

Abstract
Protein C is an important regulatory mechanism of blood coagulation. Protein C functions as an anticoagulant when converted to the active serine protease form on the endothelial cell surface. Thrombomodulin (TM), an endothelial cell surface receptor specific for thrombin, has been identified as an essential component for protein C activation. Although protein C can be activated directly by the thrombin–TM complex, the conversion is known as a relatively low-affinity reaction. Therefore, protein C activation has been believed to occur only in microcirculation. On the other hand, we have identified and cloned a novel endothelial cell surface receptor (EPCR) that is capable of high-affinity binding of protein C and activated protein C. In this study, we demonstrate the constitutive, endothelial cell–specific expression of EPCR in vivo. Abundant expression was particularly detected in the aorta and large arteries. In vitro cultured, arterial endothelial cells were also found to express abundant EPCR and were capable of promoting significant levels of protein C activation. EPCR was found to greatly accelerate protein C activation by examining functional activity in transfected cell lines expressing EPCR and/or TM. EPCR decreased the dissociation constant and increased the maximum velocity for protein C activation mediated by the thrombin–TM complex. By these mechanisms, EPCR appears to enable significant levels of protein C activation in large vessels. These results suggest that the protein C anticoagulation pathway is important for the regulation of blood coagulation not only in microvessels but also in large vessels.
Protein C pathway is an indispensable regulatory mechanism of blood coagulation, since deficiencies in this pathway lead to thrombosis (1, 2). Protein C is a γ-carboxyglutamic acid containing protein (3) and circulates as a zymogen form of serine protease (4). Protein C functions as an anticoagulant when converted to its active form by thrombin (5), and activation of protein C was demonstrated to be greatly enhanced on the endothelial cell surface (6). Thrombomodulin (TM)1 has been identified as an essential component on the endothelial cell surface (7, 8), and direct activation of protein C by the thrombin–TM complex has been demonstrated. However, the activation by the complex was observed as a relatively low-affinity reaction and the K d value was calculated as 0.7–1.0 μM (9– 11), which is 15 times higher than that of the protein C concentration (65 nM) in the circulation (12). Under these conditions, protein C activation appears unlikely to occur under normal physiological conditions. One of the proposed explanations for this has been that protein C activation mediated by the thrombin–TM complex occurs only in the microcirculation because of the greater ratio of endothelial cell surface area to blood volume (13).
On the other hand, we (14) and others (15) found that protein C and activated protein C (APC) bound to cultured endothelial cells with a relatively high affinity (K d = 30 nM). By expression cloning, we identified a novel endothelial cell surface receptor that is capable of protein C–APC binding in the presence of physiological concentrations of calcium (14) and magnesium (16). We designated this molecule as an endothelial cell protein C–APC receptor (EPCR). EPCR is a type 1 transmembrane glycoprotein containing two domains in the extracellular region that are homologous to the α1 and α2 domains of CD1/MHC class 1 molecules (17). The highly conserved structural features and ligand binding function of EPCR that are found between species suggests an important physiological function for the receptor molecule (18). In in vitro cultured human cells, significant levels of EPCR expression has been demonstrated only in human umbilical vein endothelial cells (14). In this study, we demonstrate in vivo expression of EPCR by immunohistochemical analysis. The abundant expression of EPCR was found on the endothelial cells of the aorta where protein C activation has been considered unlikely to occur. In addition, we found that protein C activation mediated by the thrombin–TM complex was promoted as a relatively high-affinity reaction in the presence of EPCR.
Materials and Methods
Cells.
Human primary venous endothelial cells (VECs) and arterial endothelial cells (AECs) were obtained from Cell Systems (Kirkland, WA) and maintained according to the manufacturer's protocol. Human kidney 293 cells (CRL 1573; American Type Culture Collection, Rockville, MD) were maintained in DMEM containing 10% fetal bovine serum. Stable transfected cell lines of 293 cells expressing human EPCR and/or TM were established as follows. Cells were transfected with a human EPCR cDNA construct in a mammalian expression vector, pEF-BOS (19), and/or a human TM cDNA (a gift from J.F. Parkinson, Lilly Research Lab., Indianapolis, IN) constructed in a mammalian expression vector pBK-EF (a gift from T. Fujimoto, Hiroshima University, Hiroshima, Japan) by the calcium/phosphate method as described (14). Cell lines were established by G-418 selection, followed by subcloning. T2 cells are positive for TM. ET1 cells and ET2 cells are positive for both EPCR and TM. Establishment of these cell lines was done in the Oklahoma Medical Research Foundation (Oklahoma City, OK) under the support of C.T. Esmon (Oklahoma Medical Research Foundation). Negative control N1 cells and EPCR positive E7 cells were established as described (16).
Histology.
Sections (4 μm) of human aorta and lung were fixed in cold acetone, paraffin embedded processed, and then stained with JRK-1, an anti-EPCR mouse monoclonal antibody, or CTM1009, an anti-TM mouse monoclonal antibody (a gift from C. Carson, Oklahoma Medical Research Foundation, Oklahoma City, OK). Antigen expression was then detected using biotinylated anti– mouse Ig, streptavidin-conjugated horseradish peroxidase, and 3,3′-diaminobenzidine as previously described (20).
Flow Cytometry.
Cells were harvested and stained with JRK-1 or CTM1009 as described (14). The antigen expression was detected on a FACScan® flow cytometer (Becton Dickinson, Sunnyvale, CA) by using FITC-conjugated goat anti–mouse IgG (Southern Biotechnology Associates, Inc., Birmingham, AL). Living cells were gated by staining with propidium iodide. Data analysis was performed by using the Win MDI program (J. Trotter, Scripps Research Institute, La Jolla, CA).
Protein C Activation Assay.
Monolayers of AECs or VECs were formed by overnight culture in 48-well microplates coated with gelatin. After washing with PBS containing 0.5 mM EDTA, the cells were incubated with various concentrations of human protein C in 100 μl of HBSS containing 0.1% BSA in the absence or presence of human thrombin (0.1 U final concentration). The plates were incubated at 37°C for 30 min. To stop the reaction, antithrombin III, at a final concentration of 1 μg/ml, and EDTA, at a 5 mM final concentration, were added and the plates were incubated for a further 5 min. Aliquots were transferred to 96-well microplates and a chromogenic substrate, S-2366 (AB Kabi Diagnostica, Stockholm, Sweden), was added at a final concentration of 200 μM. The catalytic rate of the substrate was measured by absorbance at 405 nm by using a Vmax kinetic microplate reader (Molecular Devices Co., Sunnyvale, CA). Generated APC was estimated by using a standard curve of purified APC. The same experiments were also carried out by using monolayers of transfected human 293 cells (see above) formed in microplates coated with poly-L-lysine (Sigma Chemical Co., St. Louis, MO). The K d and Vmax values were calculated using the Hyper program (J.S. Easterby, University of Liverpool, Liverpool, UK).
Results and Discussion
In a previous study, we demonstrated that the message expression of EPCR was demonstrable only with HUVECs among in vitro cultured human cells (14). The messenger RNA was also detectable in normal tissues by Northern blot analysis (data not shown). Abundant expression was detected in heart and placenta, and the message was also present in lung, kidney, and pancreas, although brain was negative for the message. To investigate the detailed expression pattern of EPCR, we performed immunohistochemical analysis of human tissues using an anti-EPCR monoclonal antibody and anti-EPCR goat Ig (16). Constitutive, endothelial cell–specific expression of EPCR was demonstrated with these antibodies. The most abundant expression of EPCR was detected on the endothelial cells of the aorta (Fig. 1 A). Moderate expression was also detected on the endothelial cells of relatively large-sized vessels (data not shown). On the other hand, little or no expression could be detected in microvessels such as the lung capillaries (Fig. 1 C).
Figure 1.

Immunoperoxidase staining of EPCR. Sections of human aorta (A) and lung (C) were stained with an anti-EPCR monoclonal antibody, JRK-1, by using biotinylated anti–mouse goat Ig and avidin-linked peroxidase. In control experiments, the sections were stained with the same reagents, but without the first antibody (B and D). The endothelial cells in aorta were positive for the antigen (A), and those in lung capillary were negative (C).
We also examined serial sections using an anti-TM monoclonal antibody. TM was expressed widely in both arterial and venous vessels as described (21), and strong expression was detected in the capillaries (data not shown). These results indicated that EPCR and TM coexpress on the endothelial cells in vessels with relatively large size. Coexpression of EPCR and TM was also demonstrated by the flow cytometer analysis using in vitro cultured endothelial cells (Fig. 2). Strong expression of EPCR was detected on cultured AECs. EPCR was also detectable in cultured VECs; however, the expression level was several times lower than that in the AECs. TM was expressed on both AECs and VECs. In contrast to EPCR, strong expression of TM was detected on VECs and a lower and heterogeneous expression was detected on AECs.
Figure 2.

Surface expression of EPCR and TM on cultured endothelial cells. In vitro cultured AECs (top) and VECs (bottom) were stained with JRK-1 (an anti-EPCR monoclonal antibody, left) or CTM1009 (anti-TM monoclonal antibody, right). The antigens expression was detected by flow cytometer analysis using FITC-conjugated anti–mouse Ig. The x-axis represents the fluorescence intensity (log scale) and the y-axis represents relatively cell number.
Since the dissociation constant between protein C and EPCR is 30 nM, which is approximately half of the blood concentration of protein C, the majority of EPCRs exposed to the blood stream should be holding protein C under physiological conditions. These findings encouraged us to reinvestigate the mechanism of protein C activation, which has been believed to be mediated by only the thrombin–TM complex. We compared activities of protein C activation by AECs and VECs, which express different amounts of EPCRs and TM on their surface (Fig. 2). In both cases, protein C was converted to APC only in the presence of thrombin. Interestingly, promotion of protein C activation by AECs was comparable to VEC-mediated activation (Fig. 3), despite the lower level of TM expression (Fig. 2). In addition, activation of protein C on AECs was observed as a relatively high-affinity reaction with a K d value of 125 ± 72 nM, suggesting contribution by the high-affinity protein C receptor to the reaction. The K d value for the reaction by VECs was slightly lower (162 ± 54 nM) than that of AECs. This could be due to the low level of expression of EPCR on VECs.
Figure 3.

Protein C activation by cultured endothelial cells. Protein C was incubated with venous (A and B) or arterial (C and D) endothelial cells in the presence of thrombin, and generated APC was measured. The y-axis represents velocity of APC generation (pM/min) in the presence of indicated concentrations of protein C (A and C). The Vmax and K d values were calculated by the Eadie-Hostee plot (B and D).
To analyze in detail the activation mechanism, we established transfected human kidney 293 cell lines that express receptor molecules. The 293 cells are negative for EPCR and TM. Mock-transfected N1 cells are used as a negative control, E7 cells are EPCR positive as described previously (16), T2 cells are positive for TM alone, and ET1 and ET2 are dual-positive cell lines expressing both EPCR and TM. Stable antigen expression was demonstrated by flow cytometer analysis as shown in Fig. 4. Protein C was incubated with these cell lines in the presence of thrombin and the subsequently generated APC was measured. Thrombin-mediated protein C activation was not detectable on the negative control N1 cells. EPCR-positive E7 also could not promote activation. On the other hand, thrombin-dependent protein C activation was detected with cells positive for TM. T2 cells promoted activation only in the presence of thrombin. The activation was completely inhibited by a functional blocking anti-TM monoclonal antibody, CTM1009 (Fig. 5 A). These results indicated that activation of protein C on T2 cells was mediated by the thrombin–TM complex. However, the conversion rate was quite low when the rate per cell was calculated. In contrast, dramatic activation was demonstrated with EPCR/TM dual-positive cells (Fig. 5 A). Five to six times more activation was detected in the case of dual-positive cells as compared with cells positive for TM alone.
Figure 4.

Surface expression EPCR and TM on transfected cell lines. Cells were stained with CTM1009 (left) or JRK-1 (right) monoclonal antibodies. The antigen expression was analyzed by flow cytometer analysis. N1 is a negative control cell line. E7 was established from cells transfected with EPCR. T2 was from TM-transfected cells. ET1 and ET2 were from dual-transfected cells with both EPCR and TM.
Figure 5.

Protein C activation by transfected cells. Monolayers of indicated cells were incubated with 300 nM of protein C and 0.1 U of thrombin in the absence or presence of 10 μg/ ml of CTM1009, and then the generated APC was measured (A). ET1, T2, and N1 cells were incubated with indicated concentrations of protein C in the presence of thrombin, and the generated APC was measured (B). The K d and Vmax values of protein C activation by ET1 cells (C) or T2 cells (D) were calculated by the Eadie-Hostee plot.
We next compared APC generation by T2 cells and ET1 cells in the presence of increasing protein C concentration (Fig. 5 B). Protein C activation by T2 cells was greatly dependent on the protein C concentration. On the other hand, dual positive ET1 cells could promote significant activation, even at the lowest concentration of protein C. The K d value of the reaction was calculated as 869 ± 396 nM for T2 cells (Fig. 5 D) and 140 ± 43 nM for ET1 cells (Fig. 5 C). The low-affinity reaction by cells positive for TM alone and the high-affinity reaction by dual-positive cells were also demonstrated with the other cell lines (data not shown). The K d value obtained using T2 cells was identical with that calculated in a reconstitution experiment using solubilized TM in phospholipid membranes (11). In contrast, the K d value for ET1 cells was almost identical with that of AEC (Fig. 3). Therefore, EPCR/TM dual-positive cells, but not cells positive for TM alone, might mimic the protein C activation mechanism observed in in vitro cultured endothelial cells. TM appears to be an essential component for activation even in the presence of EPCR, because complete inhibition of protein C activation could be demonstrated again using the anti-TM monoclonal antibody (Fig. 5 A).
To analyze the function of EPCR on endothelial cells, we established several functional blocking monoclonal antibodies against EPCR (Ye X., N. Tsuneyoshi, K. Fukudome, and M. Kimoto, unpublished data). One of these, RCR-252 (rat IgG1), was found to inhibit APC binding to EPCR-expressing cells (Fig. 6 A). RCR-252 also inhibited protein C activation mediated not only by EPCR/TM dual-transfected ET1 cells (Fig. 6 B), but also by primary cultured AECs (Fig. 6 C) in the same manner. The inhibitory effect of RCR-252 was specific for EPCR function since protein C activation mediated by TM single-positive T2 cells was not affected (Fig. 6 B). Therefore, EPCR on cultured AECs appears to function for protein C activation. Inhibition of protein C activation by RCR-252 was only partial (∼60%), in sharp contrast with the complete inhibition by anti-TM antibody (Fig. 6 C). This would be partially due to the fact that EPCR is a regulatory, but not an essential, component for the catalytic reaction in such a way that EPCR amplifies dramatically the reaction mediated by the thrombin–TM complex (Fig. 5). In addition, binding of APC to ET1 cells by RCR-252 is not blocked completely. Approximately 10% of binding still remains, even in the presence of the highest concentration of the antibody (Fig. 6 A). Therefore, actual contribution of EPCR on protein C activation would be greater than that shown in Fig. 6.
Figure 6.

Effects of anti-EPCR monoclonal antibodies on APC binding and protein C activation. ET1 cells were treated with indicated concentrations of a functional blocking anti-EPCR monoclonal antibody, RCR-252 (closed circles) or a nonblocking RCR-122 (open circles), and then incubated with 300 nM of APC. After washing, amidolytic activity of bound APC was measured by using S-2366 and indicated as the maximum velocity (A). ET1(closed circles), T2 (closed squares), and N1 (closed triangles) cells were treated with indicated concentrations of RCR-252. As a negative control, RCR-122 was used (open circles). Cells were then incubated with 300 nM of protein C and 0.1 U of thrombin. Generated APC was measured and indicated as described above (B). After incubation of AECs with RCR-252 (circles), CTM1009 (squares), or RCR-252 (triangles), APC generation was measured (C).
Since the presence of EPCR reduced the K d value for protein C activation mediated by the thrombin–TM complex, EPCR appears to concentrate protein C on the endothelial cell surface. Protein C appears to change conformation when it binds to EPCR because the velocity of the catalytic reaction mediated by the thrombin–TM complex increased in the presence of EPCR. Although EPCR can bind protein C with high affinity, the binding has been demonstrated as a reversible reaction (15). Therefore, bound protein C should be replaced continuously in circulation. By these mechanisms, protein C activation appears to be promoted effectively on the endothelial cell surface. Whether EPCR and TM exist as a complex on the endothelial cell surface or the complex is formed after binding of protein C to EPCR remains to be investigated.
Clinical studies related to the protein C pathway have been mainly focused on thrombosis in microvessels, since until now protein C activation has been considered to be restricted to the microcirculation (Fig. 7 A). However, in this study, we have demonstrated a novel mechanism for APC generation involving EPCR (Fig. 7 B). The high-affinity protein C receptor is expressed in large vessels in which protein C activation has been believed unlikely to occur. EPCR appears to enable significant levels of protein C activation in large vessels under physiological conditions. Generation of APC might function as an important regulatory mechanism for blood coagulation in large vessels. In fact, some clinical reports of arterial thrombosis appear to be related to defects in the protein C pathway (22–24). In the microvessels, we could not detect EPCR. Whether the physical condition of increasing the endothelial cell surface area to blood volume make up the low-affinity reaction or another unidentified molecule is involved in the activation remains to be investigated.
Figure 7.
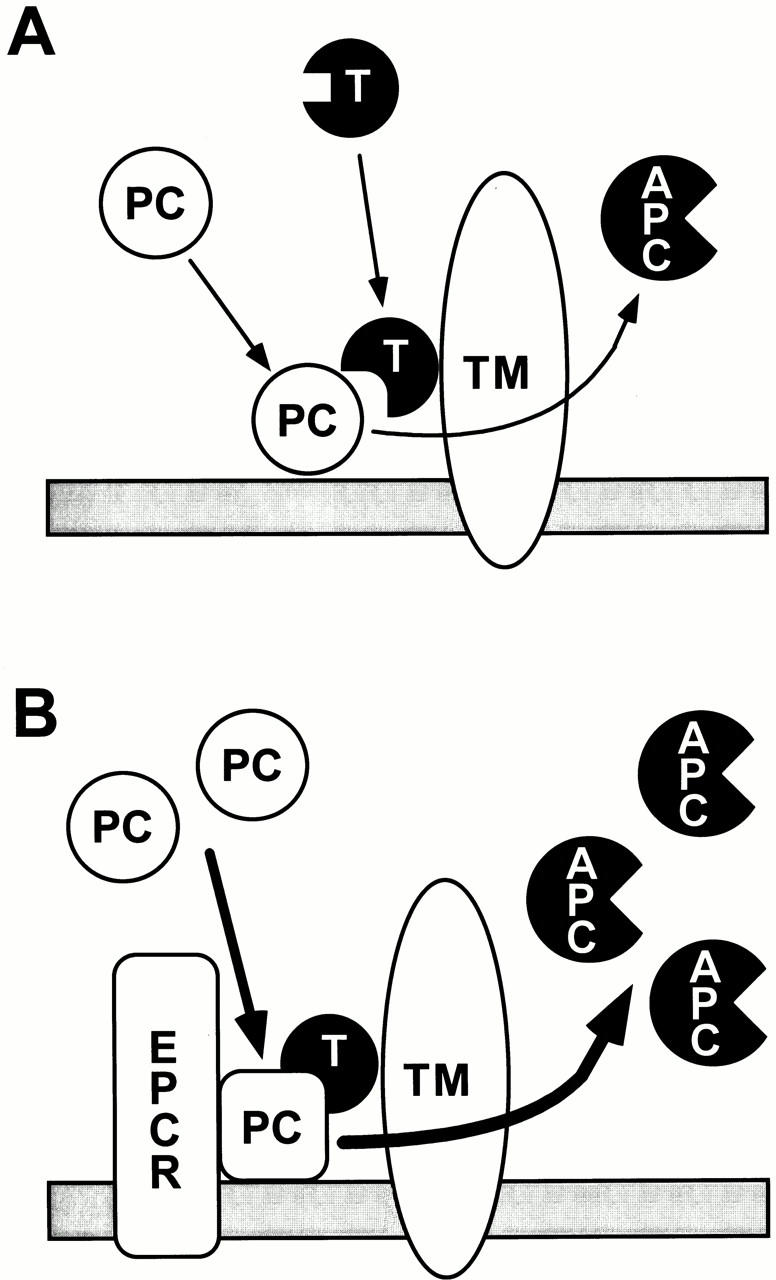
Hypothetical models for protein C activation on the endothelial cell surface. In microvessels, EPCR is not detectable. In the absence of EPCR, protein C activation mediated by the thrombin–TM complex is a relatively low-affinity reaction. For significant levels of protein C activation to occur, an increasing ratio of endothelial cell surface area to the blood volume is required (A). In the large vessels, the high-affinity binding of protein C to EPCR on the endothelial cells is a critical step for activation (B). After binding to EPCR, the structure of protein C is modulated to enable rapid conversion to the active form.
Acknowledgments
The authors thank Drs. K. Miyake and Y. Kamikubo for their helpful discussion; Dr. F. Nestel for critical reading of the manuscript; and S. Chen, F. Mutho, and C. Brown for technical support.
This work was supported by grants from the Ministry of Education, Science, Sports, and Culture of Japan to K. Fukudome and M. Kimoto, and from the Ryouichi Naitoh Foundation for Medical Research to K. Fukudome.
Footnotes
1 Abbreviations used in this paper: AEC, arterial endothelial cell; APC, activated protein C; EPCR, endothelial cell protein C-APC receptor; TM, thrombomodulin; VEC, venous endothelial cell.
References
- 1.Malm J, Laurell M, Nilsson IM, Dahlback B. Thromboembolic disease: critical evaluation of laboratory investigation. Thromb Haemostasis. 1992;68:7–13. [PubMed] [Google Scholar]
- 2.Dahlback B. Physiological anticoagulation. Resistance to activated protein C and venous thromboembolism. J Clin Invest. 1994;94:923–927. doi: 10.1172/JCI117458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenflo J. A new vitamin K–dependent protein. Purification from bovine plasma and preliminary characterization. J Biol Chem. 1976;251:355–363. [PubMed] [Google Scholar]
- 4.Esmon CT, Stenflo J, Suttie JW. A new vitamin K–dependent protein. A phospholipid-binding zymogen of a serine esterase. J Biol Chem. 1976;251:3052–3056. [PubMed] [Google Scholar]
- 5.Kisiel W, Canfield WM, Ericsson LH, Davie EW. Anticoagulant properties of bovine plasma protein C following activation by thrombin. Biochemistry. 1977;16:5824–5831. doi: 10.1021/bi00645a029. [DOI] [PubMed] [Google Scholar]
- 6.Esmon CT, Owen WG. Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc Natl Acad Sci USA. 1981;78:2249–2252. doi: 10.1073/pnas.78.4.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackman RW, Beeler DL, VanDeWater L, Rosenberg RD. Characterization of a thrombomodulin cDNA reveals structural similarity to the low density lipoprotein receptor. Proc Natl Acad Sci USA. 1986;83:8834–8838. doi: 10.1073/pnas.83.23.8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki K, Kusumoto H, Deyashiki Y, Nishioka J, Maruyama I, Zushi SM, Kawahara S, Honda G, Yamamoto S, Horiguchi S. Structure and expression of human thrombomodulin, a thrombin receptor on endothelium acting as a cofactor for protein C activation. EMBO (Eur Mol Biol Organ) J. 1987;6:1891–1897. doi: 10.1002/j.1460-2075.1987.tb02448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owen WG, Esmon CT. Functional properties of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1981;256:5532–5535. [PubMed] [Google Scholar]
- 10.Galvin JB, Kurosawa S, Moore K, Esmon CT, Esmon NL. Reconstitution of rabbit thrombomodulin into phospholipid vesicles. J Biol Chem. 1987;262:2199–2205. [PubMed] [Google Scholar]
- 11.Nishioka J, Ido M, Hayashi T, Suzuki K. The Gla26 residue of protein C is required for the binding of protein C to thrombomodulin and endothelial cell protein C receptor, but not protein S and factor Va. Thromb Haemostasis. 1996;75:275–282. [PubMed] [Google Scholar]
- 12.Miletich J, Sherman L, Broze G. Absence of thrombosis in subjects with heterozygous protein C deficiency. N Engl J Med. 1987;317:991–996. doi: 10.1056/NEJM198710153171604. [DOI] [PubMed] [Google Scholar]
- 13.Esmon CT. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J Biol Chem. 1989;264:4743–4746. [PubMed] [Google Scholar]
- 14.Fukudome K, Esmon CT. Identification, cloning, and regulation of a novel endothelial cell protein C/activated protein C receptor. J Biol Chem. 1994;269:26486–26491. [PubMed] [Google Scholar]
- 15.Bangalore N, Drohan WN, Orthner CL. High affinity binding sites for activated protein C and protein C on cultured human umbilical vein endothelial cells. Independent of protein S and distinct from known ligands. Thromb Haemostasis. 1994;72:465–474. [PubMed] [Google Scholar]
- 16.Fukudome K, Kurosawa S, Stearns-Kurosawa DJ, He X, Rezaie AR, Esmon CT. The endothelial cell protein C receptor: cell surface expression and direct ligand binding by the soluble receptor. J Biol Chem. 1996;271:17491–17498. doi: 10.1074/jbc.271.29.17491. [DOI] [PubMed] [Google Scholar]
- 17.Aruffo A, Seed B. Expression of cDNA clones encoding the thymocyte antigens CD1a, b, c demonstrates a hierarchy of exclusion in fibroblasts. J Immunol. 1989;143:1723–1730. [PubMed] [Google Scholar]
- 18.Fukudome K, Esmon CT. Molecular cloning and expression of murine and bovine endothelial cell protein C/activated protein C receptor (EPCR). The structural and functional conservation in human, bovine, and murine EPCR. J Biol Chem. 1995;270:5571–5577. doi: 10.1074/jbc.270.10.5571. [DOI] [PubMed] [Google Scholar]
- 19.Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokunaga, O. 1995. Endothelial cell adhesion molecules. Atherosclerosis III: Recent Adv. Atherosclerosis Res. 748:498–500. [PubMed]
- 21.Maruyama I, Bell CE, Majerus PW. Thrombomodulin is found on endothelium of arteries, veins, capillaries, and lymphatics, and on syncytiotrophoblast of human placenta. J Cell Biol. 1985;101:363–371. doi: 10.1083/jcb.101.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohler J, Kasper J, Witt I, Reutern GM. Ischemic stroke due to protein C deficiency. Stroke. 1990;21:1077–1080. doi: 10.1161/01.str.21.7.1077. [DOI] [PubMed] [Google Scholar]
- 23.Stefano VD, Leone G, Micalizzi P, Teofili L, Falappa PG, Pollari G, Bizzi B. Arterial thrombosis as clinical manifestation of congenital protein C deficiency. Ann Hematol. 1991;62:180–183. doi: 10.1007/BF01703145. [DOI] [PubMed] [Google Scholar]
- 24.Kazui S, Kuriyama Y, Sakata T, Hiroki M, Miyashita K, Sawada T. Accelerated brain infarction in hypertension complicated by hereditary heterozygous protein C deficiency. Stroke. 1993;24:2097–2103. doi: 10.1161/01.str.24.12.2097. [DOI] [PubMed] [Google Scholar]