

Abstract
The polypeptide (p)50 molecule, a subunit of nuclear factor (NF)-κB, is produced after proteolytic processing of the p105 precursor (NF-κB1). Although the p105 precursor has been postulated to play a role in the regulation of the Rel/NF-κB activity, its physiological relevance remains unclear. To investigate that, we generated mutant mice lacking the COOH terminal half of the p105 precursor, but expressing the p50 product (p105−/−). These mutant mice displayed an inflammatory phenotype composed of lymphocytic infiltration in lungs and liver, and an increased susceptibility to opportunistic infections. Enlargement of multiple lymph nodes, splenomegaly due to erythrocytic extramedullary hematopoiesis, and lymphoid hyperplasia were also observed in p105−/− mice. Cytokine production in p105−/− macrophages was severely impaired, whereas proliferative responses of p105−/− B cells were increased. T cell functions were only moderately impaired in mutant mice. Loss of p105 also led to enhanced constitutive p50 homodimer and inducible NF-κB activities in unstimulated and stimulated cells, respectively. As several genes regulated by Rel/NF-κB were upregulated in p105−/− thymus but downregulated in p105−/− macrophages, the enhanced p50 homodimers appear to function as transcriptional activators or repressors, depending on the cell type. Thus, the p105 precursor is indispensable in the control of p50 activity, and lack of the precursor has distinct effects on different cells.
Nuclear factor (NF)-κB plays an important role in regulating a wide variety of genes that encode proteins involved in immune, acute phase, and inflammatory responses (1–4). Several NF-κB subunit molecules have been identified that share extensive similarity to the rel protooncogene product and the Drosophila maternal gene product dorsal. In mammals, members of the Rel/NF-κB family include NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), RelA, RelB, and c-Rel. They share a conserved NH2 terminus termed the Rel homology domain (RHD)1 responsible for DNA binding, dimerization, and association with IκB inhibitors. The unconserved COOH terminus of RelA, RelB, and c-Rel mediates transcriptional activation, whereas p50 and p52 do not contain transcriptional activation domains. Mature p50 and p52 products are generated by removal of the COOH terminus of the p105 and p100 precursors, respectively (3–9). The genes of the Rel/NF-κB family are differentially expressed in lymphoid tissues (10–12) and studies of mice lacking either p50, RelA, RelB, or c-Rel demonstrate that individual members of this family have distinct functions in vivo (13–17).
In most cells, the Rel/NF-κB complexes are associated with inhibitory molecules, IκBs, in the cytoplasm. The Rel/NF-κB complexes can be rapidly induced by a variety of stimuli leading to modifications of IκB proteins that allow dissociation of NF-κB-IκB complexes and subsequent translocation of Rel/NF-κB complexes into the nucleus (5–9, 18, 19). In mammals, members of the IκB family include IκBα, IκBβ, IκBγ, Bcl-3, p105, p100, and the recently cloned IκBε (20), which share the conserved ankyrin-like repeats responsible for the interaction with the Rel/NF-κB complexes. The trans-acting IκBα and IκBβ may play a major role in controlling Rel/NF-κB activity, whereas the p105 and p100 precursors can be regulators by dimerizing with individual members of the Rel/NF-κB family including their products p50 and p52 (4–7, 9, 19). In the case of IκBα, phosphorylation of the NH2-terminal serine residues induces complete degradation of the protein, whereas phosphorylation of NH2-terminal tyrosine residues allows to release the Rel/NF-κB complexes without protein degradation (21). Proteolytic cleavage and presumably subsequent degradation of the COOH-terminal ankyrin region of the precursors (p105 and p100) also result in Rel/NF-κB activation (9, 19). Degradation of both IκBα and the COOH terminus of p105 is mediated by a ubiquitin–proteasome pathway, which requires previous phosphorylation (22, 23). Recently, two IκB kinases have been described; one of them is constitutive and requires ubiquitination for being active (24) and the other, independent of ubiquitination, is rapidly stimulated by cytokines as TNF-α and IL-1 (25).
The murine nfkb1 gene produces two transcripts: one 4.0-kb messenger RNA (mRNA) encoding the full-length polypeptide (p)105 precursor that is expressed ubiquitously, and another 2.6-kb mRNA encoding the IκBγ protein identical to the COOH terminus of p105 that is found predominantly in lymphoid cells (26, 27). These two transcripts are likely to be generated by differential promoter usage.
To investigate the physiological role of p105, we generated mice lacking the precursor but still containing the p50 product by gene targeting. Mice homozygous for a deletion of the COOH terminus of the p105 precursor developed an inflammatory phenotype in the lungs and liver, and also had splenomegaly due to extramedullary hematopoiesis and lymphoid hyperplasia, enlarged lymph nodes, and were susceptible to opportunistic infections. Cytokine secretion in mutant macrophages was severely impaired, whereas that in activated T cells appeared to be only slightly decreased. Proliferative responses of p105−/− T cells were only moderately impaired, whereas those of p105−/− B cells were increased. The p50 homodimer DNA-binding activity was increased in the absence of p105, showing the regulatory role of the precursor for the p50 homodimer activity. Moreover, the deregulated p50 homodimer activity is likely to function as a transcriptional activator or repressor because the expression of several NF-κB–regulated genes was augmented in thymus but reduced in macrophages from mutant mice.
Materials and Methods
Targeting Vector and Generation of Mutant Mice.
To generate the targeting vector plasmid PGK promoter neomycin thymidine kinase (pPNT)/IκBγ II, a small intronic sequence within the SV40 polyadenylation recognition sequences [SV40 p(A)] was deleted in pPNT/IκBγ (28).
CJ7 embryonic stem (ES) cells were electroporated with the NotI-linearized pPNT/IκBγ II, and subsequent double resistant clones were selected as previously described (29). Homologous recombination was screened by Southern blot using a 5′ external probe. 5′,3′ internal or neo-specific probes were used to demonstrate that the targeted ES cell clones (5) did not contain any random integration of the vector. Targeted clones were identified by a novel 9-kb recombinant band in addition to the wild-type band (>25 kb) in EcoRV-digested DNA when 5′ external or internal probes were used. A 6.5-kb recombinant band in addition to the 11.2-kb wild-type band in EcoRI-digested DNA was also observed in targeted ES clones when 5′ internal or neo-specific probes were used. The generation of chimeric mice, hetero-, and homozygous mutant animals was performed as previously described (29).
Histology, Immunohistochemistry, and Flow Cytometry.
Complete postmortem examinations were performed on 3 wk-, 6 wk-, 12 wk-, and 5-mo-old mice. Organs were immersion-fixed in 10% neutral buffered formalin, processed by standard methods, and paraffin embedded. Tissue sections were stained with hematoxylin and eosin for light microscopy. Immunohistochemical phenotyping of hepatic and pulmonary inflammatory infiltrates was performed using established avidin-biotin methods on serial, paraffin-embedded, and frozen tissue sections with the following antibodies: B220 (B cells; PharMingen, San Diego, CA), 7/4 (granulocytes; Serotec Ltd., Oxford, UK), F4/80 (Serotec), Mac-2 (macrophages; Cedar Lane, Hornby, Canada), and CD4 and CD8 (T cells; PharMingen). Flow cytometry analysis was performed as previously described (17).
Western Blot Analysis and Electrophoretic Mobility Shift Assay.
Whole tissue extracts isolated from several organs of 3-wk-old mice were prepared as described (30). Splenocytes or thymocytes from 3-wk-old animals were isolated as described (31). Peripheral B cells were purified from 3-wk-old mouse splenocyte suspension by mouse B cell enrichment immunocolumns (Biotex Laboratories Inc., Edmonton, Canada). Peripheral T cells were purified from spleen or lymph nodes by murine T cell enrichment columns (R&D Systems, Inc., Minneapolis, MN). Macrophages were collected from 4-wk-old mice by peritoneal lavage and purified as adherent cells to plastic dishes. B cells, T cells, and macrophages were treated with 10 μg/ml of LPS (Sigma Chemical Co., St. Louis, MO), 10 ng/ml of TNF-α (Sigma Chemical Co.), and 10 μg/ml of LPS, respectively. Nuclear extracts isolated from thymocytes, B cells, T cells, or macrophages were prepared as described (32). Western blot analysis using whole tissue extracts and electrophoretic mobility shift assay (EMSA) using nuclear extracts were performed as previously described (33). In brief, for EMSA 2–10 μg of nuclear extract were incubated at room temperature for 30 min with a labeled probe in 20 μl of a solution containing 1 μg of poly dI:dC in 20 mM Hepes, pH 7.9, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 100 mM KCl, and protease inhibitor. The DNA probes and Rel/NF-κB antibodies used in these studies were described previously (28, 34).
Reverse Transcriptase-PCR Analysis.
Total RNA from 3-wk-old mouse thymuses or macrophages was prepared using TRIzol (GIBCO BRL, Gaithersburg, MD). Reverse transcriptase PCR (RT-PCR) was performed as previously described (35). The primer sequences consisted of: GM-CSF, 5′-CCC ATC ACT GTC ACC CGG CCT TGG-3 and 5′-GTC CGT TTC CGG AGT TGG GGG GC-3′; IL-2, 5′-GTC AAC AGC GCA CCC ACT TCA AGC-3′ and 5′-GCT TGT TGA GAT GAT GCT TTG ACA-3′; TNF-β, 5′-TCC TGA AAC CTG CTG CTC ACC TTG TTG G-3′ and 5′-CTG GGG CTG AAG TGT AGA TGG GAG ATG C-3′; alpha inhibitor of κB (ikba), 5′-TGG CTG ACT GAC TCA CTG ACT GAC TGA CTC GTG CCT TGC-3′ and 5′-TGG CCG TTG TAG TTG GTG GC-3′; intercellular adhesion molecule (ICAM)-1, 5′-CCG CTT CCG CTT CCG CTA CCA TCA CCG TGT ATT C-3′ and 5′-GCC TTC CAG GGA GCA AAA CAA CTT CTG C-3′; vascular adhesion molecule (VCAM)-1, 5′-AAC AGA CAG GAG TTT TC-3′ and 5′-GTC AAC AAT AAA TGG TT-3′. The sequences of the primers for β-actin and G-CSF were previously described (29, 35). The primers for M-CSF and TNF-α were obtained commercially (Stratagene Corp., La Jolla, CA).
Proliferation Assays.
Purified B cells were treated with either anti-IgM antibody (Jackson ImmunoResearch, Bar Harbor, ME) or LPS, whereas purified T cells were treated with either coated anti-CD3 antibody (PharMingen), coated anti-CD3 plus anti-CD28 antibodies (PharMingen), or 7 ng/ml of PMA (Sigma Chemical Co.) plus 1 μg/ml of PHA (Sigma Chemical Co.). Proliferative responses were determined by [3H]thymidine incorporation as described previously (29).
Cytokine Production in T Cells and Macrophages.
Purified T cells (5 × 105/ml) were incubated with or without coated anti-CD3 antibody or coated anti-CD3 plus anti-CD28 antibodies for 24 h. Purified peritoneal macrophages (5 × 105/ml) were incubated in the presence or absence of 1 μg/ml of LPS and 100 U/ml of IFN-γ (Genzyme Corp., Cambridge, MA) for 72 h. Cytokine levels for IL-1β, IL-4, IL-6, IL-10, GM-CSF, and TNF-α in supernatants were measured by ELISA (R&D Systems, Inc.); levels of IFN-γ and IL-2 were also determined by ELISA (BIOSOURCE, Camarillo, CA).
Analysis of Germinal Center Formation.
Three 6-wk-old mice of each genotype were immunized intraperitoneally with 100 μl of phosphate-buffered saline containing 10% SRBCs. 10 d later, spleens were harvested, embedded in O.C.T. compound (Miles Laboratories Inc., Rexdale, Canada), and frozen in liquid nitrogen. Frozen tissue sections were incubated with biotinylated PNA (Vector Labs, Burlingame, CA) for centrocyte detection and with rat anti–mouse IgD as previously described (36).
Immunoglobulin Isotype Assays.
Sera were prepared from 5-wk-old sex-matched animals, and levels of Ig isotype were determined using a sandwich ELISA using a pan-specific capture antibody (Southern Biotechnology Assoc., Birmingham, AL) and isotype-specific antibodies conjugated to horseradish peroxidase (Southern Biotechnology Assoc.). For IgE levels, rat anti–mouse IgE capture antibody (PharMingen), biotinylated rat anti–mouse IgE (PharMingen), and avidin peroxidase (Sigma Chemical Co.) were used.
T Cell–dependent and –independent Humoral Immune Responses.
5-wk-old sex-matched animals were immunized by intraperitoneal injection of either 100 μg of KLH (Calbiochem Corp., La Jolla, CA) coupled to (4-hydroxy-3-nitro-phenyl) acetyl (NP; Biosearch Technologies, San Diego, CA) precipitated in alum (T cell– dependent response) or 10 μg of LPS (Escherichia coli 0111: B4; Difco, Detroit, MI) coupled to NP (T cell–independent response). Conjugates at ratios of 17:1 (NP-KLH) and 10:1 (NP-LPS) and alum precipitates were prepared as described (37, 38). Sera were collected before immunization and at 7-d intervals after immunization for a period of 3 wk. Levels of NP-specific Ig isotypes were determined by ELISA using NP-BSA (17:1) as a capture agent and goat anti–mouse isotype-specific sera directly conjugated to horseradish peroxidase (Southern Biotechnology Assoc).
Results
Generation of Mice Lacking the COOH Terminus of NF-κB1.
The vector used to generate the p105−/− was described previously (27), except for the deletion of a small intron within the SV40 p(A) to avoid an alternative RNA splicing, which produced a p50 isoform lacking the nuclear localization signal (NLS; Fig. 1 A).
Figure 1.


Generation of p105−/− mice. (A) Targeting strategy of the ankyrin-encoding region of the nfkb1 gene. The relevant structure of the murine nfkb1 gene is shown at the top. Targeting vector pPNT/IκBγ II and the targeted allele are shown at the middle and bottom, respectively. Closed boxes, exons of nfkb1 gene; open boxes, the SV40 p(A), phosphoglycerate kinase (PGK)-neo, and PGK-tk cassettes. The position of EcoRI and EcoRV sites are indicated by I and V, respectively. The diagnostic restriction fragments used for Southern blot analysis are indicated at the top (wild-type allele) and bottom (targeted allele). The DNA fragments used as 5′ external (EE), 5′ internal (XX), 3′ internal (E), and neo-specific (PP620) probes are indicated at the bottom. (B) Genotype analysis of pups generated from p105+/− intercrosses. Tail DNAs were digested with EcoRI, followed by Southern blot analysis using the 5′ internal probe XX. The wild-type allele is indicated by a 11.2-kb band, whereas the recombinant allele is represented by a 6.5-kb band. (C) Lack of p105 in homozygous mutants. Whole tissue extracts from thymus were subjected to Western blot analysis using a p50 antibody. Specific signals for the p50 isoform, p50, and p105 proteins are indicated by arrows.
The new targeting vector (PNT/IκBγII) was electroporated into CJ7 ES cells and double resistant clones were screened by Southern blot analysis as described previously (28). Chimeric mice generated by standard procedures transmitted the mutation of the nfkb1 locus to their offspring, and subsequent intercrosses between heterozygotes generated homozygous mutant (p105−/−) mice. The three different genotypes are shown by Southern blot analysis using genomic DNA isolated from mouse tails (Fig. 1 B).
Western blot analysis using whole tissue extracts isolated from the thymus demonstrated a lack of p105 but the presence of p50 in homozygous mutant mice (Fig. 1 C). A faster migrating molecule reactive to a p50 antibody was detected in p105−/+ or p105−/− extracts, despite a deletion of SV40 p(A) intronic sequences; however, the expected p50 molecule of 415 amino acids was very abundant in the mutants. The signal corresponding to the IκBγ protein could not be detected in control extracts when an IκBγ-specific antibody was used (data not shown). This is in agreement with previous observations indicating the extremely low abundance of the IκBγ protein and expression of the IκBγ transcript in a restricted population of cells (26, 27).
Increased Mortality and Histological Abnormalities of p105−/− Mice.
Heterozygotes mating produced the normal number of homozygous mutant mice (21.5%, n = 297) according to mendelian ratios. During the first 3 wk of age, homozygous mutant mice were similar in size and weight to littermates, although 20% of p105−/− pups died. After weaning, some p105−/− pups appeared sick (rough haircoat and lethargic) and showed growth retardation (data not shown). Later, some of these pups maintained in conventional housing developed eye and/or nasal infections produced by Staphylococcus xylosis and Pasteurella haemolytica (opportunistic bacteria), whereas mice maintained in microisolators did not show infections. p105−/− mice started to die at an increased rate after 2 mo of age, resulting in a 50% survival (n = 15) at 100 d old, whereas control littermates (p105+/+ and p105−/+, n = 22) showed almost 100% survival by 7 mo of age (Fig. 2 A).
Figure 2.


Gross abnormalities of p105−/− mice. (A) Mortality of control and heterozygotes (circles, n = 22) and p105−/− (triangles, n = 15) mice. Survival is shown as a percentage of the total initial number of control or p105−/− mice. (B) Histopathology of p105−/− mice. Spleen (a and b), lymph node (LN; c and d), lung (e and f), and liver (g and h) from 6-wk-old, and bone marrow (BM; i and j) from 12-wk-old control (+/+; a, c, e, g, and i) and p105−/− (−/−; b, d, f, h, and j) animals stained with hematoxylin and eosin. Marked enlargement of the spleen and lymph node in mutant mice (inset, a and b, increased erythrocytic extramedullary hematopoiesis; the white pulp was also expanded). The paracortical area of lymph nodes are expanded in mutant mice, but overall lymphocyte numbers are decreased (inset, c and d). The liver and lung contain perivascular lymphoid infiltration in mutant mice. Relative hyperplasia in the bone marrow with an associated decrease in erythrocytic precursor cells in mutant mice. (C) Splenic sections from 6-wk-old control (+/+; a and c) and p105−/− (−/−; b and d) mice immunized with SRBCs. Sections were incubated with rat anti–mouse IgD (a and b) and biotinylated PNA (c and d). Mantle zones in mutant mice are less compact (compare a and b). The PNA positive cell area corresponding to centrocytes is markedly expanded in the mutant mice as compared to the control animals (compare c and d). Original magnifications: 2 B, a and b, 62.5; c and d, 125; e and f, 250; g and h, 500; i and j, 1,000; inset, a and b, 500; and inset, c and d, 2,500; 2 C, 125.
Although 3-wk-old p105−/− mice had unremarkable histology, 6-wk-old animals developed an abnormal phenotype, which worsened with age. Histological alterations consistently occurred in the spleen, lymph nodes, lung, liver, and bone marrow. p105−/− mice had a variable degree of splenic and lymph node enlargement at postmortem examination. In young mice, splenomegaly was not only due to erythrocytic extramedullary hematopoiesis, but also to an expansion of the follicular areas with a loss of mantle and marginal zone definition (Fig. 2 B, compare a with b). All lymph nodes examined had variable degrees of paracortical zone expansion without an increase in lymphoid cells (Fig. 2 B, compare c with d). In addition, lungs and liver of p105−/− mice had mild–moderate perivascular, periairway, and periportal mixed infiltration of inflammatory cells, respectively (Fig. 2 B, e–h), which were mainly composed of lymphocytic cells (CD4+ and B220+ cells; data not shown). Myeloid hyperplasia was present in the bone marrow associated with a decrease in erythrocytic cells (Fig. 2 B, compare i and j).
To analyze in more detail the splenic B cell compartment, immunohistochemical evaluation of the spleens from mice immunized with SRBCs was performed. There were prominent peanut agglutinin (PNA)-staining cells in the central area (centrocytes), and these areas were surrounded by a compact zone of IgD+ cells (Fig. 2 C, compare a with b). The B cell area was markedly expanded in immunized mutant mice, but discrete germinal centers were not present. The PNA-staining cell area was enlarged, and the IgD+ cell area was less compact compared to wild-type mice (Fig. 2 C, c –d).
Flow cytometry revealed alterations in the hematopoietic cell population in 6-wk-old p105−/− mice (Fig. 3). The thymus from 6-wk-old mice contained a normal distribution of T cells as compared to wild-type animals (Fig. 3 A, a and b). However, there was a significant decrease of CD4+ and CD8+ T cells in both spleen and lymph node (Fig. 3 A, compare c with d; lymph node, data not shown). The reduction of the peripheral T cell population was more evident when the Thy 1.2 antibody was used (Fig. 3 B). The number of the Thy 1.2+ cells in the mutant mice were reduced by 50% as compared to the wild-type mice. Erythroblasts accumulated in mutant spleen (Fig. 3 A, e and f ) consistent with the histological results showing increased extramedullary hematopoiesis in the spleen (Fig. 2 B). The bone marrow had a decrease in the erythroblasts and only a slight increase in the granulocyte population (Fig. 3 A, compare g with h), also consistent with the histological findings.
Figure 3.
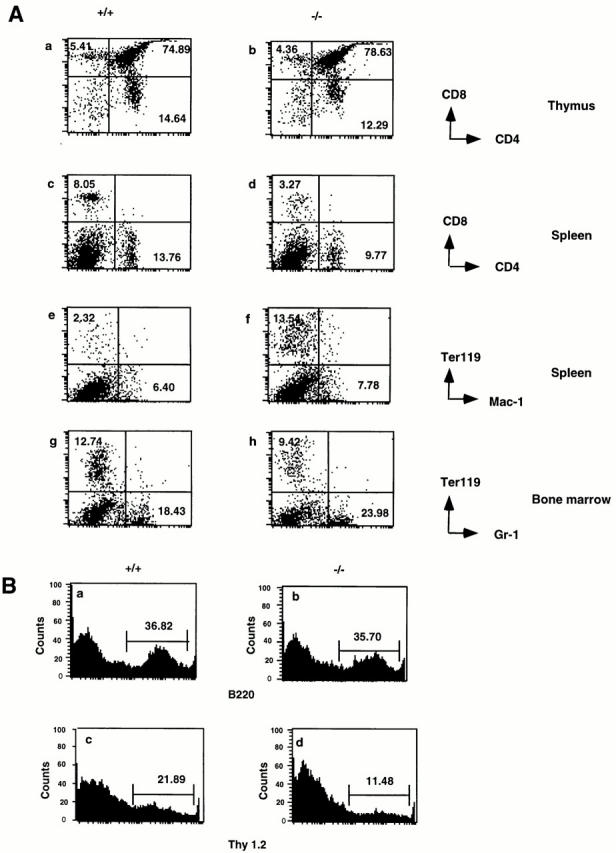
Flow cytometry analysis of hematopoietic cells in p105−/− mice. (A) Thymocytes stained for CD4 and CD8 (a and b), splenocytes stained for CD4 and CD8 (c and d) or Mac-1 and Ter 119 (e and f ), and bone marrow cells stained for Gr-1 and Ter 119 (g and h) from 6-wk-old control (+/+; a, c, e, and g), and p105−/− (−/−; b, d, f, and h) mice. Percentages of positive cells are indicated. (B) Histogram of the relative number of splenocytes stained with B220 (a and b) or Thy 1.2 (c and d) from 6-wk-old control (+/+; a and c), and p105 −/− (−/−; b and d).
Enhanced κB-binding Activities in p105− /− Mice.
The p105 precursor, as well as p100 (NF-κB2) have been shown to act as an inhibitor of NF-κB activities by trapping members of Rel/NF-κB family in the cytoplasm (39–42). Therefore, we investigated the effect of the lack of p105 on nuclear kB-binding activities in several tissues of 3-wk-old animals by EMSA (Fig. 4 A, top). The p105−/− mice had an increase in p50 homodimer κB-binding activity in some tissues, such as spleen, thymus, and liver, compared to wild-type tissues. Oct-1–binding activities were used as a loading control (Fig. 4 A, bottom). By Western blot analysis, constitutive expression of p50 was more abundant in lymphoid tissues (spleen, thymus, lymph node, and bone marrow) and in higher concentrations in extract from mutant mice (data not shown). To analyze the composition of the different migrating bands present in spleen extracts, EMSA was performed in the presence of antibodies directed against the different Rel/NF-κB proteins. Anti-p50 antiserum was able to remove the faster migrating complexes (band I), suggesting that this band is composed of p50 homodimers. The slower complexes (band II) show two different bands; the lower one was eliminated with anti-p50 and anti-RelA antiserum suggesting that it corresponds to p50/RelA heterodimers. The fact that the upper band reacted with anti-p50 antiserum but not anti-RelA, -RelB, and -p52 antisera, lead us to postulate that it represents p50/cRel complexes (Fig. 4 B).
Figure 4.
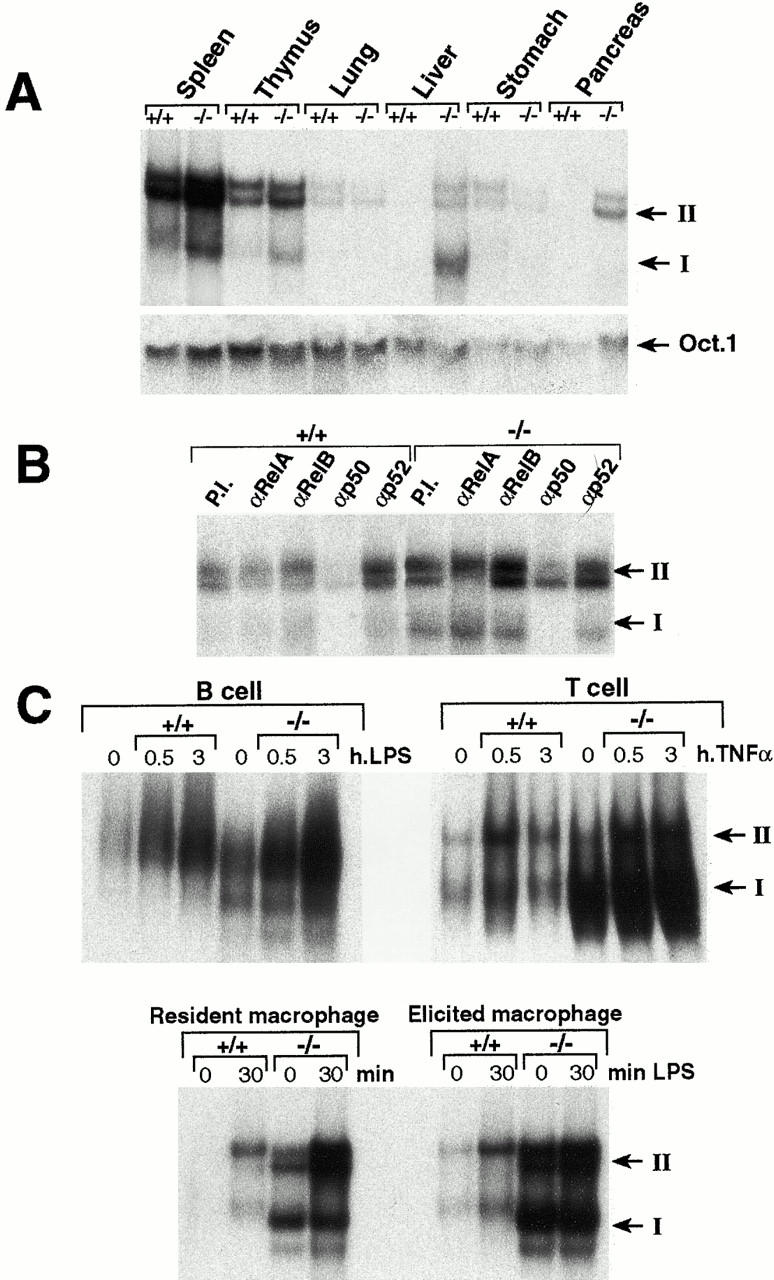
Accumulation of the p50 homodimer activity in p105−/− mice. Increased κB-binding activity in several tissues of p105−/− mice. (A) Nuclear extracts (2 μg) from spleen, thymus, lung, liver, stomach, and pancreas were used for EMSA. A κB palindromic sequence was used to detect the κB-binding activities. The octamer-specific motif was used as a control. (B) Nuclear extracts from spleen were incubated with a palindromic κB-binding site and treated with specific antisera against the different members of the Rel/NF-κB family before EMSA. P.I., preimmune serum. (C) Induction of NF-κB activity after stimulation of different cells. Nuclear extracts from untreated control and p105−/− B cells, T cells, and macrophages or treated with LPS (B cells and macrophages) or TNF-α (T cells) for the indicated period were analyzed by EMSA using a κB probe. NF-κB heterodimers and p50 homodimers are indicated as band II and I, respectively.
We also analyzed the nuclear κB-binding activities in purified B cells, T cells, and macrophages. As shown in Fig. 4 C, although the absence of p105 augmented the constitutive κB-binding in all three cell types, the p50 homodimers were markedly increased in p105−/− T cells and macrophages relative to B cells. Compared to wild type, the induced NF-κB activity (band II) was also augmented in p105−/− cells after stimulation, probably because of the degradation of IκBα. This is expected because the increase of p50 may lead not only to an augment in p50 homodimers, but also of p50 heterodimers, which can associate with IκBα.
Expression of Genes Regulated by Rel/NF-κB.
The mRNA levels of genes assumed to be regulated by Rel/NF-κB were examined to determine whether their expression was effected by the increased p50 homodimer activity present in p105−/− mice. Semiquantitative RT-PCR analysis using total RNA isolated from the thymuses of 3-wk-old animals showed that G-CSF, GM-CSF, M-CSF, IL-2, and TNF-β expression was upregulated in p105−/− mice (Fig. 5 A), whereas the mRNA levels of ICAM-1, ikba, IFN-β, IL-6, class I MHC, macrophage inflammatory protein (MIP)-2, c-myc, nfkb2, TNF-α, and VCAM-1 (Fig. 5 A and data not shown) were similar between control and p105−/− mice. Interestingly, none of the genes analyzed was downregulated in thymus of p105−/− mice, but some were upregulated, suggesting that p50 homodimers could activate gene transcription in certain cell types. However, we cannot eliminate the possibility that the increased gene expression observed in some of these cytokines is due to the small augment in heterodimeric complexes.
Figure 5.
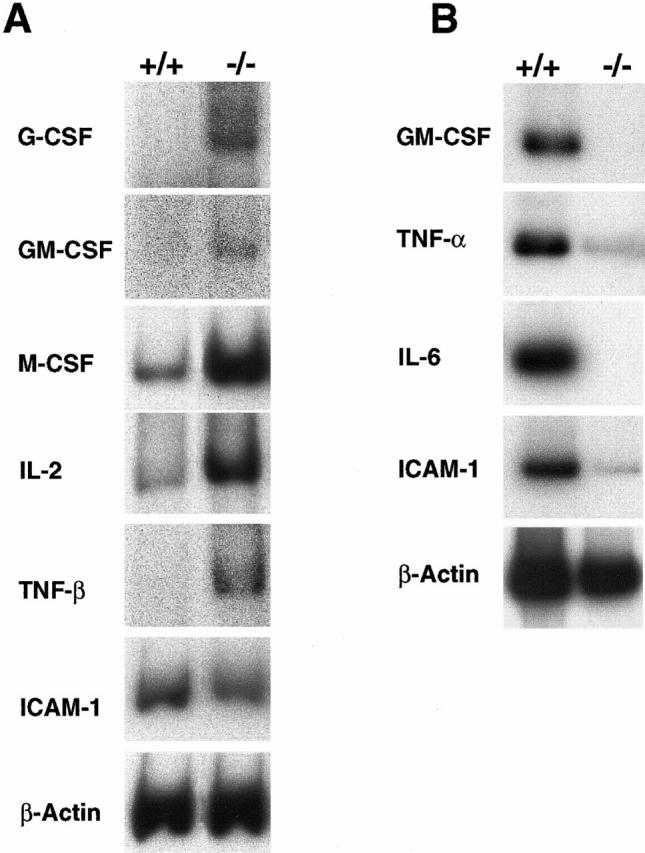
Dual role of p50 homodimers in gene expression in p105−/− mice. (A) Total RNA (0.5 μg) isolated from thymus of 3-wk-old animals or (B) total RNA (2 μg) isolated from resident macrophages of 4-wk-old animals was used for RT-PCR analysis using specific primers as indicated.
In contrast, mRNA levels of some cytokines in resident peritoneal macrophages revealed significant differences between wild-type and p105−/− cells as shown in Fig. 5 B. The levels of GM-CSF, TNF-α, IL-6, and ICAM-1 in resident macrophages were decreased compared with control. Interestingly, there also was a difference in the mRNA levels of cytokines in elicited macrophages between mutant and wild-type mice. Expression levels of genes such as IL-6, ICAM-1, and VCAM were downregulated, whereas expression levels of TNF-α and G-CSF were similar to wild-type cells (data not shown). These findings suggest that p50 homodimers can activate or inhibit gene transcription of some cytokines in a cell type–dependent manner.
Proliferative Responses of p105− /− Lymphocytes.
Proliferative responses of B or T cells purified from 3-wk-old mouse spleens were analyzed by [3H]thymidine incorporation. Both anti-IgM antibody and LPS promoted proliferation of mutant and wild-type B cells. However, at low concentrations of the stimuli, there was an increased proliferative responses of p105−/− B cells (four to fivefold; Fig. 6 A). In contrast, p105−/− T cells stimulated with anti-CD3 or anti-CD3 plus anti-CD28 did not proliferate as efficiently as control T cells (two to threefold reduction), but p105−/− T cells stimulated with PMA plus PHA exhibited similar proliferation rates to controls (Fig. 5 B). The defects of proliferative responses observed in p105−/− T cells were not found in p105+/− T cells (data not shown). These results indicate that loss of the p105 precursor has a different effect on B and T cells.
Figure 6.

Proliferative responses of lymphocytes. (A) B cell proliferation. [3H]Thymidine incorporation of splenic B cells from 3-wk-old control (closed circles) or p105−/− (open circles) mice treated for 48 h with the indicated concentrations of either anti-IgM or LPS. Values of [3H]thymidine incorporation are shown by mean ± SD. (B) T cell proliferation. [3H]Thymidine incorporation of splenic T cells from 3-wk-old control (closed bars) or p105−/− (open bars) mice treated for 48 h with either PMA plus PHA, anti-CD3, or anti-CD3 plus anti-CD28.
Cytokine Production in p105− /− T Cells and Macrophages.
Cytokine production in T cells isolated from spleen and in macrophages by peritoneal lavage of 3-wk-old mice was analyzed by ELISA. The levels of IFN-γ, IL-10, and GM-CSF released from T cells stimulated with anti-CD3 or anti-CD3 plus anti-CD28 antibodies were comparable between wild-type and p105−/− T cells, whereas the levels of IL-2, IL-4, and TNF-α produced in mutant T cells were decreased (three to sixfold) compared to control cells (Fig. 7 A).
Figure 7.
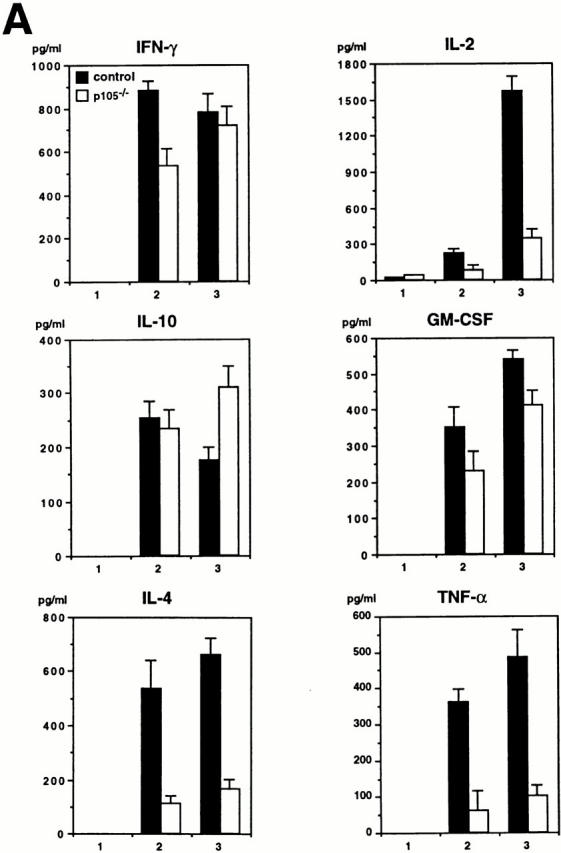
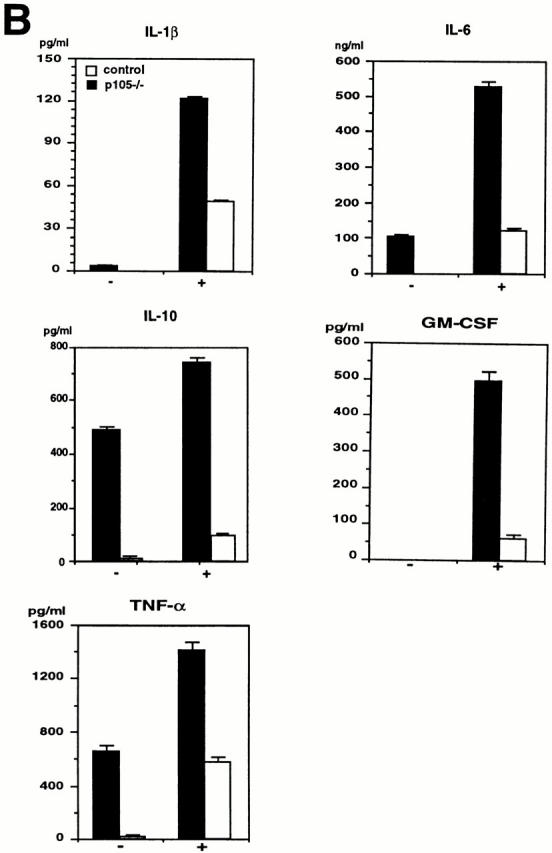
Cytokine production in T cells or macrophages. (A) Secretion of IL-2, IL-4, and TNF-α is decreased in p105−/− T cells. Splenic T cells purified from 3-wk-old control (closed bars) or p105−/− (open bars) untreated (1) or treated with anti-CD3 (2) or anti-CD3 plus anti-CD28 (3) for 24 h. The cytokine levels in the supernatants were determined by ELISA. (B) Cytokines released from macrophages. Peritoneal macrophages purified from 3-wk-old mice untreated (−) or treated with (+) LPS and IFN-γ for 72 h. The cytokine levels are indicated by mean values ± SD.
Both unstimulated and stimulated p105−/− resident peritoneal macrophages showed lower cytokine production compared to wild-type cells (17–86-fold reduction in unstimulated; 2–8-fold reduction in stimulated mutant cells relative to control). Some of the cytokine levels were undetectable in unstimulated mutant macrophages (Fig. 7 B). Cytokine production in elicited peritoneal macrophages from mutant mice was also impaired (data not shown).
Basal and Specific Antibody Production.
To assess basal production of antibodies in naive mice, we measured resting serum immunoglobulin levels in p105−/− mice and control littermates. Levels of total serum Ig were increased fivefold in p105−/− mice (Fig. 8 A). These levels resulted from a significant increase in most Ig isotypes, except for IgG3. The levels of IgE and IgG1 were especially increased in p105−/− sera (50- and 18-fold relative to wild type, respectively), whereas those of IgG3 were significantly reduced (4-fold). These results indicate that heavy chain class switching of p105−/− B cells is not impaired, confirming the findings from nfkb1 −/− mice showing that p50 is required for normal Ig isotype levels and class switching (16, 43).
Figure 8.

Basal and specific Ig production in vivo. (A) Ig isotype concentrations. The concentration of serum Ig in unimmunized control (closed circles), heterozygotes (closed triangles), and p105−/− (open circles) mice was determined by isotype-specific ELISA. (B) T cell–dependent antibody production. Mice were immunized with NP-KLH and the levels of NP-specific IgG1 were determined 1, 2, and 3 wk after immunization. (C) T cell–independent antibody production. Mice were immunized with NP-LPS and the levels of NP-specific IgG3 were determined 1, 2, and 3 wk after immunization. Each symbol represents the value obtained from one animal. Horizontal bars, mean values.
The tendency of increased basal immunoglobulin levels prompted us to examine whether humoral immune responses were affected in p105−/− animals. Mice were immunized with T cell–dependent antigen NP-KLH or T cell–independent antigen NP-LPS, and production of specific antibodies was measured at 1, 2, and 3 wk after immunization. The production of IgG1 in response to NP-KLH was increased in p105−/− mice two to threefold compared to wild-type animals at 2 and 3 wk after injection (Fig. 8 B) showing that the lack of the p105 precursor does not negatively affect the T cell–dependent immune response. When we analyzed the production of IgG3 in response to NP-LPS, the levels of specific immunoglobulin obtained were similar to those of the wild-type mice at 1 or 2 wk after immunization; however, at 3 wk, the levels of specific IgG3 were reduced threefold (Fig. 8 C). These results suggest that the lack of p105 and IκBγ does not seriously affect the production of basal and specific antibody production.
Discussion
In this study we showed that targeted disruption of the COOH-terminal ankyrin domain of NF-κB1, which eliminates both p105 and IκBγ but not p50, induced multiple abnormalities, such as inflammation in lungs and liver, myeloid hyperplasia in bone marrow, enlarged lymph nodes, and splenomegaly, presumably due to the accumulated p50 homodimer κB-binding activity. Moreover, p105−/− mice were apparently susceptible to bacterial infections, which may result from the impaired cytokine production in macrophages. In addition, p105−/− mice had different abnormalities from mice lacking the COOH terminus of NF-κB2 (p100−/−; reference 29), demonstrating the distinct physiological roles between NF-κB1 and NF-κB2.
Mice that lacked the p105 precursor and IκBγ but mainly expressed a nuclear localization signal–deficient p50 isoform instead of an expected p50 of 415 amino acids (28), did not have any of the abnormalities described here for the p105−/− mice, and showed almost 100% survival by 1 yr of age (Ishikawa, H., unpublished observations). These findings indicate that the absence of p105 by itself is not sufficient to trigger the phenotype observed in p105−/− mice and that the presence of active p50 molecules is necessary. This is further supported by the fact that p50-deficient mice (16) that lack both the p105 precursor and the p50 molecules do not present the alterations observed in p105−/− mice, strongly suggesting that the increased p50 homodimer activity contributes to the abnormalities in p105−/− mice.
Inflammatory Phenotype and Hematopoietic Abnormalities of p105− /− Mice.
Inflammation consisting of perivascular lymphocytic infiltrates was developed in lungs and liver of p105−/− mice at 6 wk of age. The perivascular region in the lung and the periportal area in the liver are the topographical sites for initial antigen exposure; therefore, the inflammatory infiltrates observed in p105−/− mice in this study may represent an exaggerated response to innocuous antigens and a defect in the natural immune response. A similar mechanism has been proposed to explain the inflammatory response in IL-2, GM-CSF, and RelB knockout mice (17, 44, 45).
Enlargement of multiple lymph nodes and splenomegaly was consistently observed in p105−/− mice from 6 wk of age onwards. In both of these secondary lymphoid organs, B and T cell compartments had alterations; however, mutant animals were still able to produce germinal centers. In fact, the PNA-positive regions in p105−/− mice were enlarged due to marked hyperplasia of centrocytes. The increase in the proliferation rate of some B cell populations in p105−/− mice may be due to the allowance of greater numbers of p50 to associate with cofactors such as Bcl-3. The results obtained in previous studies examining the association of p50 and Bcl-3 also suggest that Bcl-3 is a cofactor for p50 (12, 46, 47). On the other hand, the analyses of Bcl-3 knockout mice suggest a functional interaction between Bcl-3 and p50 in vivo (48, 49). Both p50−/− and Bcl-3−/− mice develop susceptibility to infections and they are not able to produce specific antibodies (16, 48, 49); however, only Bcl3−/− mice show a deficient germinal center formation.
The bone marrow changes observed in p105−/− mice after 12 wk of age were interpreted histologically as a myeloid hyperplasia due to the predominance of myeloid precursors relative to megakaryocytes and erythroid precursors. However, by flow cytometry analysis, the percentage of myeloid cells does not show a significant increase compared to control mice, whereas the erythroid cell number was decreased. Taken together, the histological and flow cytometry findings are consistent with a primary erythroid hypoplasia. The increased erythrocytic extramedullary hematopoiesis in spleen may have been caused, in part, by the decrease of erythroid cells in bone marrow, although the cause of this decrease is unknown. Indeed, peripheral blood cell counts were consistently normal in mutant mice at all ages examined, presumably due to the extramedullary hematopoiesis.
It is also interesting to note that some of the abnormalities in p105−/− mice resemble those observed in RelB-deficient mice, such as inflammation in lungs and liver, myeloid hyperplasia in bone marrow, and splenomegaly (17). The augmented p50 homodimers would be able to have a displacement effect on the RelB-containing NF-κB transcription complexes that are shown to contribute to the constitutive κB-binding activities (12).
Immune Functions in p105− /− Mice.
The susceptibility to opportunistic infections in the mucocutaneous areas in mutant mice at 6 wk of age and older suggests defects in normal killing and clearing of bacteria by neutrophils and macrophages. As both cell types are known to be primarily responsible for protection from most bacterial infections by nonspecific phagocytosis, their altered functions contribute to inefficient immune responses. The impaired recruitment of macrophages and PMNs after peritonitis induction in mutant mice (data not shown) suggests also a defect in the function of these cells. Moreover, the ability of p105−/− B cells to produce antibodies, which is also important for killing bacteria, was normal. T cells had alterations in the production of some cytokines such as IL-2, IL-4, and TNF-α. As cytokines have a pluripotent effect on the immune network, the impaired production in T cells may also contribute to the immune defects observed in p105−/− mice.
Interestingly, the proliferative rate of B cells from mutant mice in response to several stimuli was increased, but that of T cells was decreased. The differences between different kinds of cells may imply a dual role for p50 in vivo. This was also found in Bcl-3−/− mice (48, 49).
The analysis of basal and specific Ig production showed Ig heavy chain class switching was not impaired in the absence of p105. Interestingly, in contrast to p50−/− mice, which have markedly reduced levels of some Ig, all of those isotypes were markedly increased in p105−/− mice, implying that the p50 homodimers play a role in enhancing Ig heavy chain class switching. Alternatively, the increase of total number of B cells in peripheral organs may result in a relative accumulation of Ig levels in p105−/−. In addition, T cell–dependent and –independent immune responses in p105−/− mice are not seriously impaired; on the contrary, the specific Ig production in p50−/− mice is completely impaired (16).
The Roles of the p105 Precursor.
The p105 precursor, like IκB, is postulated to regulate Rel/NF-κB activities by dimerizing with subunits of Rel/NF-κB and retaining them in the cytoplasm, and also its proteolytic processing controls the availability of p50 molecules (2, 4, 7, 8). Elimination of the precursor simultaneously results in accumulation of p50 that may contribute to forming both homodimers and heterodimers. In the absence of p105, the p50 homodimers appeared fully active, whereas the p50-containing heterodimers seemed to be inactivated by IκB molecules. Indeed, the induced NF-κB activities were increased relative to control after stimulation of the cells (see Fig. 4 C). Moreover, subcellular localization of RelA was not altered in p105−/− thymocytes and splenocytes (data not shown), indicating that in the case of RelA-containing NF-κB complexes, such as p50-RelA, loss of p105/IκBγ could be completely compensated by the other IκB proteins. This is in agreement with our previous observation that overexpression of RelA in thymocytes does not significantly increase κB-binding activity due to a concomitant increase in IκBα levels (50). It is also important to note that mice lacking IκBα exhibit skin defects, granulocytosis, and activation of NF-κB activity composed of p50–RelA heterodimers, resulting in neonatal lethality (51, 52). This suggests that IκBα is mainly involved in the regulation of the p50–RelA heterodimer activity, and that the constitutively activated p50–RelA complex found in IκBα-deficient mice, presumably causes the abnormalities. As p105 forms dimers with its product p50 and retains it in the cytoplasm, the precursor is important for controlling, in particular, the p50 homodimer activity that is unlikely to be regulated by IκBα or IκBβ. The p105 precursor shares structural characteristics with another member of Rel/NF-κB family, the p100 precursor. Both proteins contain in the COOH terminus an ankyrin-like domain mediating κB inhibitor function and by proteolytic cleavage generate the functional p50 and p52 subunits, respectively. However, the distinct expression pattern of nfkb1 and nfkb2 transcripts in the embryo and adult mouse may indicate different roles for these molecules (12). Moreover, the findings obtained from the mutant mice lacking either p100 (28) or p105, described here, reinforce the functional differences between these proteins. Mutant mice lacking the p100 precursor developed a marked gastric hyperplasia and an increase in the κB-binding activities composed of mainly heterodimers containing p52, whereas mice lacking p105 had an inflammatory phenotype and an increase in κB-binding activities consisting mainly of the p50 homodimers.
The Biological Activity of the p50 Homodimers.
The answer to the question of whether the p50 homodimers can be transcriptional activators or repressors still remains controversial. Most previous findings demonstrate that they have an antagonistic effect on the classical NF-κB transcription factor activity through the κB motif in cells (2, 8), whereas others demonstrated that they can activate transcription in cell-free systems (53, 54). The p50 homodimer, in association with Bcl-3, which is structurally related to IκBs, has been shown to have the ability to activate transcription in cells (46, 47). More recently, nfkb1 −/− mice were shown to have reduced IL-6 but augmented IFN-β gene expression (16). However, since nfkb1 −/− mice contain neither the p50 homodimers nor the p50-containing heterodimers, most typically p50–RelA, those animals do not provide a clear answer to this question.
In this study, we show that the p50 homodimers may function as transcriptional activators or repressors depending on the cell types. In vitro analysis, such as proliferative responses and cytokine production, revealed that the absence of p105 had a dual effect on different cell types. The induced NF-κB, most likely p50–RelA heterodimers, may have a much stronger transcriptional activity than the p50 homodimers based on numerous previous in vitro studies. In p105−/− B cells, the induced NF-κB may overcome the small amount of p50 homodimer activity, whereas in p105−/− T cells and macrophages, the high levels of p50 homodimers may still efficiently compete with the induced NF-κB activity. According to this hypothesis, in stimulated cells the p50 homodimers may act like antagonists because of the weak transcriptional activity relative to the heterodimeric NF-κB. Alternatively, there may be cell-specific cofactors that interact with the p50 homodimers and can activate gene expression. This may also explain a dual effect of the p50 homodimer activity on different cell lineages. As described above, it is reasonable to speculate that Bcl-3 is a possible candidate as a cofactor, although expression of the Bcl-3 protein in different cell types has not been extensively examined. Mice lacking both p105 and Bcl-3 may be helpful to gain an insight into this possibility.
Acknowledgments
We would like to thank S. Lira, M. Swerdel, and A. Lee for generating mutant mice; C. Rizzo for cell culture; A. Lewin for tissue sectioning; K. Class for flow cytometry; N. Thomson and T. Nelson for DNA sequencing; W. Kratil for computer grafics assistance; and the staff of Veterinary Sciences at Bristol-Myers Squibb (Princeton, NJ) for excellent support. We thank R. Attar, J. Caamaño, D. Carrasco, J. Cheng, and R.-P. Ryseck for their valuable comments.
Footnotes
Abbreviations used in this paper: EMSA, electrophoretic mobility shift assay; ES, embryonic stem; ICAM, intercellular adhesion molecule; mRNA, messenger RNA; NF, nuclear factor; p, polypeptide; p(A), polyadenylation recognition sequence; PNA, peanut agglutinin; pPNT, plasmid PGK promoter neomycin thymidine kinase; RT-PCR, reverse transcriptase PCR; VCAM, vascular adhesion molecule.
References
- 1.Grilli M, Chiu J-S, Lenardo MJ. NF-κB and Rel-participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 3.Kopp EB, Ghosh S. NF-κB and Rel proteins in innate immunity. Adv Immunol. 1995;58:1–27. doi: 10.1016/s0065-2776(08)60618-5. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto S, Verma IM. Rel/NF–κB/IκB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 5.Beg AA, Baldwin AS., Jr The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev. 1993;7:2063–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 6.Gilmore TD, Morin PJ. The IκB proteins: members of a multifunctional family. Trends Genet. 1993;9:427–433. doi: 10.1016/0168-9525(93)90106-r. [DOI] [PubMed] [Google Scholar]
- 7.Liou H-C, Baltimore D. Regulation of the NF-κB/rel transcription factor and IκB inhibitor system. Curr Opin Cell Biol. 1993;5:477–487. doi: 10.1016/0955-0674(93)90014-h. [DOI] [PubMed] [Google Scholar]
- 8.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 9.Finco TS, Baldwin AS. Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity. 1995;3:263–272. doi: 10.1016/1074-7613(95)90112-4. [DOI] [PubMed] [Google Scholar]
- 10.Carrasco D, Ryseck R-P, Bravo R. Expression of relBtranscripts during lymphoid organ development: specific expression in dendritic antigen-presenting cells. Development. 1993;118:1221–1231. doi: 10.1242/dev.118.4.1221. [DOI] [PubMed] [Google Scholar]
- 11.Carrasco D, Weih F, Bravo R. Developmental expression of the c-relprotooncogene in hematopoietic organs. Development. 1994;120:2991–3004. doi: 10.1242/dev.120.10.2991. [DOI] [PubMed] [Google Scholar]
- 12.Weih F, Carrasco D, Bravo R. Constitutive and inducible Rel/NF-κB activities in mouse thymus and spleen. Oncogene. 1994;9:3289–3297. [PubMed] [Google Scholar]
- 13.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA components of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 14.Burkly L, Hesslon C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relBis required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- 15.Kontgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-relproto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 16.Sha WC, Liou H-C, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 17.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hemopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 18.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 19.Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 20.Whiteside ST, Epinat J-C, Rice NR, Israël A. I kappa B epsilon, a novel member of the IκB family, controls RelA and cRel NF-κB activity. EMBO (Eur Mol Biol Organ) J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imbert V, Rupec RA, Livolsi A, Phal HL, Traenckner EB-M, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron J-F. Tyrosine phosphorylation of IκB-α activates NF-κB without proteolytic degradation of IκB-α. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 22.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin–proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 23.Chen ZJ, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T. Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin– proteasome pathway. Genes Dev. 1995;9:1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 24.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IκBα by novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 25.Didonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 26.Inoue J-I, Kerr LD, Kakizuka A, Verma IM. IκBγ, a 70 kd protein identical to the C-terminal half of p110 NF-κB: a new member of the IκB family. Cell. 1992;68:1109–1120. doi: 10.1016/0092-8674(92)90082-n. [DOI] [PubMed] [Google Scholar]
- 27.Liou H-C, Nolan GP, Ghosh S, Fujita T, Baltimore D. The NF-κB p50 precursor, p105, contains an internal IκB-like inhibitor that preferentially inhibits p50. EMBO (Eur Mol Biol Organ) J. 1992;11:3003–3009. doi: 10.1002/j.1460-2075.1992.tb05370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa H, Ryseck R-P, Bravo R. Characterization of ES cells deficient for the p105 precursor (NF-κB1): role of p50 NLS. Oncogene. 1996;13:255–263. [PubMed] [Google Scholar]
- 29.Ishikawa H, Claudio E, Carrasco D, Ryseck R-P, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lernbecher T, Muller U, Wirth T. Distinct NF-κB/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365:767–770. doi: 10.1038/365767a0. [DOI] [PubMed] [Google Scholar]
- 31.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Schevach, and W. Strober. 1992. Isolation and fractionation of mononuclear cell populations. Current Protocols in Immunology. Greene Publishing Associates & Wiley-Interscience, New York.
- 32.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Claudio E, Segade F, Wrobel K, Ramos S, Bravo R, Lazo PS. Molecular mechanisms of TNFα cytotoxicity: activation of NF-κB and nuclear translocation. Exp Cell Res. 1996;224:63–71. doi: 10.1006/excr.1996.0111. [DOI] [PubMed] [Google Scholar]
- 34.Dobrzanski P, Ryseck R-P, Bravo R. Differential interactions of Rel/NF-κB complexes with IκBα determine pools of constitutive and inducible NF-κB activity, which differ in their composition. EMBO (Eur Mol Biol Organ) J. 1994;13:4608–4616. doi: 10.1002/j.1460-2075.1994.tb06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carrasco D, Rizzo CA, Dorfman K, Bravo R. The v-rel oncogene promotes malignant T-cell leukemia/ lymphoma in trangenic mice. EMBO (Eur Mol Biol Organ) J. 1996;15:3640–3650. [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto M, Mariathasan S, Nahm MH, Baranyay F, Peschon JJ, Chaplin DD. Role of lymphotoxin and the type I TNF receptor in the formation of germinal centers. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 37.Chase MW. Production of antiserum. Methods Immunol Immunochem. 1967;1:197–209. [Google Scholar]
- 38.Lalor PA, Nelson GJV, Sanderson RD, McHeyzer-Williams MG. Functional and molecular characterization of single, (4-hydroxy-3-nitrophenyl) acetyl (NP)- specific, IgG+ B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur J Immunol. 1992;22:3001–3011. doi: 10.1002/eji.1830221136. [DOI] [PubMed] [Google Scholar]
- 39.Rice NR, MacKichan ML, Israel A. The precursor of NF-κB p50 has IκB-like functions. Cell. 1992;71:243–253. doi: 10.1016/0092-8674(92)90353-e. [DOI] [PubMed] [Google Scholar]
- 40.Mercurio F, DiDonato JA, Rosette C, Karin M. p105 and p98 precursor proteins play an active role in NF-κB–mediated signal transduction. Genes Dev. 1993;7:705–718. doi: 10.1101/gad.7.4.705. [DOI] [PubMed] [Google Scholar]
- 41.Naumann M, Wulczyn FG, Scheidereit C. The NF-κB precursor p105 and the proto-oncogene product Bcl-3 are IκB molecules and control nuclear translocation of NF-κB. EMBO (Eur Mol Biol Organ) J. 1993;12:213–222. doi: 10.1002/j.1460-2075.1993.tb05647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun S-C, Ganchi PA, Beraud C, Ballard DW, Greene WC. Autoregulation of the NF-κB transactivator RelA (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc Natl Acad Sci USA. 1994;91:1346–1350. doi: 10.1073/pnas.91.4.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B Cells from p50/ NF-κB knockout mice have selective defects in proliferation, differentiation, germ-line CHtranscription, and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 44.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 45.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC. Involvement of granulocyte–macrophage colony-stimulating factor in pulmonary homeostasis. Science. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 46.Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- 47.Fujita T, Nolan GP, Liou H-C, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3encodes a transcriptional coactivator that activates through NF-κB p50 homodimers. Genes Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- 48.Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- 49.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, et al. Critical roles for the Bcl-3 oncoprotein in T cell–mediated immunity, splenic microarchitecture and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 50.Perez P, Lira SA, Bravo R. Overexpression of RelA in transgenic mouse thymocytes: specific increase in levels of the inhibitor protein IκBα. Mol Cell Biol. 1995;15:3523–3530. doi: 10.1128/mcb.15.7.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klement JF, Rice NR, Car BD, Abondanzo SJ, Powers GD, Bhatt H, Chen C-H, Rosen CA, Stewart CL. IκBα deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 53.Fujita T, Nolan GP, Ghosh S, Baltimore D. Independent modes of transcriptional activation by the p50 and p65 subunits of NF-κB. Genes Dev. 1992;6:775–787. doi: 10.1101/gad.6.5.775. [DOI] [PubMed] [Google Scholar]
- 54.Kretzschmar M, Meisterernst M, Scheidereit C, Li G, Roeder RG. Transcriptional regulation of the HIV-1 promoter by NF-κB in vitro. Genes Dev. 1992;6:761–774. doi: 10.1101/gad.6.5.761. [DOI] [PubMed] [Google Scholar]