male F1 (xid.bcl-x-87) | 6,500 | yes | − | xid | ||||
| Male F1 (xid.bcl-x-81) | 1,200 | yes | − | xid | ||||
| Male F1 (xid.bcl-x-81) | 1,999 | yes | − | xid | ||||
| Male xid 1 | 2,000 | na | − | xid | ||||
| Male xid 2 | 1,524 | na§ | − | xid |
2 × 105 B cells were stimulated for 48 h with 25 μg/ml anti-IgM and pulsed for the final 4 h with 1 μCi [3H]thymidine.
+ indicates presence of wild-type Btk; − indicates presence of mutant Btk. Since Btk is an X-linked gene, males carry only one allele.
na, not applicable.
Partial Correction of Other xid Traits.
Since the Bcl-xL trans- gene restored the proliferative response in xid B cells, we examined the male F1 (xid. bcl-x-87) mice to evaluate the overall ability of the Bcl-xL transgene to complement other immunodeficient traits. The male F1 (xid.bcl-x-87) mice, all of which expressed the bcl-x transgene along with defective Btk (F1 male Nos. 1, 2, 3, and 4, Table 2) had ∼3 times the number of splenic B cells as were detected in normal CBA/Ca mice and 15 times the number of splenic B cells found in the CBA/N (xid) mice. The effect of the bcl-x transgene was also detected in the female F1 (xid.bcl-x-87) mice carrying the bcl-x transgene (F1 female Nos. 2 and 4, Table 2) increasing the splenic B cells numbers to 2–3 times that detected in the normal CBA/Ca controls or female F1 (xid.bcl-x-87) mice that did not express the transgene. Consistent with these findings, the presence of the bcl-x transgene in the male F1 (xid.bcl-x-87) mice increased the total number of peritoneal B cells to that detected in the normal CBA/Ca control mice, which is >10 times the number of B cells detected in the peritoneal cavity of xid mice (Table 3).
Table 2.
Comparison of Splenic B Cell Numbers and Phenotype in CBA/Ca, xid, Female F1 (xid.bcl-x-87), and Male F1 (xid.bcl-x-87) Mice
| Strain and gender | No. | Total B cell number × 107 | MFI* IgM | MFI* IgD | Percentage of B cells‡ | Transgene | Btk‖ | xid | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CBA/Ca males | 1 | 4.7 | 429 | 953 | 43 | na§ | + | no | ||||||||
| 2 | 3.8 | 480 | 974 | 43 | na | |||||||||||
| xid males | 1 | 0.6 | 881 | ND | 22 | na | − | yes | ||||||||
| 2 | 0.9 | 1015 | ND | 23 | na | |||||||||||
| 3 | 0.9 | 1220 | 804 | 24 | na | |||||||||||
| 4 | 0.7 | 1182 | 934 | 22 | na | |||||||||||
| F1 (xid.bcl-x-87) females | 1 | 5.7 | 427 | 1,258 | 36 | no | −/+ | no | ||||||||
| 2 | 8.0 | 567 | 1,023 | 50 | yes | |||||||||||
| 3 | 12.2 | 440 | ND | 32 | no | |||||||||||
| 4 | 4.2 | 702 | ND | 31 | yes | |||||||||||
| F1 (xid.bcl-x-87) males | 1 | 12.5 | 731 | 965 | 50 | yes | − | yes | ||||||||
| 2 | 10.5 | 765 | 881 | 50 | yes | |||||||||||
| 3 | 14.0 | 983 | 1,020 | 54 | yes | |||||||||||
| 4 | 12.8 | 990 | 1,005 | 52 | yes | |||||||||||
Mean fluorescence intensity (MFI) was determined by flow cytometry using FITC-conjugated anti-IgM or PE-conjugated anti-IgD antibodies.
Percentage of B cells was determined for lymphoid gate only.
na, not applicable.
+ indicates wild-type Btk; − indicates mutant Btk.
Table 3.
Number and Phenotype of B Cells in the Peritoneal Cavity of CBA/Ca, xid, and male F1 (xid.bcl-x-87) Mice
| Total number of B cells × 105 | Percentage of CD5 B cells‡ | Total number of CD5 B cells × 104 | Transgene | Btk‖ | xid | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CBA/Ca | 1 | 11 | 58 | 64 | na§ | + | no | |||||||
| male | 2 | 13 | 55 | 71 | na | |||||||||
| xid | 1 | 2 | 12 | 2 | na | − | yes | |||||||
| male | 2 | 1 | 15 | 1 | na | |||||||||
| F1 male | 1 | 14 | 4 | 5 | yes | − | yes | |||||||
| (xid.bcl-x-87) | 2 | 14 | 4 | 6 | yes |
Normal peritoneal cells were recovered from 8-wk-old mice by gently washing peritoneal cavity with 10 ml of cold RPMI with 10% FCS. More than 9 ml was recovered from each mouse by this procedure.
Percentage includes only cells within the lymphoid gate.
na, not applicable.
+ indicates wild-type Btk; − indicates mutant Btk.
In addition to expanding the peripheral B cell pool, the bcl-x-87 transgene provided some correction of the unusually high levels of sIgM expressed on xid splenic B cells (Table 2). Transgene positive splenic B cells from male F1 (xid.bcl-x-87) mice expressed less IgM than the B cells from the xid mice, whereas the levels of IgD were comparable between these two groups (Table 2). However, the levels of IgM on B cells from the male F1 (xid.bcl-x-87) was still higher than that detected on either the CBA/Ca mice or the female F1 (xid.bcl-x-87). Additionally, though the peritoneal compartment of the male F1 (xid.bcl-x-87) mice was expanded, most of the cells did not express the typical cell surface phenotype of CD5 B cells, which represent the major B cell subset in a normal peritoneal cavity (Table 3). The transgene did increase the number of CD5 B cells to fourfold that detected in the xid mice. However, this still reflected a 90% reduction when compared with wild type animals.
To determine the effect of the bcl-x transgene on viability, splenic B cells from xid, CBA/Ca controls, male and female F1 (xid.bcl-x-87) mice that carried the bcl-x transgene were cultured in vitro and viability compared. As expected, viability of xid B cells was less than B cells from normal control CBA/Ca (Fig. 8), however, the bcl-x-87 transgene substantially enhances the viability of the B cells in both the male and female F1(xid.bcl-x-87) mice.
Figure 8.

Comparison of in vitro viability. Splenic B cells from CBA/Ca, xid, male F1 (xid.bcl-x-87), and female F1 (xid.bcl-x-87) mice were cultured in 96-well flat-bottomed wells at 106 cells/ml in RPMI with 10% FCS for times indicated. From days 1 to 6, viability of spleen B cells was assessed by Trypan blue exclusion. Results are representative of three independent experiments.
Although the bcl-x-87 transgene increased the number and viability of B cells from xid F1 male mice, Bcl-x was incapable of correcting the reduced levels of serum IgM and IgG3 levels in the male F1 (xid.bcl-x-87) mice compared with xid mice (Fig. 9). Curiously, the levels of IgM in the F1 male (xid.bcl-x-87) mice were consistently lower than levels detected in the male xid mice. Serum IgM and IgG3 levels in the female F1 (xid.bcl-x-87) mice were indistinguishable from that detected in the parent C57BL/6 strain.
Figure 9.

Serum immunoglobulin levels of IgM and IgG3. Sera was collected from 8-wk-old xid (n = 5), CBA/Ca (n = 4), F1 female (n = 5), and F1 male (n = 6) mice. Biotin-conjugated anti-IgG3 and anti-IgM reagents were used in combination with horseradish peroxidase–conjugated streptavidin in ELISAs to quantitate serum Ig levels. Serial dilutions of each sample were made and compared with standard curves generated by IgM or IgG3 preparations of known concentrations. Asterisks indicate mice carrying the bcl-x transgene.
Interestingly, only the F1 offspring from the bcl-x-87 transgenic subline showed any correction of the xid phenotype. Table 1 indicates that anti-Ig–stimulated B cells from male F1 (xid.bcl-x-81) mice showed little difference in [3H]thymidine incorporation when compared with the xid mice. The bcl-x-81 transgene expresses at high levels in developing B cells (26), but at low levels in peripheral B cells (Behrens, T., et al., unpublished data). Additionally, there was minimal increase in the peripheral B cell pool, little enhanced viability, and no difference in serum Ig levels between the F1 male (xid.bcl-x-81) in comparison with the xid males (data not shown).
Discussion
Our initial experiments focused on the hypothesis that the observed inability of xid B cells to proliferate to antigen receptor cross-linking stemmed from a failure to induce components of the cell cycle machinery. However, although there was an ∼50% reduction in cyclin, cdk, and kinase activity in anti-Ig–stimulated xid B cells compared with normal controls, the complete complement of cyclins and cdks were induced in anti-Ig–stimulated xid B cells. Additionally, the kinetics of protein induction and active kinase induction in stimulated xid B cells was comparable to that detected in anti-Ig–treated control B cells. In data not shown, we confirm that the destruction of the cdk inhibitor, p27, after stimulation with anti-Ig antibodies was also indistinguishable in xid and control B cells, as was the appearance of the INK4 inhibitors, p18 and p19. Together these results suggest that the signals inducing cell cycle machinery after antigen receptor cross-linking in B cells from xid mice are intact although insufficiently amplified due to mutations in Btk.
The decision to undergo cellular replication occurs late in G1 at the restriction point, R, after which the cell is committed to undergo DNA replication and cellular division. Despite the diminished induction of cell cycle regulatory proteins in anti-Ig–stimulated xid B cells, these cells passed the restriction point and began entry into S phase, albeit some 12 h after control B cells. The ability of anti-Ig–stimulated xid B cells to pass the restriction point suggests that the insufficient amplification of the Ig-generated signal simply resulted in a delay in passage through R, rather than an inability to pass R. Typically, unless severe damage to DNA is encountered, the entry into S phase guarantees completion of cell cycle progression. Indeed, there was significant cdk2- and cdc2-associated kinase activity present at time points during which the S and G2/M phases should have occurred; however, at this time at least 85% of the cells were apoptotic, indicating that the cell cycle machinery was still engaged despite the fact that the cells were dying.
Our finding that Bcl-x induction was diminished in anti-Ig–stimulated xid B cells is consistent with reports from Anderson et al. (29) and Choi et al. (28). We extend these observations by demonstrating that transgene driven expression of Bcl-xL substantially corrects the inability of xid B cells to proliferate in response to stimulation with anti-Ig antibodies in vitro. It is noteworthy that we could detect some Bcl-xL induction in anti-Ig–stimulated B cells from xid mice, indicating again that the signaling pathway leading to Bcl-xL induction is not entirely disrupted. It seems likely that some critical level of Bcl-xL protein is needed to permit anti-Ig–induced proliferation since reconstitution of the proliferative response was only detected in xid crosses with the bcl-x-87 transgenic mouse, which expresses high levels of Bcl-xL in the peripheral B cell compartment. Despite the difference in phenotype, both the bcl-x-87 and bcl-x-81 transgenics are carried on the C57BL/6 background. Thus, it is unlikely that the correction in the xid phenotype observed with the male F1 (xid.bcl-x-87) mice is due to C57BL/6 background genes.
It is significant that the proportion of anti-Ig–responsive B cells in the male F1 (xid.bcl-x-87) mice and the rate of entry into S phase was still reduced compared with female F1 (xid.bcl-x-87) mice. We suspect this results from the still incompletely amplified signal through Btk leading to half-maximal induction of cell cycle regulatory proteins. These results suggest that the crippling effect of mutant Btk on the proliferative response is the failure to properly amplify signals generated by the antigen receptor. This is supported by our data showing the induction of both cell cycle regulatory proteins plus Bcl-xL although at levels far below those detected in wild-type mice. Thus, one would predict that if signals generated at the antigen receptor could be significantly enhanced that perhaps B cells from xid mice could be stimulated to proliferate. Indeed, reports from Lindsberg et al. (40) showed that xid B cells can proliferate in the presence of anti-Ig reagents, which induces extensive and prolonged antigen receptor cross-linking.
The physiological significance of defective Bcl-xL induction in relation to other immunodeficient traits is established in our male F1 (xid.bcl-x-87) mice, where there was a dramatic increase in the number of splenic and peritoneal B cells plus enhanced in vitro viability. The hyperreconstitution in splenic B cell numbers we observed is comparable to that reported in the original description of the bcl-x-87 transgenic line (27). We suspect that the expansion of the splenic B cell pool stems from the demonstrated enhancement of B cell viability coupled with inhibition of cell death rather than from enhancing B cell proliferation. The transgene, however, provided only minimal correction of the unusual phenotype of splenic B cells in the xid mice. Splenic B cells expressed IgM at levels intermediate between the xid and the CBA/Ca controls suggesting, at most, a partial correction of the atypical xid splenic B cell phenotype.
The number of B lymphocytes in the peritoneal cavity of male F1 (xid.bcl-x-87) mice was also expanded to numbers comparable to normal control mice. However, there was only a modest increase in the total number of CD5- expressing B cells compared with control mice. Furthermore, the expanded peritoneal B cell population in the male F1 (xid.bcl-x-87) mice was not the CD5 “sister” population since the B cells did not express Mac-1 nor the characteristic ratio of IgMhi/IgDlo (data not shown). The inability to reconstitute CD5 B cells is difficult to interpret since it has not been established that the bcl-x transgene is expressed in the CD5 B cell compartment; normal CD5 B cells express Bcl-xL constitutively (29), making the efficiency of transgene-driven Bcl-xL expression difficult to establish in these cells. Alternatively, we cannot rule out the possibility that there are additional defects in the Btk signal-transduction cascades that effect development of this particular B cell subset. Regardless of the mechanism, the poor reconstitution of the CD5 B cell population may partially explain the failure to correct the serum IgM and IgG3 deficiencies since CD5 B cells have been shown to be a major source of these isotypes in vivo (41).
Our demonstration that anti-Ig–stimulated xid B cells can induce the complete complement of cell cycle regulatory proteins while the cells continue to undergo apoptosis indicates that induction of cell cycle machinery alone is insufficient to ensure cellular replication. At least for B lymphocytes, induction of cell cycle machinery must be coupled with antiapoptotic proteins for proliferation. Furthermore, our results suggest a specific requirement for the antiapoptotic protein Bcl-x, since Woodland et al. has shown that transgene driven expression of Bcl-2 in xid mice could not support anti-Ig induced proliferation (39). Therefore, Bcl-xL may have a unique and nonredundant role in maintaining viability during antigen-driven B cell expansion.
Figure.
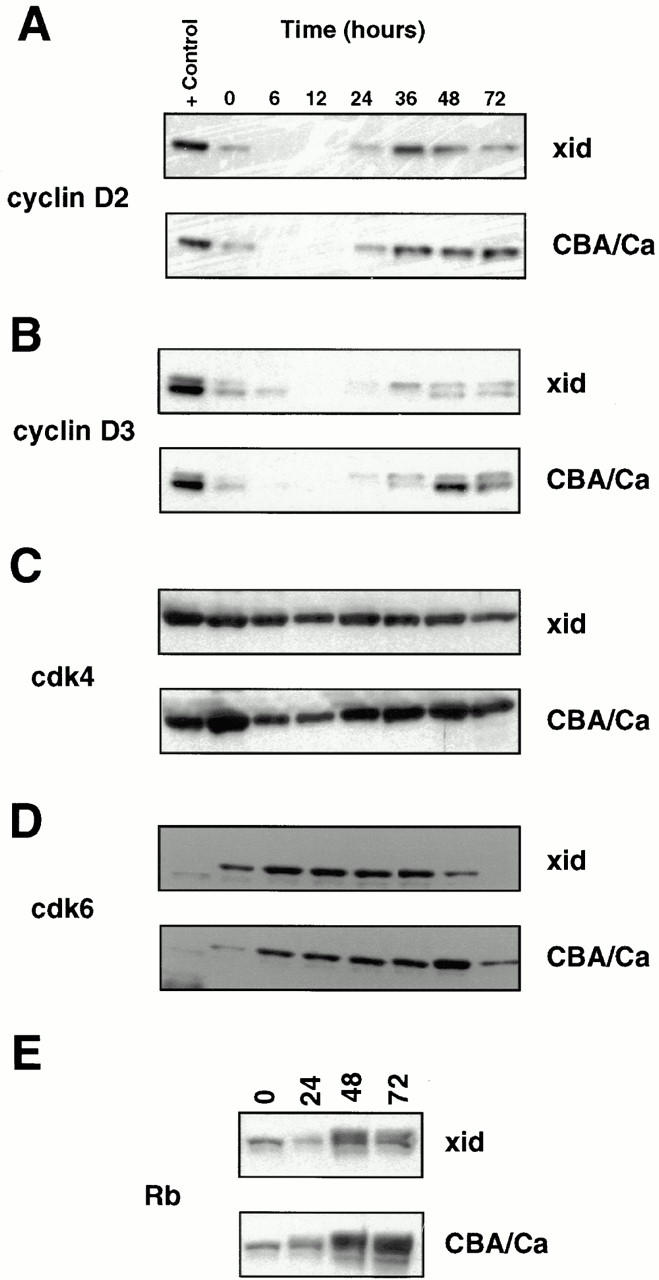

Figures 2 and 3. Induction of cell cycle regulatory proteins in anti-IgM–stimulated B cells from xid and CBA/Ca mice. B cells were stimulated with 25 μg/ml anti-IgM and pellets were collected at the indicated time points after stimulation. Figure 2: Western blots were prepared and sequentially screened with antibodies against cyclin D2 (A), cyclin D3 (B), cdk4 (C), cdk6 (D), and Rb (E). Figure 3: The same Western blot was sequentially screened with antibodies against cyclin E (A), cyclin A (B), cyclin B (C), cdk2 (D), and cdc2 (E). Horseradish peroxidase–conjugated secondary reagents were used in combination with enhanced chemiluminescence to visualize results. The cell line, P3X, was used as positive control.
Footnotes
1 Abbreviations used in this paper: BrdU, bromodeoxyuridine; Btk, Bruton's tyrosine kinase; cdc, cell division cycle; cdk, cyclin-dependent kinases; PI, propidium iodide; Rb, retinoblastoma gene; XLA, X-linked agammaglobulinemia.
DNAX Research Institute is fully funded by Schering Plough.
References
- 1.Billups LG, Lassoued K, Nunez C, Wang J, Kubagawa H, Gartland GL, Burrows PD, Cooper MD. Human B-cell development. Ann NY Acad Sci. 1995;764:1–8. doi: 10.1111/j.1749-6632.1995.tb55798.x. [DOI] [PubMed] [Google Scholar]
- 2.Satterthwaite, A., and O.N. Witte. 1996. Genetic analysis of tyrosine kinase function in B cell development. Annu. Rev. Immunol 14:131–154. [DOI] [PubMed]
- 3.Yel L, Minegishi Y, Coustan-Smith E, Buckley RH, Trubel H, Pachman L, Kitchingman GT, Campana D, Rohrer J, Conley ME. Mutations in the mu heavy-chain gene in patients with agammaglobulinemia. N Engl J Med. 1996;335:1486–1493. doi: 10.1056/NEJM199611143352003. [DOI] [PubMed] [Google Scholar]
- 4.Kitamura, D., J. Roes, R. Kurn, and K. Rajewsky. 1991. A B cell deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 350: 423–426. [DOI] [PubMed]
- 5.Thomas, J.D., P. Sideras, C.I.E. Smith, I. Vorechovsk, V. Chapman, and W.E. Paul. 1993. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 261:355–358. [DOI] [PubMed]
- 6.Rawlings DJ, Saffron D, Tsukada D, Largaespada D, Grimaldi JC, Cohen L, Morh RN, Bazan FJ, Howard M, Copeland NG, Jenkings NA, Witte ON. Mutations in unique region of Btk in immunodeficient XIDmice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 7.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Muller S, Kantor AB, Herzenberg LA, Rosen RS, Sideras P. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 8.Lerner, J.D., M.W. Appleby, R.N. Morh, S. Chien, D.J. Rawlings, C.R. Maliszewski, O.N. Witte, and R.M. Perlmutter. 1995. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity 3:301–312. [DOI] [PubMed]
- 9.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, Belmont JW, Cooper MD, Conley ME, Witte ON. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–284. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 10.Vetrie D, Vorechovsky I, Sideras P, Holand P, Davies A, Flinter A, Hammarstrom AL, Kinnon C, Levinsky R, Bobrow M, et al. The gene involved in X-linked agammaglobulinemia is a member of the srcfamily of protein- tyrosine kinases. Nature. 1993;361:226–229. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 11.Conley ME, Parolini O, Rohrer J, Campana D. X-linked agammaglobulinemia: new approaches to old questions based on the identification of the defective gene. Immunol Rev. 1994;138:5–21. doi: 10.1111/j.1600-065x.1994.tb00844.x. [DOI] [PubMed] [Google Scholar]
- 12.Scher I. The CBA/n mouse strain; an experimental model illustrating the influence of the X-chromosome on immunity. Adv Immunol. 1982;33:1–71. doi: 10.1016/s0065-2776(08)60834-2. [DOI] [PubMed] [Google Scholar]
- 13.Oka Y, Rolink AG, Andersson J, Kamanaka M, Uchida J, Yasui T, Kishimoto H, Kikutani H, Melchers F. Profound reduction of mature B cell numbers, reactivities and serum Ig levels in mice which simultaneously carry the XIDand CD40 deficiency genes. Int Immunol. 1996;8:1675–1685. doi: 10.1093/intimm/8.11.1675. [DOI] [PubMed] [Google Scholar]
- 14.Hardy RR, Hayakawa K, Parks DR, Herzenberg LA. Demonstration of B cell maturation in X-linked immunodeficient mice by simultaneous 3-color immunofluorescence. Nature. 1983;306:270–272. doi: 10.1038/306270a0. [DOI] [PubMed] [Google Scholar]
- 15.Mosier D. Are xid B lymphocytes representative of any normal B cell population? . J Mol Cell Immunol. 1985;2:70–78. [PubMed] [Google Scholar]
- 16.Hayakawa K, Hardy RR, Herzenberg LA. Peritoneal Ly1 B cells: genetic control and autoantibody production. Eur J Immunol. 1986;16:450–465. doi: 10.1002/eji.1830160423. [DOI] [PubMed] [Google Scholar]
- 17.Melchers F, Haasner D, Grawunder U, Kalberer C, Karasuyama H, Winkler T, Rolink AG. Roles of IgH and L chains and of surrogate H and L chains in the development of cells of the B lymphocyte lineage. Annu Rev Immunol. 1994;12:209–225. doi: 10.1146/annurev.iy.12.040194.001233. [DOI] [PubMed] [Google Scholar]
- 18.Wortis HH. Surface markers, heavy chain sequences and B cell lineages. Int Rev Immunol. 1992;8:235–246. doi: 10.3109/08830189209055576. [DOI] [PubMed] [Google Scholar]
- 19.Sherr CJ. Cancer cell cycle. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 20.Lees E. Cyclin dependent kinase regulation. Curr Opin Cell Biol. 1995;7:773–780. doi: 10.1016/0955-0674(95)80060-3. [DOI] [PubMed] [Google Scholar]
- 21.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 22.Evan GI, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7:825–834. doi: 10.1016/0955-0674(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 23.Pandey S, Wang E. Cells en route to apoptosis are characterized by the upregulation of c-fos, c-myc, c-jun, cdc2, and RB phosphorylation, resembling events of early cell-cycle traverse. J Cell Biochem. 1995;58:135–150. doi: 10.1002/jcb.240580203. [DOI] [PubMed] [Google Scholar]
- 24.Fanidi A, Harrington E, Evan GI. Cooperative interaction between c-mycand Bcl-2 proto-oncogenes. Nature. 1992;359:554–556. doi: 10.1038/359554a0. [DOI] [PubMed] [Google Scholar]
- 25.Fukada T, Hibi M, Tamanaka Y, Takahashi-Texuka M, Fujitani Y, Tamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT 3 in anti-apoptosis. Immunity. 1996;5:449–460. doi: 10.1016/s1074-7613(00)80501-4. [DOI] [PubMed] [Google Scholar]
- 26.Fang W, Mueller DL, Pennel CA, Rivard JJ, Li YS, Hardy RR, Schlissel MS, Behrens TW. Frequent aberrant immunoglobulin gene rearrangements in pro-B cells revealed by a bcl-xLtransgene. Immunity. 1996;4:291–299. doi: 10.1016/s1074-7613(00)80437-9. [DOI] [PubMed] [Google Scholar]
- 27.Grillot DAM, Merino R, Pena JC, Fanslow WC, Finkelman FD, Thompson CB, Nunez G. bcl-xexhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J Exp Med. 1996;183:381–391. doi: 10.1084/jem.183.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi MSK, Holman M, Atkins CJ, Klaus GGB. Expression of bcl-x during mouse B cell differentiation and following activation by various stimuli. Eur J Immunol. 1996;26:676–682. doi: 10.1002/eji.1830260325. [DOI] [PubMed] [Google Scholar]
- 29.Anderson JS, Teutsch M, Dong Z, Wortis HH. An essential role for Bruton's tyrosine kinase in the regulation of B-cell apoptosis. Proc Natl Acad Sci USA. 1996;93:10966–10971. doi: 10.1073/pnas.93.20.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motoyama N, Want F, Roth KA, Sawa H, Nakayama K, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in bcl-x–deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 31.Heath AW, Wu WW, Howard MC. Monoclonal antibodies to murine CD40 define two distinct functional epitopes. Eur J Immunol. 1994;24:1828–1834. doi: 10.1002/eji.1830240816. [DOI] [PubMed] [Google Scholar]
- 32.Solvason N, Wu WW, Kabra N, Wu X, Lees E, Howard MC. Induction of cell cycle regulatory proteins in anti-immunoglobulin–stimulated mature B lymphocytes. J Exp Med. 1996;184:407–417. doi: 10.1084/jem.184.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Draetta G, Beach D. Activation of cdc2 protein kinase during mitosis in human cells: cell cycle–dependent phosphorylation and subunit rearrangement. Cell. 1988;54:17–26. doi: 10.1016/0092-8674(88)90175-4. [DOI] [PubMed] [Google Scholar]
- 34.Carayon P, Bord A. Identification of DNA-replicating lymphocyte subsets using a new method to label the bromo-deoxyuridine incorporated into the DNA. J Immunol Methods. 1992;147:225–230. doi: 10.1016/s0022-1759(12)80012-3. [DOI] [PubMed] [Google Scholar]
- 35.Ferlin WG, Severinson E, Strom L, Heath AW, Coffman RL, Ferrick DA, Howard MC. CD40 signaling induces interleukin-4–independent IgE switching in vivo. . Eur J Immunol. 1996;26:2911–2915. doi: 10.1002/eji.1830261216. [DOI] [PubMed] [Google Scholar]
- 36.Sieckmann DG, Scher I, Asofsky R, Mosier DE, Paul WE. Activation of mouse lymphocytes by anti-IgM. II. A thymus-independent response by a mature subset of B lymphocytes. J Exp Med. 1978;148:1628–1643. doi: 10.1084/jem.148.6.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lund-Johansen F, Frey T, Ledbetter JA, Thompson PA. Apoptosis in hematopoietic cells is associated with an extensive decrease in cellular phosphotyrosine content that can be inhibited by the tyrosine phosphatase antagonist pervanadate. Cytometry. 1996;25:182–190. doi: 10.1002/(SICI)1097-0320(19961001)25:2<182::AID-CYTO7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 38.DeFranco AL, Kung JT, Paul WE. Regulation of growth and proliferation in B cell subpopulations. Immunol Rev. 1982;64:161–182. doi: 10.1111/j.1600-065x.1982.tb00423.x. [DOI] [PubMed] [Google Scholar]
- 39.Woodland RT, Schmidt MR, Korsmeyer SJ, Gravel KA. Regulation of B cell survival in xid mice by the proto-oncogene bcl-2. . J Immunol. 1996;156:2143–2154. [PubMed] [Google Scholar]
- 40.Lindsberg M-L, Brunswick M, Yamada H, Lees A, Inman J, June CH, Mond JJ. Biochemical analysis of the immune B cells defect in xidmice. J Immunol. 1991;147:3774–3779. [PubMed] [Google Scholar]
- 41.Solvason N, Lehuen A, Kearney JF. An embryonic source of Ly1 but not conventional B cells. Inter Immunol. 1991;3:543–550. doi: 10.1093/intimm/3.6.543. [DOI] [PubMed] [Google Scholar]