

Abstract
In this study, we investigated the activity of transcription factor NF-κB in macrophages infected with Yersinia enterocolitica. Although triggering initially a weak NF-κB signal, Y. enterocolitica inhibited NF-κB activation in murine J774A.1 and peritoneal macrophages within 60 to 90 min. Simultaneously, Y. enterocolitica prevented prolonged degradation of the inhibitory proteins IκB-α and IκB-β observed by treatment with lipopolysaccharide (LPS) or nonvirulent, plasmid-cured yersiniae. Analysis of different Y. enterocolitica mutants revealed a striking correlation between the abilities of these strains to inhibit NF-κB and to suppress the tumor necrosis factor α (TNF-α) production as well as to trigger macrophage apoptosis. When NF-κB activation was prevented by the proteasome inhibitor MG-132, nonvirulent yersiniae as well as LPS became able to trigger J774A.1 cell apoptosis and inhibition of the TNF-α secretion. Y. enterocolitica also impaired the activity of NF-κB in epithelial HeLa cells. Although neither Y. enterocolitica nor TNF-α could induce HeLa cell apoptosis alone, TNF-α provoked apoptosis when activation of NF-κB was inhibited by Yersinia infection or by the proteasome inhibitor MG-132. Together, these data demonstrate that Y. enterocolitica suppresses cellular activation of NF-κB, which inhibits TNF-α release and triggers apoptosis in macrophages. Our results also suggest that Yersinia infection confers susceptibility to programmed cell death to other cell types, provided that the appropriate death signal is delivered.
The genus Yersinia includes three species, which are pathogenic for rodents and humans. Y. pestis is the etiological agent of plague, Y. pseudotuberculosis and Y. enterocolitica cause gastrointestinal syndromes, lymphadenitis, and septicemia (1). Despite of different routes of infection, these three species share the common capability to resist the immune response of the host. This enables extracellular survival and proliferation of the bacteria in the host lymphoid tissue (1, 2). The three pathogenic Yersinia spp. harbor a common 70-kb virulence plasmid (pYV) that encodes the Yersinia Yop virulon, a sophisticated bacterial system that mediates delivery of Yops (Yersinia outer proteins) inside eukaryotic cells by surface-bound bacteria (1, 3, 4). The delivered Yops disrupt key functions of the host cell (1, 5). At least four Yops, the so-called effector Yops, YopH, YopE, YopM, and YpkA (YopO in Y. enterocolitica) are translocated across the eukaryotic cell plasma membrane (1). YopE disrupts the actin microfilament structure and acts synergistically with the protein tyrosine phosphatase YopH to inhibit phagocytosis and to suppress the oxidative burst of professional phagocytes (6–10). YpkA/YopO displays serine/threonine kinase activity and is, like YopH, supposed to interfere with host cell signaling pathways (11). Export and translocation of effector Yops is mediated by a virulence plasmid-encoded protein secretion system and requires control by YopB, YopD, LcrV, and YopN (1).
Studies in the murine infection model provide evidence that the Yersinia Yop virulon mediates suppression of the TNF-α and IFN-γ production in vivo (12). The cytokines TNF-α and IFN-γ play a central role in the inflammatory response to bacterial infection. They are crucial in limiting the severity of Yersinia infection (13). Consequently, inhibition of TNF-α and IFN-γ synthesis enhances the ability of Yersinia to multiply in the host (12, 13). Previous studies in our laboratories revealed that Y. enterocolitica promotes deactivation of mitogen-activated protein kinases (MAPKs)1 in cultured macrophages (14). An important role of MAPK cascades in the regulation of the macrophage TNF-α production has been widely documented (15–18), and our study suggests that Y. enterocolitica suppresses the macrophage TNF-α production by shortening MAPK activities (14). In addition, we and others recently demonstrated that interaction of Yersinia with macrophages culminates in activation of the intrinsic macrophage cell death program (19–21). Apoptosis as an innate cell suicide mechanism for removing unwanted cells from the multicellular organism appears to play a role in some infectious diseases (22). However, the mechanism by which Yersinia promotes macrophage cell death is not clear yet.
In this study, we analyzed the impact of Y. enterocolitica on activation of transcription factor NF-κB. The active heterodimer p50/p65 form of NF-κB plays a central role in immunological processes by controlling expression of a variety of genes involved in inflammatory responses (i.e., TNF-α, IL-1, IL-6, IL-8, GM-CSF; reference 23). Furthermore, there is increasing evidence that activation of NF-κB provides cells with resistance to apoptosis induced by different stimuli (24–28). NF-κB can be activated in macrophages by exposure to LPS or inflammatory cytokines such as TNF-α or IL-1, viral infection, UV radiation, and by other physiological and nonphysiological agonists (24–26). In its inactive form, NF-κB is sequestered in the cytoplasm in a complex with the inhibitory proteins IκB-α or IκB-β (23, 29). After stimulation by the different inducers, the IκB inhibitors get phosphorylated and degraded through the ubiquitin–proteasome pathway, thereby releasing NF-κB heterodimer (23, 29). Free NF-κB translocates to the nucleus, where it binds to its target sequences and activates transcription (23, 29). Duration of NF-κB activation has been found to depend on the activating stimuli, which either degrade IκB-α and IκB-β (persistent NF-κB activation), or only IκB-α (transient NF-κB activation) (30). Bacterial LPS induces persistent NF-κB activation by degrading IκB-α as well as IκB-β in responsive cells (30).
Here, we report that Y. enterocolitica impairs activation of NF-κB in murine J774A.1 and peritoneal macrophages and in human epithelial HeLa cells. Our study implies a direct link between the prevention of NF-κB activation and apoptotic cell death as well as TNF-α suppression in Yersinia-infected macrophages. Thus, interference of Yersinia with macrophage NF-κB activation may crucially contribute to subvert the host immune response and determine the outcome of Yersinia infection.
Materials and Methods
Bacterial Strains and Growth Conditions.
The bacterial strains used in this study are listed in Table 1. Overnight cultures grown at 26°C were diluted 1:20 in fresh Luria-Bertani broth and grown for 2 h at 37°C as described previously (14, 19). The bacteria were then washed once and resuspended in PBS at a concentration of 108 bacteria/ml. The desired bacterial concentration was adjusted by measuring the optical density at 600 nm and checked by plating serial dilutions of the samples on agar and counting CFUs after incubation at 26°C for 20 h.
Table 1.
Y. enterocolitica Strains Used in this Study
| Strain | Relevant characteristics | Reference | ||
|---|---|---|---|---|
| WA-314 | Serogroup O8; clinical isolate harboring virulence plasmid pYVO8 | 31 | ||
| WA-C | Plasmidless derivative of strain WA-314 | 31 | ||
| WA-C(pYV-7146) | Mutant strain, deficient in YopH secretion; insertional inactivation of sycH, | 10 | ||
| the gene for the YopH-specific chaperone SycH | ||||
| WA-C(pYV-515) | Mutant strain, deficient in secretion of Yops; insertional inactivation of lcrD, | 10 | ||
| the gene encoding LcrD, which is essential for Yop secretion | ||||
| WA-C(pLCR) | Strain harboring plasmid pLCR encoding the secretion apparatus | 32 | ||
| of Y. enterocolitica including the genes for YopD, YopB, YopN, and LcrV | ||||
| WA-C(pLCR, pB8-23) | Strain WA-C(pLCR) harboring an additional plasmid encoding the genes | 32 | ||
| for YopH, YopE, and YadA | ||||
| WA-C(pYV-OP-1) | yopO/yopP-negative mutant; insertional inactivation of the yopO/yopP operon | This study |
Construction of a Y. enterocolitica yopO/yopP Mutant Strain.
Two PCR primers were deduced from the yopO/yopP homologous ypkA/ yopJ operon in Y. pseudotuberculosis (11) to amplify an internal fragment spanning from bp 223 to 1030. Primer Ypk-Mf GAGTG CATGCTGAGGGCTGATGAAAT and Ypk-Mr TATTGCA TGCTTATCCTTAGTTTCTATTA harbor additional SphI restriction sites at their 5′ ends (underlined). Y. enterocolitica strain WA-314, bearing the virulence plasmid pYVO8 (31), was used as a template to amplify the PCR product of 815 bp. This PCR product was purified, cut with SphI, and ligated into the unique SphI site of the suicide plasmid pGPCAT (32) resulting in pGPC-KM1. Escherichia coli SM10 was used as host strain for transformation. pGPC-KM1 was mobilized into strain WA-314 by conjugation and inserted into pVYO8 by homologous recombination. Transconjugants harboring the cointegrate (pYVO8::pGPC-KM1) were selected by chloramphenicol and nalidixic acid resistance. The resulting clone WA-C(pYV-OP-1) was characterized by restriction enzyme analysis, and inability to produce YopO and YopP was verified by SDS-PAGE.
Cell Culture and Preparation of Macrophages.
The murine macrophage-like cell line J774.A1 and the human epithelial HeLa cell line were routinely grown in cell growth medium (RPMI 1640 medium supplemented with 10% heat inactivated fetal calf serum and 5 mM L-glutamine; Life Technologies, Cergy, Pontoise, France) at 37°C and 5% CO2 in a humidified atmosphere. Elicited peritoneal macrophages were obtained from male 8–10-wk-old Swiss mice 4 d after intraperitoneal inoculation of 1.5 ml of 3% thioglycolate broth. Peritoneal exudate cells were washed and cultured at 37°C in cell growth medium. After 2 h, nonadherent cells were removed by repeated washing, and remanent macrophages were further incubated in cell growth medium at 37°C and 5% CO2.
Stimulation of Cells.
Cells were either treated with bacteria, with 10 μg/ml LPS from E. coli (Sigma Chemical Co., St. Louis, MO), or with 20 ng/ml human TNF-α (National Institute for Biological Standards and Control, Hertfordshire, UK) as indicated. Infections were performed at a multiplicity of infection (MOI) of 50:1 for different periods of time as indicated. After infection, HeLa cells were centrifuged for 5 min at 400 g to facilitate contact between bacteria and cells. For incubation times >60 min (J774A.1 cells and peritoneal macrophages) or 90 min (HeLa cells), bacteria were killed by the addition of gentamicin (100 μg/ml) after 60 or 90 min of infection, respectively. In some experiments, cells were pretreated with 10 μM of the proteasome inhibitor Z-Leu-Leu-leucinal (MG-132; Biomol Research Laboratories, Plymouth Meeting, MA), the MAPK-ERK kinase 1 (MEK1) inhibitor PD098059 (50 μM; New England Biolabs, Beverly, MA), or the p38 (MAPK different from ERK or JNK) inhibitor SB203580 (10 μM; provided by J.C. Lee, SmithKline Beecham, King of Prussia, PA) for 30 min before stimulation.
Preparation of Nuclear Extracts.
Nuclear proteins from cells treated as indicated were extracted according to a modification of the procedure described by Thieblemont et al. (33). In brief, cells were washed with ice-cold PBS and lysed with hypotonic buffer (5 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1 mM PMSF, 5 μg/ml leupeptin, 4 μg/ml aprotinin, 1 μg/ml pepstatin, 2 μg/ml antipain, and 1 μg/ml chymostatin). The cells were left on ice for 20 min and subsequently centrifuged at 1,000 g. The nuclear pellets were resuspended in extraction buffer (20 mM Hepes, pH 7.9, 25% glycerol, 1 M NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 0.5 mM 1,4 dithiothreitol (DTT), 0.5 mM PMSF, 2 μg/ml leupeptin, 1.5 μg/ml aprotinin, 0.5 μg/ ml pepstatin, 1 μg/ml antipain, and 0.5 μg/ml chymostatin) and incubated on ice for 30 min. The nuclear proteins in the supernatant were recovered after centrifugation at 16,000 g, quantified by using a protein assay kit (BioRad Labs, Munich, Germany), and stored in aliquots at −80°C.
Electrophoretic Mobility Shift Assays.
For the electrophoretic mobility shift assays (EMSAs; reference 33), the NF-κB oligonucleotide probe (5′-ACAAGGGACTTTCCGCTGGGGACTTTCCAG-3′), synthesized by Eurobio (Les Ulis, France), was radiolabeled with γ-[32P]ATP using T4 polynucleotide kinase (Eurogentec, Serraing, Belgium). Nuclear proteins (10 μg) were preincubated with 5 μg of salmon sperm DNA (GIBCO BRL, Gaithersburg, MD) on ice for 15 min before addition of 2–5 ng of the radiolabeled (50,000–100,000 cpm/ng) NF-κB oligonucleotide probe. The DNA-binding reactions were performed in the presence of 25 mM Hepes, pH 7.9, 0.5 mM EDTA, 0.5 mM DTT, and 5% glycerol for 30 min at room temperature (final volume: 20 μl). Competition studies were carried out with a 50-fold molar excess of unlabeled oligonucleotides added to the reaction mixtures before addition of radiolabeled oligonucleotides. For supershift assays, 1 μl of rabbit polyclonal IgG directed to the p65 NF-κB subunit (provided by J. Imbert, Institut National de la Santé et de la Recherche Médicale [INSERM] U119, Marseille, France; reference 34) was added to the extracts before incubation with the labeled oligonucleotide probe. After incubation, the reaction products were analyzed by 6% PAGE using Tris-borate-EDTA running buffer (45 mM Tris-borate, pH 8.0, 10 mM EDTA). The gels were dried and analyzed with a PhosphorImager (Molecular Dynamics, Bondoufle, France).
Western Immunoblot Assays.
Cytoplasmic extracts were prepared from 106 cells, treated, as indicated, by lysis in buffer containing 10 mM Tris, pH 8.0, 60 mM KCl, 1 mM EDTA, 0.5% Nonidet P-40, 1 mM DTT, 1 mM PMSF, 1 mM benzamidin, 20 mM β-glycerophosphate, 5 mM p-nitrophenyl phosphate, and 0.1 mM Na3VO4. Proteins were separated by 10% SDS-PAGE, electrotransferred to polyvinylidene difluoride membrane, and blocked with 3% BSA. Immunostaining for IκB-α and IκB-β were performed with polyclonal rabbit anti–IκB-α and anti–IκB-β antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoreactive bands were visualized by incubation with donkey anti–rabbit antibodies conjugated to horseradish peroxidase (Amersham Corp., Arlington Heights, IL) using enhanced chemiluminescence reagents (New England Nuclear Life Science, Boston, MA).
Assessment of Apoptosis by Fluorescence Microscopy.
Apoptotic cells were detected and quantified by an assay based on the detection of phosphatidylserine exposed on the outer leaflet of apoptotic cells as described previously (19). Cells treated as indicated were stained with FITC-conjugated annexin V (Annexin-V-Fluos; Boehringer Mannheim GmbH, Mannheim, Germany), which binds with high affinity to membrane-exposed phosphatidylserine. The simultaneous application of the DNA stain propidium iodide (Sigma Chemical Co.) allowed discrimination of apoptotic from necrotic cells. The percentages of apoptotic cells were determined by counting a minimum of 100 cells/sample in a fluorescence microscope (DM IRB; Leica, Wetzlar, Germany). Results are expressed as mean percentages of apoptotic cells ± SD from three independent experiments.
Quantitation of TNF-α Secretion.
For TNF-α quantitation, J774A.1 cells and peritoneal macrophages were dispatched in plastic culture plates (5 × 105/sample) and treated as indicated. The cell culture supernatants were removed after a final 150-min incubation, and the TNF-α cytokine levels in the supernatants were evaluated by a cytotoxic assay performed with the TNF-α– sensitive fibroblast cell line L929 as described previously (14). Experiments were performed in quadruplicate. Results are expressed as mean percentages of picograms of TNF-α per milliliter ± SD from one representative experiment out of three performed.
Results
Y. enterocolitica Inhibits Nuclear Translocation of NF-κB in J774A.1 Macrophages.
To evaluate a role of NF-κB in the macrophage response to Y. enterocolitica infection, we analyzed nuclear translocation of NF-κB in Y. enterocolitica– infected J774A.1 macrophages by EMSA. J774A.1 cells were either infected with virulence plasmid–cured (WA-C) or with virulence plasmid–harboring wild-type (WA-314) yersiniae, or treated with LPS from E. coli. At different time points after challenge, nuclear protein extracts were assayed for NF-κB DNA-binding activities using a radiolabeled NF-κB–specific probe (33). As shown in Fig. 1 A, strong radioactive DNA binding to nuclear proteins was observed after 30 min when cells were treated with LPS or virulence plasmid–cured Y. enterocolitica. Infection with wild-type yersiniae for 30 min induced formation of a protein–DNA complex migrating at the same mobility, but the DNA-binding activity of this complex was greatly reduced as compared to virulence plasmid–cured yersiniae or to LPS. To examine the specificity of the DNA-binding capability of the complexes generated by virulence plasmid–cured and –harboring yersiniae after 30 min, a 50-fold molar excess of unlabeled NF-κB oligonucleotides was added for competition (Fig. 1 B). As expected, the unlabeled oligonucleotides prevented formation of radiolabeled protein–DNA complexes. Incubation with an antibody directed to the p65 NF-κB subunit resulted in supershift of these complexes, confirming their identities as transcription factor NF-κB (Fig. 1 B). Translocation of NF-κB in response to treatment with virulence plasmid–cured strain WA-C or with LPS was enhanced after 60 min and persisted at least up to 90 min (Fig. 1 A). In contrast, nuclear extracts from cells infected for 60 and 90 min with wild-type Y. enterocolitica did not give rise to complex formation anymore, indicating that the low nuclear translocation of NF-κB induced by wild-type yersiniae within 30 min is only short living and already completely abolished after 60 min. These data demonstrate that wild-type Y. enterocolitica suppresses translocation of NF-κB to the nucleus and, thus, prevents prolonged NF-κB activation. In addition, infection of J774A.1 cells with wild-type Y. enterocolitica for 60 min inhibited activation of NF-κB in response to subsequent challenge with LPS from E. coli (data not shown), indicating that Yersinia infection actively suppresses the NF-κB signal induced by LPS.
Figure 1.
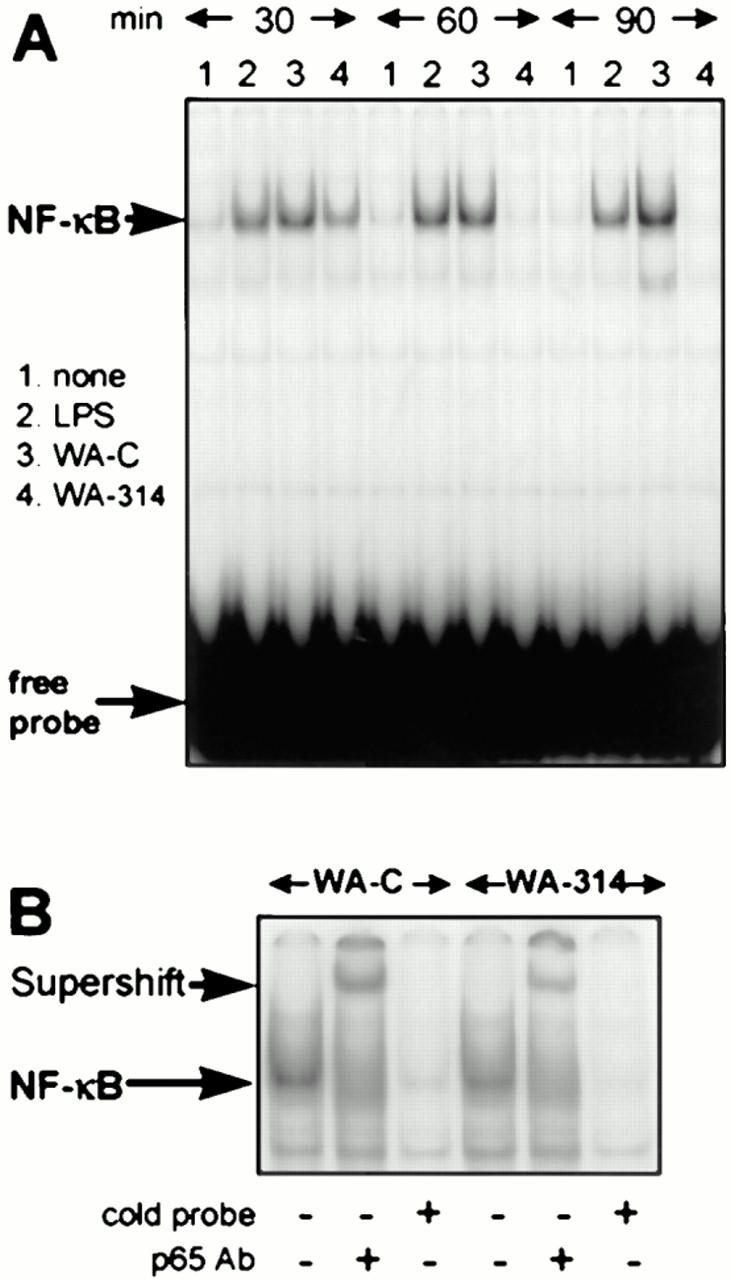
Y. enterocolitica impairs activation of NF-κB in J774A.1 macrophages. (A) Kinetics of NF-κB DNA-binding activity in J774A.1 macrophages treated with Y. enterocolitica and LPS as detected by EMSA. J774A.1 cells were untreated (lane 1) or stimulated with LPS (lane 2), with the virulence plasmid–cured Yersinia strain WA-C (lane 3) or the virulent wild-type strain WA-314 (lane 4). The NF-κB activities were determined after 30, 60, and 90 min of stimulation. (B) Investigation of the identity of NF-κB by supershift assay with an antibody directed against the p65 NF-κB subunit (p65 Ab), and by competition with a 50-fold molar excess of the unlabeled oligonucleotide (cold probe). Experiments were performed with the NF-κB complexes obtained by infections with strain WA-C and WA-314 after 30 min.
Y. enterocolitica Inhibits Prolonged Degradation of IκB-α and IκB-β.
We examined the changes in cytoplasmic IκB-α (37 kD) and IκB-β (46 kD) protein levels of J774A.1 cells to determine whether degradation of these inhibitory proteins correlates with the respective level of NF-κB activation. Immunoblotting analysis revealed that virulence plasmid–harboring yersiniae (WA-314) as well as virulence plasmid–cured yersiniae (WA-C) and LPS rapidly induced phosphorylation and proteolysis of IκB-α within 10–30 min (Fig. 2). Phosphorylation of IκB-α resulted in an upward shift of IκB-α in the SDS-PAGE because of slower electrophoretical mobility (35). IκB-α phosphorylation and degradation occured even faster in case of infection with the wild-type strain WA-314 (Fig. 2, lane 4) as compared to strain WA-C (Fig. 2, lane 3). This effect may be attributed to the presence of the virulence plasmid–encoded cell adhesin YadA, since a yadA-negative mutant (36) revealed a kinetic of IκB-α degradation similar to virulence plasmid–cured yersiniae (data not shown). This is also in line with a faster increase of the oxidative burst of polymorphonuclear leukocytes infected with YadA-bearing yersiniae as compared to leukocytes treated with yersiniae lacking YadA (10). Fig. 2 shows that, after 45 min, newly synthesized IκB-α accumulated and increasing amounts of IκB-α reappeared in the immunoblot. However, only J774A.1 macrophages infected with wild-type strain WA-314 completely restored their IκB-α level within 60 min and no phosphorylated forms of IκB-α could be detected anymore. In contrast, IκB-α in cells treated with LPS or with strain WA-C remained phosphorylated and degraded to a remarkable degree throughout the time investigated. The results obtained for IκB-β were similar to those obtained for IκB-α, although degradation of IκB-β in response to treatment with LPS and both Yersinia strains was not as dramatic as for IκB-α and occurred later (30–45 min). However, also in the case of IκB-β, infection with strain WA-314 caused complete restoration of the cytoplasmic pool of the inhibitory protein at time points when degradation of IκB-β due to treatment with LPS or virulence plasmid– cured yersiniae was still obvious (60–120 min). Thus, the correlation between the lack of persistent IκB-α/IκB-β degradation and the inhibition of NF-κB translocation suggests that Y. enterocolitica suppresses NF-κB activation by preventing prolonged proteolysis of the inhibitory proteins.
Figure 2.

Y. enterocolitica inhibits prolonged degradation of IκB-α and IκB-β in J774A.1 macrophages. The kinetics of degradation of IκB-α and IκB-β was determined in untreated J774A.1 macrophages (lane 1) and in macrophages stimulated with LPS (lane 2), with the virulence plasmid–cured Yersinia strain WA-C (lane 3), and with the virulent wild-type strain WA-314 (lane 4). The cytoplasmic lysates of stimulated J774A.1 cells were prepared at the time points indicated, subjected to SDS-PAGE, and immunoblotted with polyclonal anti–IκB-α and anti–IκB-β antibodies, respectively.
Y. enterocolitica–mediated Inhibition of NF-κB Activation Correlates with J774A.1 Cell Apoptosis and Suppression of TNF-α Production.
Since NF-κB was reported to play a central role in the regulation of apoptotic cell death and of cytokine secretion (23–29), we wondered whether the inhibition of NF-κB translocation is related to Yersinia-induced apoptosis and TNF-α suppression. We compared nuclear translocation of active NF-κB in response to infection with different Y. enterocolitica mutants (Fig. 3 A) with the capabilities of these strains to trigger J774A.1 cell apoptosis (Fig. 3 B) and to block TNF-α production (Fig. 3 C). TNF-α secretion in the culture supernatant was determined after 150 min, a time point when onset of J774A.1 cell apoptosis due to infection with wild-type yersiniae starts to be detectable (19). Previously, we demonstrated that apoptosis and TNF-α inhibition induced by Y. enterocolitica depend on a functional Yop secretion system, but occur independently of the presence of the tyrosine phosphatase YopH (14, 19). In line with these results, we now found that the YopH secretion negative strain WA-C(pYV-7146) (sycH mutant) prevented the translocation of NF-κB (Fig. 3, lane 4). On the contrary, strain WA-C(pYV-515), which is affected in the secretion of all Yops (lcrD mutant), exhibited a strong NF-κB–binding activity (Fig. 3, lane 6) similar to strain WA-C (Fig. 3, lane 2). These results show that also for the inhibition of NF-κB, a functional Yop secretion system is required, but YopH appears to be dispensable. Furthermore, we analyzed two Yersinia strains expressing only a restricted repertoire of yop genes. Strain WA-C(pLCR) secretes the translocator/regulator proteins YopD, YopB, YopN, and LcrV. The second strain, referred to as WA-C(pLCR, pB8-23), additionally secretes YopH and YopE. These two Yops confer resistance to phagocytosis and suppression of the J774A.1 cell oxidative burst to Y. enterocolitica (14). Nevertheless, neither strain was efficiently inhibiting the translocation of NF-κB, nor promoting apoptosis or TNF-α suppression. Fig. 3 also displays the results obtained with a Y. enterocolitica mutant selectively affected in the expression of the serine/threonine kinase gene yopO and the cotranscribed yopP. This mutant was remarkably impaired in its ability to induce apoptosis (52 ± 15% apoptotic cells) and to inhibit the TNF-α production (0.65 ± 0.04 ng TNF-α/ml), as compared to the virulent wild-type strain (90 ± 4% apoptotic cells, 0.14 ± 0.05 ng TNF-α/ml). Interestingly, this strain was also not as efficient as the wild-type strain to inhibit the translocation of NF-κB (intensity of NF-κB DNA-binding activity of 12 for the yopO/yopP mutant, and of 5 for the wild-type strain; Fig. 3). Thus, our data point out a close correlation between the capability of Y. enterocolitica to prevent activation of NF-κB, to trigger apoptosis, and to suppress the TNF-α production in J774A.1 macrophages. The caspase inhibitor ZVAD.fmk, which efficiently blocks execution of the Yersinia-induced apoptotic program (19), did not interfere with TNF-α suppression mediated by Yersinia (data not shown), indicating that TNF-α suppression is not a result of the apoptotic process.
Figure 3.
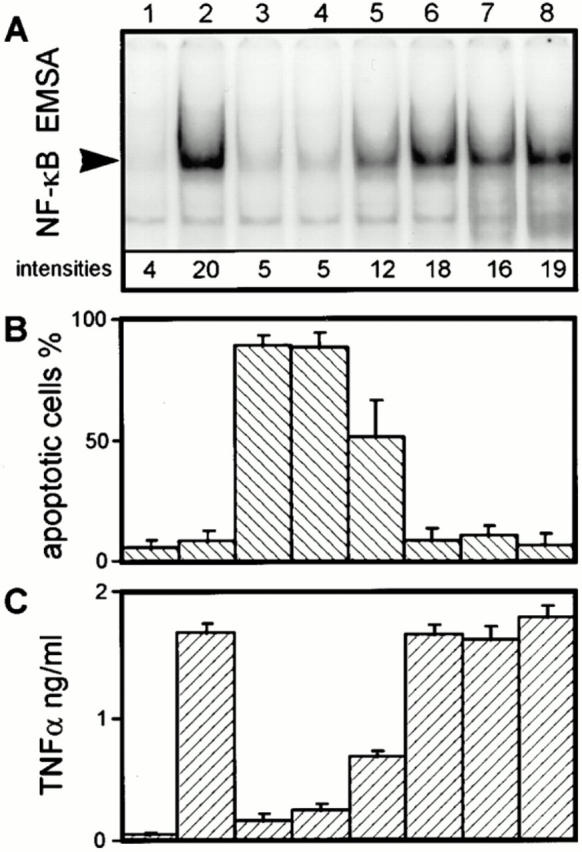
Correlation between inhibition of the NF-κB response, and apoptosis and TNF-α suppression in J774A.1 cells infected with different Y. enterocolitica mutants. (A) J774A.1 cell NF-κB activities. (B) Quantitation of J774A.1 cell apoptosis. (C) Quantitation of J774A.1 cell TNF-α production. J774A.1 cells were either untreated (lane 1), or infected with the virulence plasmid–cured strain WA-C (lane 2), the virulent wild-type strain WA-314 (lane 3), the YopH secretion-negative strain WA-C(pYV-7146) (lane 4), the yopO/yopP mutant WA-C(pYV-OP-1) (lane 5), the Yop secretion-negative strain WA-C(pYV-515) (lane 6), strain WA-C(pLCR) secreting YopD, YopB, YopN, and LcrV (lane 7), and strain WA-C(pLCR, pB8-23) secreting YopD, YopB, YopN, LcrV, YopH, and YopE (lane 8). (A) The NF-κB activities of J774A.1 cells were determined 60 min after infection by EMSA and quantified with a PhosphorImager. The radioactive intensities of the NF-κB DNA-binding activities are given below the respective NF-κB signal. This figure shows one experiment representative for three performed. Only sections of the autoradiogram containing the protein–DNA complexes are shown. (B) Apoptosis of J774A.1 cells was assayed 4 h after onset of infection by staining the cells with fluorescein-conjugated annexin V and propidium iodide, and counting apoptotic cells in a fluorescence microscope. (C) The TNF-α activities in the cell culture supernatants (dilution 1:40) were measured 150 min after the onset of infection, using a cytotoxic assay performed with the TNF-α–sensitive fibroblast cell line L929.
Murine Peritoneal Macrophages Respond to Y. enterocolitica Infection as J774A.1 Cells.
To determine whether Y. enterocolitica interferes with the activation of NF-κB also in primary macrophages, we investigated the effects of Yersinia infection on murine peritoneal macrophages. EMSAs revealed that virulent wild-type yersiniae impaired nuclear translocation of NF-κB in peritoneal macrophages within 90 min (Fig. 4 A). In contrast, virulence plasmid–cured yersiniae induced an increasing NF-κB response, similar to J774A.1 cells. Supershift experiments with the anti-p65 antibody identified the detected protein–DNA complexes as NF-κB (data not shown). Analyzing the IκB-α and IκB-β protein levels, we found that IκB-α as well as IκB-β were efficiently degraded upon infection with both Yersinia strains (Fig. 4 B). However, as in J774A.1 cells, substantial amounts of IκB-α and IκB-β were regenerated within 60– 90 min, specifically in macrophages infected with the wild-type strain. Additionally, apoptosis and inhibition of the TNF-α production were found only in peritoneal macrophages treated with wild-type Y. enterocolitica, but not with the virulence plasmid–cured strain (Table 2). Thus, the effects observed on J774A.1 cells appear to hold true also for primary peritoneal macrophages, validating J774A.1 cells as an appropriate infection model for analyzing Yersinia–macrophage interaction.
Figure 4.

Y. enterocolitica impairs activation of NF-κB in murine peritoneal macrophages. (A) Kinetics of NF-κB DNA-binding activity. (B) Kinetics of degradation of IκB-α and IκB-β. Murine peritoneal macrophages were untreated (lane 1) or stimulated with LPS (lane 2), with the virulence plasmid–cured Yersinia strain WA-C (lane 3) or the virulent wild-type strain WA-314 (lane 4). After 30, 60, and 90 min, (A) the NF-κB activities were determined by EMSA and (B) the IκB-α and IκB-β protein levels were determined in cytoplasmic lysates subjected to SDS-PAGE and immunoblotted with polyclonal anti–IκB-α and anti–IκB-β antibodies, respectively.
Table 2.
Wild-type Y. enterocoliticia Inhibits the TNF-α Production and Triggers Apoptosis in Murine Peritoneal Macrophages
| Stimulation | TNF-α production* | Percent apoptotic cells‡ | ||
|---|---|---|---|---|
| pg/ml | ||||
| None | 5 ± 1 | 3.5 ± 2 | ||
| LPS | 88 ± 9 | ND | ||
| WA-C | 66 ± 4 | 4.5 ± 2 | ||
| WA-314 | 11 ± 3 | 81 ± 8 |
The TNF-α activity in the cell culture supernatant (dilution 1:25) was measured 150 min after the onset of stimulation using a cytotoxic assay performed with the TNF-α–sensitive fibroblast cell line L929.
Apoptosis was assayed 4 h after onset of stimulation by staining the cells with fluorescein-conjugated annexin V and propidium iodide, and counting apoptotic cells in a fluorescence microscope.
Prevention of NF-κB Activation by Proteasome Inhibitor MG-132 Triggers Apoptosis in J774A.1 Cells Treated with Virulence Plasmid–cured Yersiniae or LPS.
To confirm a possible relation between the blockage of NF-κB activation and the onset of apoptosis, we sought a compound inhibiting efficiently and selectively the translocation of NF-κB. Since NF-κB translocation depends on the protease activity in the proteasome, which degrades IκBs (23, 29, 37), we used the peptide Z-Leu-Leu-leucinal (MG-132), which specifically inhibits the proteasome pathway (37, 38). Pretreatment of J774A.1 cells with this compound substantially inhibited IκB-α degradation (data not shown) and NF-κB activation due to infection with virulence plasmid–cured yersiniae (Fig. 5, lane 3). Interestingly, after 4 h of incubation, >90% of the cells pretreated with MG-132 and infected with virulence plasmid–cured yersiniae underwent apoptosis (Table 3). The same held true for J774A.1 cells stimulated with LPS after pretreatment with MG-132. These results once again suggest a direct link between the level of NF-κB activation and the readiness of J774A.1 cells to undergo apoptotic cell death upon stimulation. When we analyzed the impact of MG-132 on the TNF-α response, we found that the TNF-α production induced by virulence plasmid–cured yersiniae and by LPS was completely abolished by pretreatment with MG-132 (Table 3). These data imply a key role of NF-κB activity not only in the prevention of apoptosis, but also in the regulation of the TNF-α production.
Figure 5.

The NF-κB response induced by virulence plasmid–cured Y. enterocolitica (WA-C) in J774A.1 cells is reduced by pretreatment with the proteasome inhibitor MG-132, but not by pretreatment with SB203580 and PD098059. J774A.1 cells were untreated (lanes 1–2), pretreated for 30 min with either 10 μM of the proteasome inhibitor MG-132 (lane 3), or a combination of the p38 inhibitor SB203580 (10 μM) and the MEK1 inhibitor PD098059 (50 μM; lane 4). Thereafter, cells were infected with the virulence plasmid– cured Yersinia strain WA-C (lanes 2–4). After 45 min, NF-κB activities were analyzed by EMSA.
Table 3.
Pretreatment with the Proteasome Inhibitor MG-132 Triggers Apoptosis and Suppresses the TNF-α Production in J774A.1 Macrophages Stimulated by LPS or Virulence Plasmid–cured Y. enterocolitica (WA-C)
| Stimulation* | Percent apoptotic cells‡ | TNF-α production§ | ||||||
|---|---|---|---|---|---|---|---|---|
| −MG-132 | +MG-132 | −MG-132 | +MG-132 | |||||
| None | 6 ± 3 | 7 ± 2 | 31 ± 5 | 36 ± 10 | ||||
| LPS | 8 ± 4 | 96 ± 3 | 1,725 ± 96 | 61 ± 42 | ||||
| WA-C | 9 ± 4 | 94 ± 4 | 1,630 ± 68 | 17 ± 5 | ||||
| WA-314 | 90 ± 4 | 95 ± 6 | 144 ± 53 | 21 ± 6 | ||||
J774A.1 cells remained unstimulated or were treated with either LPS, virulence plasmid–cured yersiniae (WA-C), or wild-type yersiniae (WA-314) under conditions with or without pretreatment with the proteasome inhibitor MG-132 (10 μM).
Apoptosis was assayed 4 h after onset of stimulation by staining the cells with fluorescein-conjugated annexin V and propidium iodide, and counting apoptotic cells in a fluorescence microscope.
The TNF-α activity in the cell culture supernatant (dilution 1:40) was measured 150 min after the onset of stimulation, using a cytotoxic assay performed with the TNF-α–sensitive fibroblast cell line L929.
Y. enterocolitica-mediated Inhibition of NF-κB Promotes Apoptosis of HeLa Cells in Response to TNF-α.
In a previous study, we demonstrated that Y. enterocolitica infection leads to apoptosis selectively in macrophages, but not in epithelial HeLa cells (19). To reevaluate a possible role of NF-κB in Yersinia-mediated apoptosis, we studied whether NF-κB activation was also affected in Y. enterocolitica–infected HeLa cells. Fig. 6 shows that the virulence plasmid–cured strain WA-C (lane 6) induced substantial NF-κB–binding activity, but not as strong as treatment of HeLa cells with TNF-α (lane 4). Supershift assays with the anti-p65 subunit antibody identified the detected nuclear complexes as NF-κB (Fig. 6, lanes 3, 5, and 7). In contrast, the virulent wild-type strain WA-314 (Fig. 6, lane 8) prevented nuclear translocation of NF-κB. However, strain WA-314 did not induce HeLa cell apoptosis (Table 4). When we treated HeLa cells either with strain WA-C or WA-314, and restimulated thereafter with TNF-α, strain WA-314 exclusively caused a marked decrease of the resulting NF-κB signal (compare in Fig. 6 lane 1 with lanes 2 and 4). Strikingly, this mode of treatment led to apoptotic cell death of 62 ± 15% of the HeLa cell population after 6 h (Table 4). Similarly, pretreatment of HeLa cells with the proteasome inhibitor MG-132 and subsequent stimulation with TNF-α strongly induced apoptosis, but not the addition of MG-132 alone. These results support previous studies that demonstrated that the fate of epithelial HeLa cells upon treatment with TNF-α depends on the ability of NF-κB to be activated (28, 39). Y. enterocolitica, by inhibiting the activation of NF-κB, promotes apoptosis of HeLa cells in response to TNF-α.
Figure 6.

Y. enterocolitica impairs activation of NF-κB in epithelial HeLa cells. Treatment of epithelial HeLa cells was performed in two steps. During the first step, the HeLa cells were untreated (lanes 3, 4, and 9) or infected with either virulence plasmid–cured yersiniae (lanes 2, 5, and 6), or wild-type yersiniae (lanes 1, 7, and 8) for 90 min. Thereafter, half of the samples were challenged with 20 μg/ml human TNF-α (lanes 1–4) for 30 min. Nuclear extracts were prepared after the first incubation step (lanes 5–9) or after additional TNF-α treatment (lanes 1–4), and analyzed for NF-κB DNA-binding activity by EMSA. Supershift assays with an antibody directed against the p65 NF-κB subunit identified the detected complexes as NF-κB (lanes 3, 5, and 6).
Table 4.
TNF-α Triggers Apoptosis of Epithelial HeLa Cells after Pretreatment with Wild-type Y. enterocolitica or the Proteasome Inhibitor MG-132
| Pretreatment* | Percent apoptotic cells‡ | |||
|---|---|---|---|---|
| −TNF-α | +TNF-α | |||
| None | 10 ± 6 | 9 ± 6 | ||
| WA-C | 11 ± 2 | 7 ± 6 | ||
| WA-314 | 13 ± 4 | 62 ± 15 | ||
| MG-132 | 11 ± 3 | 57 ± 12 | ||
HeLa cells were either not pretreated, or pretreated for 90 min with virulence plasmid–cured yersiniae (WA-C), with wild-type yersiniae (WA-314), or with the proteasome inhibitor MG-132 (10 μM). Thereafter, half of the samples were challenged with human TNF-α (20 μg/ml).
Apoptosis was assayed 6 h after onset of stimulation with TNF-α by staining the cells with fluorescein-conjugated annexin V and propidium iodide, and counting apoptotic cells in a fluorescence microscope.
Inhibitors of p38 MAPK (SB203580) and MAPK-ERK-kinase 1 (PD098059) Do Not Interfere with Yersinia-mediated Apoptosis, but Cumulatively Block the TNF-α Production Induced by Virulence Plasmid–cured Yersiniae.
In a previous study, we found that Y. enterocolitica initially stimulates and subsequently deactivates the macrophage MAPKs extracellular signal–regulated kinase (ERK)1/2, p38, and c-Jun NH2-terminal kinase (JNK) (14). There is increasing evidence that MAPK cascades serve to regulate cell proliferation and cell death (40). To verify involvement of MAPKs in Yersinia-induced apoptosis of J774A.1 cells, we analyzed the effects of two specific inhibitors of MAPK pathways. The compound SB203580 specifically inhibits p38/CSBP kinase activities (41), whereas PD098059 selectively blocks activation of the ERK kinase MEK1, which in turn prevents activation of ERK1/2 (42). There is no drug available yet to selectively inhibit the JNK pathway. J774A.1 cells were pretreated with SB203580 and PD098059, and thereafter challenged with yersiniae. Our experiments revealed that neither compound, alone or in combination, blocked J774A.1 cell apoptosis induced by wild-type yersiniae; in each case, 85–95% of cells became apoptotic after 4 h of incubation. Thus, neither the p38 nor the ERK1/2 MAPK pathways seem to be crucially engaged in Yersinia-mediated apoptosis. In addition, we analyzed the impact of SB203580 and PD098059 on the J774A.1 cell TNF-α production. Table 5 shows that both compounds substantially inhibited the TNF-α release of J774A.1 cells in response to infection with virulence plasmid–cured yersiniae. These effects seem to be cumulative, since addition of the two compounds together blocked the TNF-α production completely. However, under the same experimental conditions, neither compound prevented NF-κB activation due to infection with strain WA-C (Fig. 5, lane 4), which demonstrates that these inhibitors do not exert their effects on TNF-α production via the NF-κB signaling pathway. These results indicate that not only NF-κB, but also the p38 and ERK1/2 MAPK pathways, are important regulators of the J774A.1 cell TNF-α production in response to bacterial infection.
Table 5.
Inhibitors of p38 (SB203580) and MEK1 (PD098059) Cumulatively Inhibit the TNF-α Production of J774A.1 Macrophages Stimulated with Virulence Plasmid–cured Y. enterocolitica (WA-C).
| Pretreatment* | TNF-α production‡ | |||
|---|---|---|---|---|
| No bacteria | WA-C | |||
| Nothing | 37 ± 4 | 1,625 ± 126 | ||
| SB203580 | 59 ± 16 | 752 ± 121 | ||
| PD098059 | 47 ± 10 | 680 ± 150 | ||
| SB203580 + PD098059 | 45 ± 8 | 49 ± 5 | ||
J774A.1 cells were either not pretreated or pretreated for 30 min with the compounds SB203580 (10 μM), PD098059 (50 μM), or the two compounds together. Thereafter, half of the samples were challenged by the addition of virulence plasmid–cured yersiniae (WA-C).
The TNF-α activity in the cell culture supernatant (dilution 1:120) was measured 4 h after the onset of infection with strain WA-C, using a cytotoxic assay performed with the TNF-α–sensitive fibroblast cell line L929.
Discussion
Pathogenic Yersinia spp. counteract host defense mechanisms by interfering with eukaryotic signal transduction pathways. In this study, we investigated the impact of Y. enterocolitica on nuclear translocation of NF-κB, which is a critical regulator of cytokine expression (23), but also plays an intriguing role in promoting cell survival (24–28). Our present data show that Y. enterocolitica strongly impairs activation of NF-κB in eukaryotic cells. In murine J774A.1 and peritoneal macrophages, wild-type Y. enterocolitica initially induced a weak NF-κB signal. This signal was completely abolished after 60 min in J774A.1 macrophages and almost completely inhibited after 90 min in peritoneal macrophages, respectively. In contrast, virulence plasmid– cured yersiniae and LPS promoted persistent NF-κB activation for at least 90 min. Analysis of the cytoplasmic levels of the inhibitory proteins IκB-α and IκB-β showed that infection with wild-type yersiniae caused an initial degradation of IκB-α and IκB-β similar to nonvirulent yersiniae and LPS. After 60–90 min, however, when NF-κB activity was inhibited, the cytoplasmic pools of IκB-α and IκB-β were already completely restored. On the contrary, cells treated with virulence plasmid–cured yersiniae or with LPS exhibited a substantial degree of phosphorylation and degradation of IκB-α and IκB-β throughout the time investigated. These results suggest that Y. enterocolitica impairs activation of NF-κB by preventing prolonged phosphorylation and degradation of IκB-α and IκB-β inhibitory proteins. The fact that the suppression of the NF-κB activity occured after only 60–90 min of infection can be explained by a delay necessary for Yersinia to exert its effects on the host cell after host cell contact (1).
To substantiate a possible relation between NF-κB inhibition and induction of apoptosis and blockage of TNF-α production, we checked a set of genetically designed Y. enterocolitica mutants for these activities in J774A.1 cells. Indeed, we found a striking correlation between the abilities of these strains to inhibit activation of NF-κB, to promote apoptosis, and to suppress TNF-α production. The strains capable of inhibiting translocation of NF-κB also efficiently blocked TNF-α secretion and strongly induced apoptosis. Conversely, the strains that elicited a strong NF-κB signal were not able to trigger apoptosis, but generated a marked TNF-α response. The experiments on these strains showed that inhibition of NF-κB is distinct from the effects mediated by YopH and YopE, but depends on a functional Yop secretion system. This implies that indeed one or several secreted virulence factors are involved. Recently, the YopP protein of Y. enterocolitica (YopJ in Y. pseudotuberculosis) was reported to play a role in the promotion of macrophage apoptosis (20, 21). In this study, we analyzed a Y. enterocolitica mutant affected in expression of the serine/threonine kinase gene yopO and the cotranscribed yopP. Indeed, this yopO/yopP double mutant was markedly impaired in its ability to induce apoptosis. Simultaneously, this mutant was not as efficient as the wild-type strain to block the secretion of TNF-α. And, most interestingly, the translocation of NF-κB was also not as efficiently inhibited. Thus, our data suggest a direct link between activation of NF-κB and cell survival/TNF-α production, or, conversely, inhibition of NF-κB and subsequent cell death/TNF-α suppression in Yersinia-infected macrophages. This coherence was further confirmed by the finding that inhibition of NF-κB activation by compound MG-132, which selectively blocks the proteasome pathway, triggered apoptosis and TNF-α suppression in J774A.1 cells treated either with LPS or with nonvirulent yersiniae. These results suggest a dual function of NF-κB in macrophages infected with nonvirulent yersiniae, which is prevention of cell death on the one hand, and induction of TNF-α production on the other. Wild-type yersiniae, by counteracting the activation of NF-κB, impair cell survival and suppress the macrophage TNF-α production.
The situation in epithelial HeLa cells appears different. Our previous work showed that HeLa cells do not undergo apoptotic cell death upon Yersinia infection, in contrast to macrophages (19). Nevertheless, Yersinia-infected HeLa cells are severely affected by the actin depolymerizing activity of YopE (19). This indicates that Yersinia also exerts its effects on HeLa cells, but the response of HeLa cells apparently differs from that of macrophages. Our study demonstrated that wild-type Y. enterocolitica also blocked activation of NF-κB in HeLa cells without triggering apoptosis. However, when HeLa cells were pretreated with wild-type yersiniae or with the proteasome inhibitor MG-132, subsequent stimulation with the cytokine TNF-α strongly promoted HeLa cell apoptosis. In line with the fact that the activity of NF-κB determines the fate of TNF-α–treated HeLa cells (28, 39), infection with virulent yersiniae substantially suppressed the NF-κB response induced by TNF-α. This signals the cell to undergo apoptosis.
An essential role of NF-κB in preventing cell death has been demonstrated for different stimuli, such as TNF-α, ionizing radiation, or the genotoxic agent daunorubicin (24–26). Activation of NF-κB appears to provide protection against apoptotic killing induced by these stimuli (24– 26). TNF-α, which is the best studied agonist of this group, simultaneously activates cytotoxic and death-preventing pathways. The balance between these two pathways determines the fate of the cell. The pathways that prevent cell death depend on protein synthesis, but not the cytotoxic pathways (43). NF-κB functions to transcriptionally upregulate a gene or group of genes encoding proteins that mediate cell survival, such as proteins of the IAP (inhibitor of apoptosis protein) family (44, 45). Y. enterocolitica suppresses NF-κB activity, which in turn may affect expression of the genes that are necessary for cell survival. This leads to domination of death-inducing pathways and rapid apoptosis. This concept exactly fits to our previous finding that Yersinia triggers apoptosis at a level upstream or apart from the actual core apoptotic program (caspase activities) activated by death-inducing pathways (19). Furthermore, this mechanism explains why Y. enterocolitica triggers apoptosis not only in macrophages, but also in epithelial HeLa cells, provided that the appropriate death signal (TNF-α in our experiments) is present. Interestingly, LPS alone appears to be sufficient to signal the macrophage to undergo apoptosis if activation of NF-κB is blocked by the proteasome inhibitor MG-132. Cell death under these conditions is not mediated by traces of TNF-α secreted by the macrophage, since increasing amounts of TNF-α–neutralizing antibodies added to the medium did not reduce the rate of apoptosis (data not shown). This finding confirms that even during treatment with LPS, activation of NF-κB is indispensable for cell survival.
The signaling pathways that induce programmed cell death under conditions where NF-κB is inhibited are less clear. Several lines of evidence indicate that MAPK modules are involved in the activation of apoptosis-inducing pathways (46–50). The MAPK and NF-κB signaling systems represent two distinct but interactive signal transduction pathways (39, 51) and it may be possible that apoptosis by Yersinia requires inhibition of NF-κB, as well as activation of MAPK cascades. Indeed, we found that Yersinia infection initially activates the macrophage MAPK pathways p38, ERK, and JNK (14). Using drugs that are presently available and selectively block MAPK activities (SB203580, PD098059), we analyzed the role of p38 and ERK in Yersinia–macrophage interaction. Our experiments showed that these drugs did not prevent cell death mediated by wild-type Yersinia, which indicates that p38 and ERK are at least not crucially implicated in Yersinia-induced apoptosis. On the other hand, these two compounds cumulatively blocked the J774A.1 cell TNF-α production induced by virulence plasmid–cured yersiniae without interfering with activation of NF-κB. This proves that besides NF-κB, the p38 and ERK MAPK pathways are also involved in J774A.1 cell TNF-α production. Indeed, MAPK as well as NF-κB signaling pathways play an important role in the regulation of the macrophage TNF-α production, either at the transcriptional (NF-κB, ERK, JNK) and/or posttranscriptional level (p38, JNK) (15–18, 52, 53). Activities of these pathways contribute to TNF-α production rather additively than competitively, since maximal activation of the TNF promotor requires at least two cis-acting regulatory elements, such as NF-κB and c-Jun (52). In agreement with these findings, our results postulate that Y. enterocolitica blocks the macrophage TNF-α production in a bimodal manner, by inhibiting both the MAPK as well as the NF-κB signaling pathways. The simultaneous affection of the two signal transduction systems implies that Yersinia mediates blockage of common upstream activators, such as the signaling cascades of the Ras superfamily of small G proteins, namely Cdc42, Rac-1, and Rho, which are implicated in activation of NF-κB (54), as well as of MAPK modules (55). Studies addressing this question are presently underway.
In summary, our study provides new insights into the mechanisms by which Y. enterocolitica subverts eukaryotic cells and modulates the immune response of the host. Y. enterocolitica interferes with the activation of transcription factor NF-κB in eukaryotic cells. This strategy serves to suppress the TNF-α production of macrophages and contributes to trigger macrophage cell death by apoptosis. Epithelial HeLa cells undergo apoptosis upon Yersinia infection only when the cytokine TNF-α is present simultaneously. Since yersiniae block the TNF-α secretion in vivo, this may indicate a strategy of Yersinia to focus apoptosis selectively towards macrophages, whereas nonphagocytic cells escape programmed cell death due to Yersinia infection.
Acknowledgments
We thank Dr. J.C. Lee (SmithKline Beecham) for the gift of SB 203580 and Drs. Jacques Dornand, Astrid Haeffner, François Hirsch, Brigitte Kahn-Perles, Jean Imbert, Christian Roy, and Marie-Luce Vignais for interesting discussions. We are grateful to Sophie Lotersztajn for her suggestion to use the MG-132 proteasome inhibitor.
This work was supported by grant ARC 6497 from the Association pour la Recherche contre le Cancer, by grant CHRX-CT94-0451 of the Human Capital and Mobility Program from the European Union, by grant SFB 217 from the Deutsche Forschungsgemeinschaft, and by the French–German exchange program PROCOPE. K. Ruckdeschel was supported by a postdoctoral fellowship from the Fondation pour la Recherche Médicale and by the Bundesministerium für Forschung und Technologie. B. Rouot was supported by a fellowship from OECD (Biological Resource Management for Sustainable Agricultural Systems).
Footnotes
Abbreviations used in this paper: DTT, 1,4 dithiothreitol; EMSA, electrophoretic mobility shift assay; ERK, extracellular signal–regulated kinase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase; MEK1, MAPK-ERK kinase 1.
References
- 1.Cornelis GR, Wolf-Watz H. The YersiniaYop virulon: a bacterial system for subverting eukaryotic cells. Mol Microbiol. 1997;23:861–867. doi: 10.1046/j.1365-2958.1997.2731623.x. [DOI] [PubMed] [Google Scholar]
- 2.Simonet M, Richard S, Berche P. Electron microscopic evidence for in vivo extracellular localisation of Yersinia pseudotuberculosisharboring the pYV plasmid. Infect Immun. 1990;58:841–854. doi: 10.1128/iai.58.3.841-845.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heesemann J, Gross U, Schmidt N, Laufs R. Immunochemical analysis of plasmid-encoded proteins released by enteropathogenic Yersiniasp. grown in calcium-deficient media. Infect Immun. 1986;54:561–567. doi: 10.1128/iai.54.2.561-567.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Straley SC, Plano GV, Skrzypek E, Bliska JB. Yops of Yersiniaspp. pathogenic for humans. Infect Immun. 1993;61:3105–3110. doi: 10.1128/iai.61.8.3105-3110.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bliska JB, Galán JE, Falkow S. Signal transduction in the mammalian cell during bacterial attachment and entry. Cell. 1993;73:903–920. doi: 10.1016/0092-8674(93)90270-z. [DOI] [PubMed] [Google Scholar]
- 6.Rosqvist R, Bölin I, Wolf-Watz H. Inhibition of phagocytosis in Yersinia pseudotuberculosis: a virulence plasmid-encoded ability involving the Yop2b protein. Infect Immun. 1988;56:2139–2143. doi: 10.1128/iai.56.8.2139-2143.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosqvist R, Forsberg A, Rimpiläinen M, Bergman T, Wolf-Watz H. The cytotoxic protein YopE of Yersiniaobstructs the primary host defence. Mol Microbiol. 1990;4:657–667. doi: 10.1111/j.1365-2958.1990.tb00635.x. [DOI] [PubMed] [Google Scholar]
- 8.Rosqvist R, Forsberg A, Wolf-Watz H. Intracellular targeting of the YersiniaYopE cytotoxin in mammalian cells induces actin microfilament disruption. Infect Immun. 1991;59:4562–4569. doi: 10.1128/iai.59.12.4562-4569.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bliska JB, Black DS. Inhibition of the Fc receptor–mediated oxidative burst in macrophages by the Yersinia pseudotuberculosistyrosine phosphatase. Infect Immun. 1995;63:681–685. doi: 10.1128/iai.63.2.681-685.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruckdeschel K, Roggenkamp A, Schubert S, Heesemann J. Differential contribution of Yersinia enterocoliticavirulence factors to evasion of microbicidal action of neutrophils. Infect Immun. 1996;64:724–733. doi: 10.1128/iai.64.3.724-733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galyov EE, Håkansson S, Forsberg Å, Wolf-Watz H. A secreted protein kinase of Yersinia pseudotuberculosisis an indispensable virulence determinant. Nature. 1993;361:730–732. doi: 10.1038/361730a0. [DOI] [PubMed] [Google Scholar]
- 12.Nakajima R, Brubaker RR. Association between virulence of Yersinia pestisand suppression of gamma interferon and tumor necrosis factor alpha. Infect Immun. 1993;61:23–31. doi: 10.1128/iai.61.1.23-31.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Autenrieth IB, Heesemann J. In vivo neutralization of tumor necrosis factor alpha and interferon-gamma abrogates resistance to Yersinia enterocoliticain mice. Med Microbiol Immunol. 1992;181:333–338. doi: 10.1007/BF00191545. [DOI] [PubMed] [Google Scholar]
- 14.Ruckdeschel K, Machold J, Roggenkamp A, Schubert S, Pierre J, Zumbihl R, Liautard J-P, Heesemann J, Rouot B. Yersinia enterocolitica promotes deactivation of macrophage mitogen-activated protein kinases extracellular signal-regulated kinase-1/2, p38, and c-Jun NH2-terminal kinase: correlation with its inhibitory effect on tumor necrosis factor α production. J Biol Chem. 1997;272:15920–15927. doi: 10.1074/jbc.272.25.15920. [DOI] [PubMed] [Google Scholar]
- 15.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 16.Trotta R, Kanakaraj P, Perussia B. FcγR-dependent mitogen-activated protein kinase activation in leukocytes: a common signal transduction event necessary for expression of TNF-α and early activation genes. J Exp Med. 1996;184:1027–1035. doi: 10.1084/jem.184.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng ZM, Specter S. Dynamic production of tumour necrosis factor-α (TNF-α) messenger RNA, intracellular and extracellular TNF-α by murine macrophages and possible association with protein tyrosine phosphorylation of STAT1α and ERK2 as early signal. Immunology. 1996;87:544–550. doi: 10.1046/j.1365-2567.1996.513591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose DM, Winston BW, Chan ED, Riches DWH, Gerwins P, Johnson GL, Henson PM. Fcγ receptor cross-linking activates p42, p38, and JNK/SAPK mitogen-activated protein kinases in murine macrophages: role for p42 MAPK in Fcγ receptor–stimulated TNF-α synthesis. J Immunol. 1997;158:3433–3438. [PubMed] [Google Scholar]
- 19.Ruckdeschel K, Roggenkamp A, Lafont V, Mangeat P, Heesemann J, Rouot B. Interaction of Yersinia enterocoliticawith macrophages leads to macrophage cell death through apoptosis. Infect Immun. 1997;64:4813–4821. doi: 10.1128/iai.65.11.4813-4821.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monack DM, Mecsas J, Ghori N, Falkow S. Yersiniasignals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci USA. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mills SD, Boland A, Sory M-P, Smissen PVD, Kerbourch C, Finlay BB, Cornelis GR. Yersinia enterocoliticainduces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc Natl Acad Sci USA. 1997;94:12638–12643. doi: 10.1073/pnas.94.23.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zychlinsky A, Sansonetti P. Apoptosis in bacterial pathogenesis. J Clin Invest. 1997;100:493–495. doi: 10.1172/JCI119557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 24.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α–induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 25.Wang C-Y, Mayo MW, Baldwin AS., Jr TNF– and cancer therapy–induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 26.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Vermat IM. Suppression of TNF-α–induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 27.Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, FitzGerald MJ, Rothstein TL, Sherr DH, Sonenshein GE. Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO (Eur Mol Biol Organ) J. 1996;15:4682–4690. [PMC free article] [PubMed] [Google Scholar]
- 28.Zong W-X, Farrell M, Bash J, Gélinas C. v-Rel prevents apoptosis in transformed lymphoid cells and blocks TNFα-induced cell death. Oncogene. 1997;15:971–980. doi: 10.1038/sj.onc.1201266. [DOI] [PubMed] [Google Scholar]
- 29.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 30.Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 31.Heesemann J. Chromosomal-encoded siderophores are required for mouse virulence of enteropathogenic Yersiniaspecies. FEMS Microbiol Lett. 1987;48:229–233. [Google Scholar]
- 32.Roggenkamp A, Schubert S, Jacobi CA, Heesemann J. Dissection of the Yersinia enterocoliticavirulence plasmid pYVO8 into an operating unit and virulence genes modules. FEMS Microbiol Lett. 1996;134:69–73. doi: 10.1111/j.1574-6968.1995.tb07916.x. [DOI] [PubMed] [Google Scholar]
- 33.Thieblemont N, Haeffner-Cavaillon N, Haeffner A, Cholley B, Weiss L, Kazatchkine MD. Triggering of complement receptors CR1 (CD35) and CR3 (CD11b/ CD18) induces nuclear translocation of NF-κB (p50/65) in human monocytes and enhances viral replication in HIV-infected monocytic cells. J Immunol. 1995;155:4861–4867. [PubMed] [Google Scholar]
- 34.Kahn-Perlès B, Lipcey C, Lécine P, Olive D, Imbert J. Temporal and subunit-specific modulations of the Rel/NF-κB transcription factors through CD28 costimulation. J Biol Chem. 1997;272:21774–21783. doi: 10.1074/jbc.272.35.21774. [DOI] [PubMed] [Google Scholar]
- 35.Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IκBα kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–222. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 36.Roggenkamp A, Neuberger H-R, Flügel A, Schmoll T, Heesemann J. Substitution of two histidine residues in YadA protein of Yersinia enterocoliticaabrogates collagen binding, cell adherence and mouse virulence. Mol Microbiol. 1995;16:1207–1219. doi: 10.1111/j.1365-2958.1995.tb02343.x. [DOI] [PubMed] [Google Scholar]
- 37.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin–proteasome pathway is required for processing the NF-κB1 precurser protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 38.Rock, K.L., C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang, and A.L. Goldberg. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 78:761–771. [DOI] [PubMed]
- 39.Liu Z, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 40.Anderson P. Kinase cascades regulating entry into apoptosis. Microbiol Mol Biol Rev. 1997;61:33–46. doi: 10.1128/mmbr.61.1.33-46.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JC, Young PR. Role of CSBP/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J Leukocyte Biol. 1996;59:152–157. doi: 10.1002/jlb.59.2.152. [DOI] [PubMed] [Google Scholar]
- 42.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallach D. Cell death induction by TNF: a matter of self control. Trends Biochem Sci. 1997;22:107–109. doi: 10.1016/s0968-0004(97)01015-3. [DOI] [PubMed] [Google Scholar]
- 44.Chu Z-L, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor–induced cell death by inhibitor of apoptosis c-IAP2 is under NF-κB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.You M, Ku P-T, Hrdlickova R, Bose HR., Jr ch-IAP1, a member of the inhibitor-of-apoptosis protein family, is a mediator of the antiapoptotic activity of the v-Rel oncoprotein. Mol Cell Biol. 1997;17:7328–7341. doi: 10.1128/mcb.17.12.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 47.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 48.Chen Y-R, Wang Y, Templeton D, Davis RJ, Tan T-H. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation: duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 49.Goillot E, Raingeaud J, Ranger A, Tepper RI, Davis RJ, Harlow E, Sanchez I. Mitogen-activated protein kinase–mediated Fas apoptotic signaling pathway. Proc Natl Acad Sci USA. 1997;94:3302–3307. doi: 10.1073/pnas.94.7.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwenger P, Bellosta P, Vietor I, Basilico C, Skolnik EY, Vilcek J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor–induced c-Jun N-terminal kinase/stress-activated kinase activation. Proc Natl Acad Sci USA. 1997;94:2869–2873. doi: 10.1073/pnas.94.7.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song HY, Régenier CH, Kirsching CJ, Goeddel DV, Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-κB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor–associated factor 2. Proc Natl Acad Sci USA. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao J, Mackman N, Edgington TS, Fan S-T. Lipopolysaccharide induction of the tumor necrosis factor-α promotor in human monocytic cells: regulation by Egr-1, c-Jun, and NF-κB transcription factors. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 53.Swantek JL, Cobb MH, Geppert TD. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-α) translation: glucocorticoids inhibit TNF-α translation by blocking JNK/SAPK. Mol Cell Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perona R, Montaner S, Saniger L, Sánchez-Pérez I, Bravo R, Lacal JC. Activation of the nuclear factor-κB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1996;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- 55.Symons M. Rho family GTPases: the cytoskeleton and beyond. Trends Biochem Sci. 1996;21:178–181. [PubMed] [Google Scholar]