

Abstract
Cytolytic T cells use two mechanisms to kill virally infected cells, tumor cells, or other potentially autoreactive T cells in short-term in vitro assays. The perforin/granule exocytosis mechanism uses preformed cytolytic granules that are delivered to the target cell to induce apoptosis and eventual lysis. FasL/Fas (CD95 ligand/CD95)–mediated cytolysis requires de novo protein synthesis of FasL by the CTL and the presence of the death receptor Fas on the target cell to induce apoptosis. Using a CD8+ CTL clone that kills via both the perforin/granule exocytosis and FasL/Fas mechanisms, and a clone that kills via the FasL/Fas mechanism only, we have examined the requirement of intra- and extracellular Ca2+ in TCR-triggered cytolytic effector function. These two clones, a panel of Ca2+ antagonists, and agonists were used to determine that a large biphasic increase in intracellular calcium concentration, characterized by release of Ca2+ from intracellular stores followed by a sustained influx of extracellular Ca2+, is required for perforin/granule exocytosis. Only the sustained influx of extracellular Ca2+ is required for FasL induction and killing. Thapsigargin, at low concentrations, induces this small but sustained increase in [Ca2+]i and selectively induces FasL/Fas-mediated cytolysis but not granule exocytosis. These results further define the role of Ca2+ in perforin and FasL/Fas killing and demonstrate that differential Ca2+ signaling can modulate T cell effector functions.
Upon recognition of Ag/MHC, a cytolytic T cell (CTL) is activated to perform many effector functions, including cytokine secretion, receptor modulation, cytolysis and eventually cell division and proliferation, or apoptosis and death. CTLs use at least three mechanisms of killing to lyse virus-infected cells, tumor cells, or potentially autoreactive T cells. The perforin and Fas ligand (FasL)/Fas1 mechanisms account for all of the killing detected in short term assays in vitro (1) with the TNF-α mechanism requiring 24–48 h (2). The perforin/granule exocytosis pathway is primarily used to kill virus infected and tumorigenic cells (3–7) and is characterized by the pore-forming protein, perforin, and several proteases or granzymes that are stored in the CTL's cytolytic granules (8). Upon Ag/MHC recognition, these proteins are delivered to the target cell to induce membrane damage, apoptosis, and eventually lysis (7, 9, 10). This lethal hit is delivered with a t1/2 of 7–10 min and is both temperature and Ca2+ dependent (11). The FasL/Fas mechanism of killing appears not to be involved in eradication of virally infected cells, but instead plays an important role in eliminating autoreactive T cells (12–15). This is evident in mice or humans that lack FasL or Fas and which develop lymphadenopathy and lupus-like autoimmunity (13, 16). Both the perforin and the FasL/Fas mechanism require TCR–Ag/MHC interactions, which trigger perforin/granule exocytosis and induce FasL expression respectively. Because FasL expression requires de novo protein synthesis in the T cell, it takes longer to lyse the target cell than the perforin/granule exocytosis mechanism (17, 18). Once FasL is expressed on the surface of the T cell it can kill Fas-expressing cells in an MHC-unrestricted manner (19, 20). Since perforin-mediated cytolysis involves release of preformed granules whereas FasL/ Fas cytolysis requires induction of gene expression, we asked whether these effector functions are regulated by different TCR signaling pathways.
The TCR signal transduction pathways that regulate perforin and FasL/Fas killing are less well defined than the multiple pathways identified that regulate IL-2 production in CD4+ Th1 cells. The PI3kinase, protein kinase C, Ras/ Raf/Erk, JNK, and Ca2+ signaling pathways have all been implicated in regulating IL-2 production in CD4+ T cells (21–24). Calcium signaling regulates growth, death, differentiation, cytotoxicity, and cytokine secretion in T cells (25–28). Several Ca2+-sensitive transcriptional regulators, including NF-κB (29), Jun kinase (JNK) (30), and NFAT (nuclear factor of activated T cells; reference 31) participate in varying combinations to regulate growth cytokines such as IL-2, IL-4, and GM-CSF and inflammatory cytokines such as IL-1, IL-6, IL-8, and TNF (28–31). More recently, several groups have reported that tyrosine kinases such as ZAP70 and PI3K are involved in regulating FasL expression (32–34). A sustained rise in intracellular Ca2+ concentration ([Ca2+]i) can activate calcineurin, a Ca2+-dependent, cyclosporin A (CsA)–sensitive serine/threonine phosphatase that dephosphorylates the transcription factor NFAT (28). Once dephosphorylated, NFAT migrates to the nucleus, where it associates with Jun and Fos to promote transcription of a host of immunoregulatory genes (31). Calcium regulation of NFAT translocation in and out of the nucleus and transcription of an IL-2 reporter gene has been visualized at the single cell level (35). The importance of Ca2+ in lymphocyte activation is evident from the effectiveness of the immunosuppressant CsA (BIOMOL Research Labs., Plymouth Meeting, PA) and the finding that patients with lymphocytes defective in Ca2+ signaling suffer from primary immunodeficiency (36).
Classical studies performed by Gray and co-workers recorded the spatial and temporal aspects of Ca2+ signaling in CTLs after TCR engagement (37–40). This TCR triggering led to a two component increase in [Ca2+]i due from an initial release of Ca2+ from intracellular stores followed by a sustained influx of extracellular Ca2+ (37). Additional studies demonstrated that extracellular Ca2+ was required for TCR-triggered serine esterase release (41), and CTLs with ζ chain mutations, defective in Ca2+ signaling, were incapable of killing target cells (42). Confounding these studies were reports of a Ca2+-independent mechanism of killing (43, 44), which was eventually shown to be mediated by FasL/Fas interactions (17). Although discovering Fas solved the mystery concerning an alternative mechanism of killing used by CTLs, it did not resolve the role of Ca2+ used by these two mechanisms of cytotoxicity.
We have generated a series of IL-2–dependent, Ag-independent clones (CTL-FDs) that have lost the ability to kill via the perforin/granule exocytosis mechanism of killing, although they contain functional perforin and granzymes. These variants have retained the ability to kill via the FasL/ Fas mechanism of killing and they secrete IFN-γ. Using these clones or an altered peptide ligand, we have shown that distinct TCR signaling events activate perforin versus FasL/Fas killing (45, 46). In examining the parental 14-7 and the variant 14-7FD clones, we discovered that bypassing early TCR signaling events with phorbol ester and Ca2+ ionophore could trigger granule exocytosis in the variant clone. In addition, CsA inhibited FasL/Fas killing but not perforin killing. This evidence and the fact that 14-7FD did not display the characteristic biphasic Ca2+ response after TCR engagement led us to further examine the role of intracellular and extracellular Ca2+ in perforin and FasL/Fas mediated cytotoxicity.
In this paper, we report that a large biphasic Ca2+ response is required for perforin killing, whereas a smaller sustained Ca2+ influx is sufficient for FasL induction and killing. We also show that replacing the large [Ca2+]i flux in 14-7FD with ionomycin or thapsigargin while stimulating with PMA is enough to trigger perforin granule/exocytosis cytotoxicity. Extracellular Ca2+ was required for both perforin and FasL/Fas killing. Lastly, we demonstrate that differential Ca2+ signaling can activate FasL/Fas versus perforin killing. These findings should provide insight into the role of Ca2+ in activation of differential TCR signaling pathways.
Materials and Methods
Cell Lines.
The L1210Fas+ (H-2d) (17) and the L1210Fas- (H-2d) (47), target cells were maintained in DMEM (4.5 g/liter glucose) supplemented with 10% (vol/vol) heat inactivated FCS (GIBCO BRL, Gaithersburg, MD), 2 mM glutamine, 5 × 10−5 M 2-ME, and antibiotics (10 U/ml penicillin G and 10 μg/ml streptomycin sulfate).
T Lymphocyte Clones.
T lymphocyte clones, 14-7, 11-1, and 14-13, (48) were stimulated in vitro with influenza A/JAPAN/57 virus infected, γ-irradiated (2,000 rad) BALB/c spleen cells every 7 d in the presence of 10 U/ml human rIL-2 (Biosource International, Inc., Camarillo, CA) in complete media: Iscove's media (GIBCO BRL, Gaithersburg, MD), 10% heat inactivated FCS (HIFCS, Hyclone Laboratories, Logan, UT), 2 mM glutamine and 5 × 10−5 M 2-ME. The 14-7FD clone was seeded once a week at 0.5 × 106 cells/5ml of complete medium and split 1:2 after 3.5 d in medium containing 30 U/ml human rIL-2.
Reagents.
Ionomycin (1 M stock in DMSO), thapsigargin (1 M stock in DMSO; reference 49), PMA (1 M stock in DMSO), nordihydroguaiaretic acid (NDGA; reference 50) (1M stock in DMSO), EGTA (100 mM stock in dH20, pH 7.8), NiCl2 (10 M stock in BSS buffer [140 mM NaCl, 3 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 0.1% glucose, 1% NBCS, 10 mM Hepes, at a final pH of 7.4]), and emetine (1 mg/ml stock in EtOH) were all obtained from Sigma Chemical Co., (St. Louis, MO). SK&F-96365 (1 M stock in dH2O; reference 51) and nifedipine (1 M stock in DMSO; reference 52) were obtained from Calbiochem (La Jolla, CA).
Anti-FasL antibody, #F37720, was purchased from Transduction Laboratories (Lexington, KY).
Flow Cytometry.
106 cells/ml were mock stimulated and were stimulated with plate-bound anti-CD3ε (145-2C11; reference 53) at 5 μg/ml in the presence or absence of various Ca2+ antagonists for 6 h in MEM, 1% FCS, 2 mM Gln, and 50 μM 2-ME. T cells were collected, transferred to a round-bottomed microtiter plate, centrifuged, and resuspended in 50 μl of Fas.Fc at 10 μg/ml for 30 min on ice in PBS with 1% newborn calf serum (NBCS). T cells were washed two times, incubated for 30 min with a biotinylated goat anti–human IgG (5 μg/ml; Southern Biotechnology Associates, Birmingham, AL), washed two more times, and incubated in the dark for 30 min with Streptavidin R-PE (5 μg/ml; Caltag Labs., Burlingame, CA), and then washed three times in PBS with 1% NBCS and analyzed on a FACScan® (Becton Dickinson, Mountain View, CA).
Intracellular Ca2+ Mobilization Studies.
CTL clones were Ficoll purified 4 d after in vitro stimulation and 106 CTL/ml were incubated with 1 μM Indo-1 for 1 h, and then washed 3 times in BSS buffer to remove free dye. CTLs were stimulated with 90 μl of 1.0 mg/ml anti-CD3ε mAb (145-2C11) or with various concentrations of thapsigargin or ionomycin at 37°C and the ratio of 398:480 was measured over time. Excitation was at 340 nm in an SLM 8000 Spectrafluorometer (SLM Aminco, Urbana, IL) in the T format. Calibration was conducted as previously described (40).
N-α-benzyloxycarbonyl-l -lysine thiobenzyl Esterase Assay.
Exocytosis of cytolytic granules was measured by the N-α-benzyloxycarbonyl-l-lysine thiobenzyl (BLT) esterase (granzyme A) activity in the supernatant of stimulated T cells. 96-well PVC round-bottomed plates (Dynatech, Chantilly, VA) were coated with 50 μl of 5 μg/ml mAb to CD3ε (145-2C11) for 30 min at room temperature. Plates were washed twice with PBS+2% NBCS before plating 14-7 and 14-7FD at 5 × 104 cells/well in quadruplicates. PMA, ionomycin, or thapsigargin were added at various concentrations and combinations to the appropriate wells so that the total volume was 100 μl. The plate was spun for 1 min at 250 g before incubating the plate at 37°C for 4 h. 30 μl of the supernatants were harvested and assayed for BLT esterase activity as previously described (41), adapted for a 96-well microtiter plate. After 30 min, absorbance values at 405 nm were determined by reading the plate on a Bio-Tek EL-340 ELISA reader (Bio-Tek Instruments, Burlington, VT) at an OD of 405 nm. All experiments were performed in quadruplicate. Total release was determined by lysing the CTL in 1% Triton X-100. Spontaneous release was <20% for all experiments.
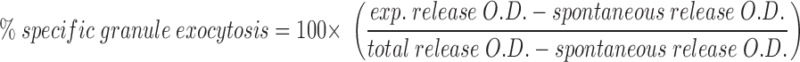 |
51Cr–release Cytotoxicity Assay.
Target cells were incubated with sodium 51chromate for 3 h at 37°C. Target cells were washed three times, mock treated, or sensitized with the hemagglutinin (HA)529–537 (IYATVAGSL) peptide (0.01 μM; reference 54) before plating at 104 target cells/well. In all experiments 14-7, 11-1, 14-13, and 14-7FD were added at an E/T of 5:1 in round-bottomed plates, spun for 1 min at 250 g, and incubated at 37°C in a CO2 incubator. Anti-FasL antibody was added to T cells at 5 mg/ml for 15 min at room temperature before plating. Supernatants (100 μl) were harvested at 4 h for the perforin killing experiments and at 6–8 h for the FasL/Fas killing experiments from each well and counted on a gamma counter (Isomedic; ICN Biomedical, Huntsville, AL). All experiments were performed in quadruplicate. Spontaneous release was <10% in all experiments. Percentage of specific lysis was calculated as follows:
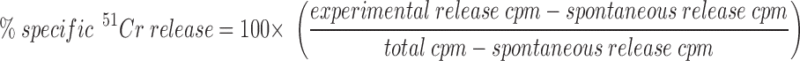 |
Results
Anti-CD3ε Effectively Stimulates Perforin/Granule Exocytosis and FasL/Fas Cytotoxicity in 14-7 and 14-7FD.
We have previously reported that distinct TCR signaling events trigger perforin versus FasL/Fas cytotoxicity and IFN-γ production (45, 46). Ag/MHC complexes trigger perforin/ granule exocytosis and induce FasL/Fas killing in 14-7, but only FasL/Fas killing in 14-7FD (46). We had also observed that plate-bound anti-CD3ε triggered granule exocytosis and IFN-γ production in 14-7, whereas it only stimulated IFN-γ production in 14-7FD. Since we planned to use anti-CD3ε in our Ca2+ measurements, we wanted to confirm that soluble anti-CD3ε paralleled the Ag/MHC response in the CTL killing assays. Fig. 1 shows that anti-CD3ε works in a soluble but not a plate-bound fashion to trigger 14-7 to kill via the perforin mechanism (Fig. 1 a). Anti-CD3ε works both in solution and when plate-bound to trigger granule exocytosis in the presence or absence of target cells in 14-7 but not in 14-7FD (Fig. 1 b). In addition, anti-CD3ε induces both 14-7 and 14-7FD to lyse targets via the FasL/Fas mechanism (Fig. 1 c). In all perforin and granule exocytosis experiments, the CTLs were pretreated with 1 μg/ml emetine, an irreversible protein synthesis inhibitor (55), for 5 min before the start of the assay to prevent FasL induction or production of granzyme A (56). In agreement with our previous work, anti-CD3ε stimulated 14-7FD to kill via the FasL/Fas mechanism only (Fig. 1, a–c). These results demonstrate that soluble anti-CD3ε could substitute for Ag/MHC to trigger perforin and FasL/Fas cytotoxicity in our model system.
Figure 1.

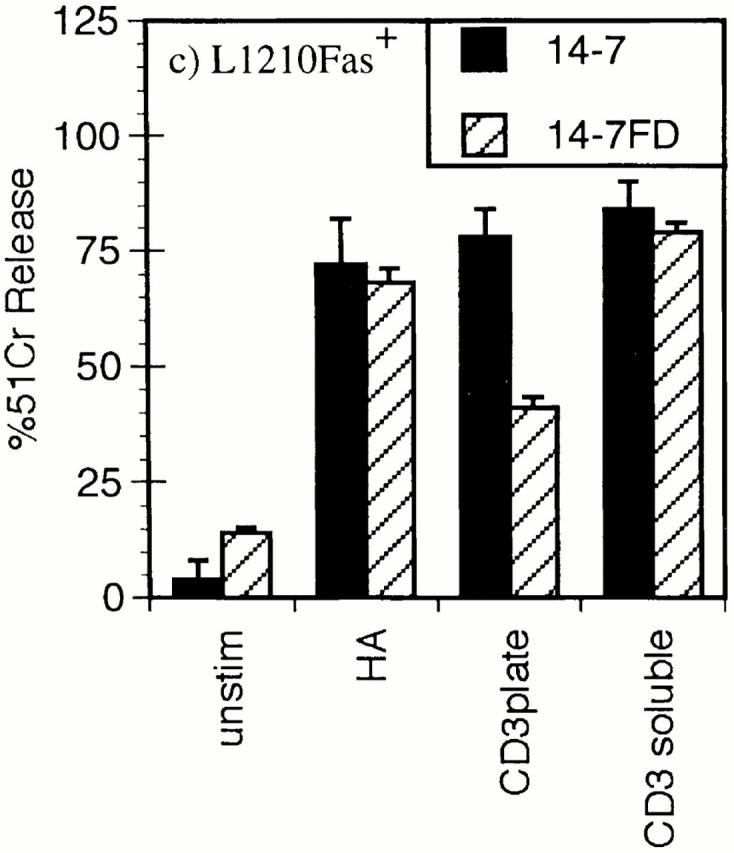
Anti-CD3ε stimulation of perforin/granule exocytosis and FasL/Fas killing. 14-7 and 14-7FD were incubated with either mock treated or peptide antigen HA529–537 (0.01 μM) pulsed target cells at an effector to target ratio of 5:1. For anti-CD3ε wells, soluble 145-2C11 (5 μg/ml) was added or the plate was coated with 5 μg/ ml 145-2C11 for 30 min at room temperature before addition of the CTL or the target. (a) 51Cr cytolysis assay with L1210Fas- target. (b) BLT-esterase assay with supernatants from a and from CTL stimulated in the absence of target cells. (c) 51Cr release assay with L1210Fas+ target.
Anti-CD3ε Stimulation Induces Qualitatively and Quantitatively Distinct Ca2+ Signals in 14-7 and 14-7FD.
We have previously reported that 14-7 and 14-7FD differ dramatically in their TCR-stimulated total Ca2+ response (46), but in that study we did not distinguish between the intracellular Ca2+ release and extracellular Ca2+ influx components of the response. We next examined the role intra- and extracellular Ca2+ played in TCR signaling in 14-7 and 14-7FD. 14-7 has a resting Ca2+ level of ∼50 nM, which peaks at 2 μM within 30 s of TCR stimulation and is sustained at ∼1 μM for at least 6 min (Fig. 2 a). This TCR-stimulated biphasic Ca2+ profile is very similar to that observed by others (37, 57, 58). In contrast, 14-7FD has a much higher resting level of [Ca2+]i than does 14-7, between 80 and 300 nM (Fig. 2 b). When stimulated with anti-CD3ε, 14-7FD does not undergo the characteristic Ca2+ response seen in 14-7, but has a slow sustained increase in [Ca2+]i levels (Fig. 2 b). To measure the contribution of released intracellular Ca2+ to the rise in [Ca2+]i, we used EGTA to chelate extracellular Ca2+. When 14-7 was stimulated through the TCR in the presence of EGTA, the intracellular release component was easily detected (Fig. 2 c). In comparison, when 14-7FD was stimulated with anti-CD3ε in the presence of EGTA, little to no intracellular release component was detected (Fig. 2 d). To determine the source of the increase in [Ca2+]i, we did the converse of the previous experiment and stimulated 14-7FD through the TCR and then added EGTA. In 14-7, addition of EGTA after stimulation led to a reduction in [Ca2+]i with a return to baseline over the course of ∼45 s (Fig. 2 e). In comparison, addition of EGTA after anti-CD3ε stimulation of 14-7FD resulted in an almost immediate reduction in [Ca2+]i (Fig. 2 f). The inorganic Ca2+ blocker Ni2+ gave very similar results (data not shown). These results suggest that for 14-7FD the increase in [Ca2+]i is almost exclusively due to influx, apparently occurring in the presence of very little to no release of intracellular Ca2+. These results demonstrate that Ca2+ signaling in 14-7 and 14-7FD are qualitatively and quantitatively distinct and provide a correlation between the differential Ca2+ response and the ability to kill via the perforin/granule exocytosis or FasL/Fas mechanism, respectively (Figs. 1 and 2).
Figure 2.
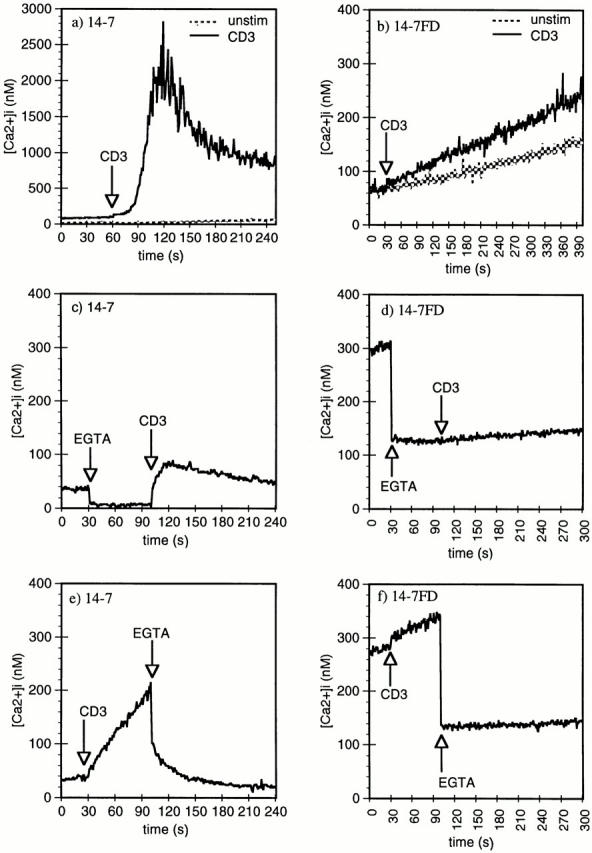
Anti-CD3ε induction of Ca2+ mobilization and influx in 14-7 and 14-7FD. Anti-CD3ε (145-2C11) (45 μg/ml) was used to trigger 14-7 and 14-7FD Ca2+ mobilization, and influx was monitored by indo-1 fluorescence. EGTA (5 mM) was used to examine the intra- and extracellular release components of the Ca2+ signaling pathways in 14-7 (c, e) and 14-7FD (d, f ). EGTA was added before (c and e) or after (d and f ) anti-CD3ε addition.
Release of Intracellular Ca2+Induces Influx of Extracellular Ca2+ in 14-7FD.
We next wanted to dissect the Ca2+ signaling mechanism in 14-7FD. We hypothesized that either there was a defect in the TCR signaling pathway leading to intracellular Ca2+ release or that there was a failure of intracellular released Ca2+ to open plasma membrane Ca2+ channels. In many cell types extracellular Ca2+ influx is initiated by the release of intracellular Ca2+ stores (59–61). To test whether intracellular Ca2+ release could activate the Ca2+ entry pathway in 14-7FD, we used thapsigargin, a microsomal Ca2+ pump inhibitor, to deplete intracellular Ca2+ stores (49, 59, 62). Thapsigargin induces Ca2+ release by inhibiting an endoplasmic reticulum(ER) membrane Ca2+ ATPase pump that constantly pumps Ca2+ back into the ER (49). Thapsigargin induced the release of intracellular Ca2+ when extracellular Ca2+ was absent (Fig. 3 a) in 14-7FD. This intracellular Ca2+ release induced influx of extracellular Ca2+, as shown by the fact that addition of EGTA inhibited extracellular Ca2+ influx (Fig. 3 b). These data provide evidence that differential TCR signaling response in 14-7FD lies upstream of ER Ca2+ release, not in intracellular Ca2+–induced extracellular Ca2+ influx.
Figure 3.

Thapsigargin (Tg) induction of intracellular Ca2+ release and Ca2+ influx in 14-7FD. 5 mM EGTA was added before (a) or after (b) stimulation of 14-7FD with 100 nM thapsigargin.
Ca2+ Channel Antagonists Inhibit Perforin/Granule Exocytosis and FasL/Fas-mediated Cytotoxicity.
This differential Ca2+ signaling in 14-7 and 14-7FD prompted us to further define the extracellular and intracellular Ca2+ requirements for perforin and FasL/Fas killing. Several groups have examined the effects of Ca2+ channel antagonists and Ca2+ signaling in CTL-mediated killing (37, 41, 42). Since the FasL/Fas-based mechanism of killing was not discovered until 1993 (17), we sought to reexamine the ability of various Ca2+ antagonists to inhibit perforin/granule exocytosis or FasL/Fas-based killing. The L type channel antagonists (nifedipine, nitrendipine, and verapamil [52]), T type channel antagonists (NDGA [50] and SK&F-96365 [51]), calmodulin antagonists (trifluoperazine, W7 [60]), Ca2+ chelator (EGTA), and Ca2+ channel blocker Ni2+ (26) were examined for their effects on CTL-mediated cytotoxicity. Trifluoperazine and W7 were toxic to the CTL in 4–8-h assays. EGTA inhibited perforin killing by 14-7 on HA529-pulsed Fas− targets (Fig. 4 a) with an IC50 between 1 and 5 mM. Nifedipine, SK&F-96365, and NDGA inhibited perforin killing (Fig. 4 a) with IC50s of 50–100 μM, 10–50 μM, and 5–10 μM, respectively (Fig. 4 a). These data demonstrate the requirement for influx of extracellular Ca2+ in perforin killing and extend the original observations to include new, more T cell–selective, T type Ca2+ channel antagonists.
Figure 4.

Extracellular Ca2+ is required for perforin/granule exocytosis and FasL/Fas-mediated cytotoxicity. 14-7 (a, b, d) and 14-7FD (c) were pretreated for 40 min with nifedipine, EGTA, SK&F-96365, NDGA, or Ni2+ at various concentrations before the start of a 51Cr cytolysis assay (a–c) or before anti-CD3ε stimulation (d). 14-7 was incubated with HA529–537 peptide (0.01 μM)–pulsed L1210Fas target in a 51Cr–release assay (a), and supernatants from a were used to measure granzyme A activity in a granule exocytosis assay (b). 14-7FD was incubated with HA529–537 peptide (0.01 μM)–pulsed L1210Fas+ targets in a 51Cr–release assay (c). 14-7 was stimulated with plate-bound anti-CD3ε for 6 h before staining for FasL with the Fas.Fc fusion protein by flow cytometry (d). All figures are representative of at least three separate experiments.
We tested the same panel of inhibitors for the ability to inhibit anti-CD3ε–triggered granule exocytosis in a target cell–independent assay. In concordance with our perforin killing assay, nifedipine, SK&F-96365, NDGA, and EGTA all inhibited granule exocytosis with similar IC50s as had been seen in the perforin-killing assay (Fig. 4 b). Ni2+ also inhibited TCR-triggered granule exocytosis (reference 40 and data not shown). These data indicate that extracellular Ca2+ is required for granule exocytosis killing and that both T and L type selective antagonists can inhibit granule exocytosis.
Historically, the FasL/Fas mechanism of killing was discovered as being a Ca2+ independent mechanism of killing (17). These initial reports were somewhat contradictory to our finding that CsA inhibited TCR induced FasL/Fas killing, since it is widely known that CsA acts on calcineurin, a Ca2+-dependent phosphatase (31, 63). Since 14-7/14-7FD provides a unique system to address this issue, we screened the same panel of Ca2+ antagonists for their ability to block TCR-induced FasL/Fas killing by 14-7FD. Normally, TCR engagement induces signals that lead to induction of the FasL gene. Once FasL is expressed, the CTL can lyse Fas-expressing targets. EGTA, NDGA, and SK&F-96365 inhibited TCR-induced FasL/Fas killing with IC50s slightly higher than those required to inhibit perforin killing (Fig. 4 c). In contrast to its effect on perforin killing, nifedipine had no effect on TCR-induced FasL/Fas killing (Fig. 4 c). To confirm that extracellular Ca2+ was required to induce FasL expression in 14-7, we screened Ni2+ and the panel of inhibitors for the ability to inhibit anti-CD3ε–induced FasL expression as detected by flow cytometry. As shown in Fig. 4 d, Ni2+, EGTA, SK&F-96365, and NDGA inhibited induction of FasL expression, whereas nifedipine was ineffective (Fig. 4 d). These data demonstrate that extracellular Ca2+ influx is required for TCR-triggered perforin lysis, granule exocytosis, and FasL expression, as well as FasL/Fas killing.
A Large Increase in [Ca2+]i Is Required for Perforin Killing.
We next wanted to test whether a Ca2+ agonist in combination either with PMA or a TCR signal could restore the large increase in [Ca2+]i in 14-7FD and therefore restore perforin/granule exocytosis killing. Using either thapsigargin or ionomycin to induce an increase in [Ca2+]i in conjunction with anti-CD3ε or Ag/MHC stimulation, we examined granule exocytosis (Fig. 5, a–d) and perforin killing (Fig. 5, e and f ). As previously seen, anti-CD3ε triggers granule exocytosis in 14-7, but not in 14-7FD (Fig. 5, a and b). Both the combination of PMA and ionomycin and that of PMA and thapsigargin trigger granule exocytosis for both 14-7 and 14-7FD (Fig. 5, a and b). Ionomycin alone induces a small yet significant secretion of granzyme A in 14-7FD (Fig. 5 b). We then tested the ability of ionomycin and thapsigargin to work in combination with anti-CD3ε or Ag/MHC to trigger granule exocytosis. Ionomycin or thapsigargin with either anti-CD3ε or Ag/MHC does not significantly trigger granule exocytosis in 14-7FD over ionomycin or thapsigargin alone (Fig. 5, b and c). These data indicate that mimicking the large [Ca2+]i increase with ionomycin or thapsigargin in combination with PMA could restore perforin/granule exocytosis killing in 14-7FD, whereas ionomycin or thapsigargin in combination with a TCR signal was not sufficient. This suggests that other TCR signals in addition to the functional TCR signaling pathways in 14-7FD and Ca2+ are required for perforin killing by 14-7FD.
Figure 5.
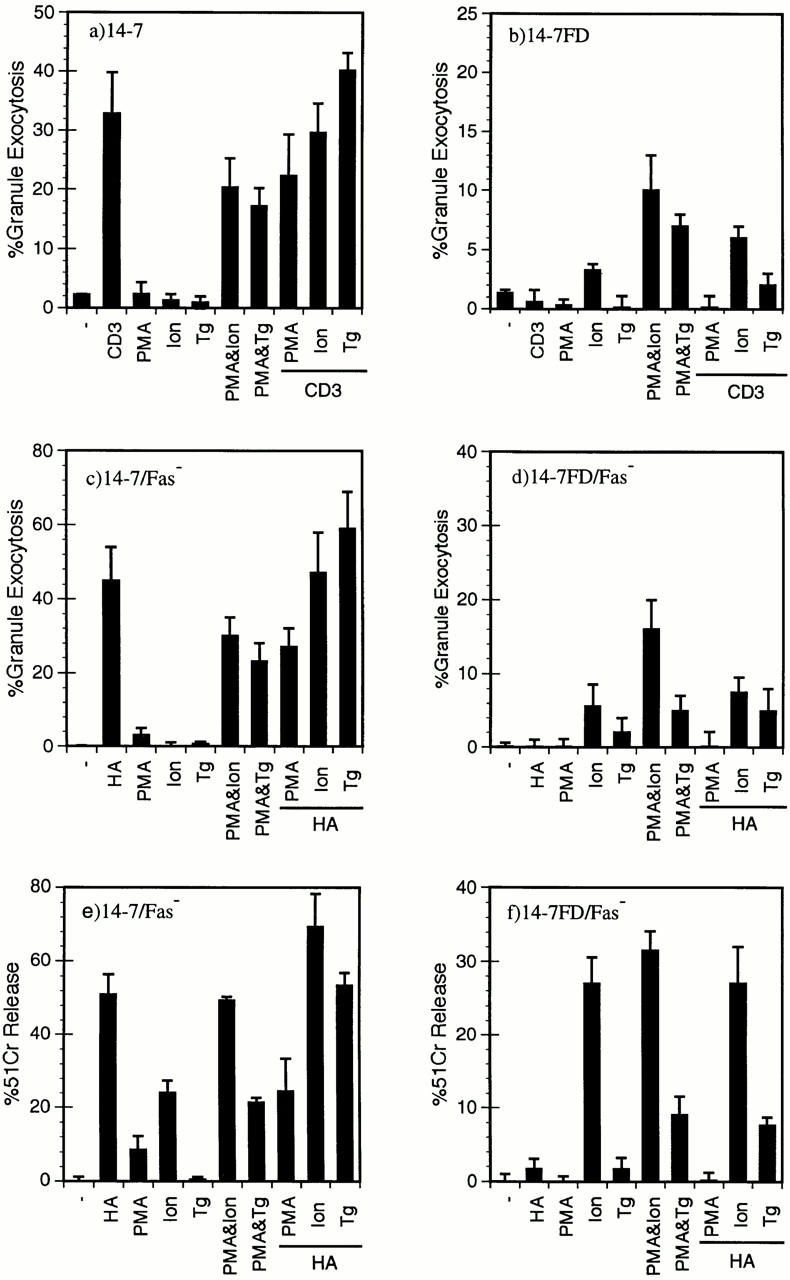
A large increase in [Ca2+]i is required for perforin/granule exocytosis killing. 14-7 (a) and 14-7FD (b) were stimulated with plate-bound anti-CD3ε, PMA (25 ng/ml), ionomycin (Ion; 1,000 nM), thapsigargin (Tg; 200 nM), or a combination of PMA and ionomycin, PMA and thapsigargin, or anti-CD3ε plus PMA, ionomycin, or thapsigargin, respectively, in the absence of target cells, and percentage of granule exocytosis was determined. 14-7 (c, e) and 14-7FD (d, f ) were incubated with HA529–537 peptide (0.01 μM)–pulsed or mock-treated L1210Fas− target cells. At the start of the assay PMA, ionomycin, or thapsigargin were added at the aforementioned concentrations. After 4 h, supernatants were collected and percentage of granule exocytosis (c and d) and of specific killing (e and f ) was determined.
To confirm that PMA plus ionomycin or thapsigargin triggered perforin killing as well as granule exocytosis in 14-7FD, we used the same combinations of ionomycin or thapsigargin with either PMA or HA529-pulsed Fas− targets in a cytotoxicity assay. As previously seen, 14-7 killed the HA529 pulsed Fas− target, but 14-7FD did not (Figs. 1 and 5, e and f ). Both PMA and ionomycin and PMA and thapsigargin activated both 14-7 and 14-7FD to kill via the perforin mechanism (Fig. 5, e and f ). Neither HA529 and ionomycin nor HA529 and thapsigargin significantly induced perforin killing over ionomycin or thapsigargin alone for 14-7FD (Fig. 5 f ). Additionally, ionomycin alone induced significant killing by both 14-7 and 14-7FD (Fig. 5, e and f ). In summary, these data suggest that an increase in [Ca2+]i is required for perforin killing.
Differential Ca2+ Signaling Activates Perforin Versus FasL/ Fas Killing.
Lastly, we wanted to determine if altering Ca2+ signaling in 14-7 with ionomycin or thapsigargin could differentially activate FasL/Fas versus perforin killing, in much the same way that TCR signaling selectively activates FasL/Fas killing in 14-7FD. First, we examined the effect of various concentrations of ionomycin or thapsigargin on Ca2+ mobilization and influx in 14-7 to see if we could find a concentration that mimicked the TCR-induced Ca2+ response in 14-7FD. Ionomycin gave a concentration-dependent decrease in the magnitude of the response (Fig. 6 a). Although the magnitude of the response decreases with lower amounts of ionomycin, the Ca2+ response maintained its biphasic appearance (Fig. 6 a). Thapsigargin, on the other hand, gives a quantitatively and qualitatively different response than ionomycin (Fig. 6 b). The maximal response with thapsigargin raises [Ca2+]i levels to 900 nM, whereas ionomycin gives a maximal [Ca2+]i response of ∼2000 nM (Fig. 6, a and b). The ionomycin [Ca2+]i response in 14-7 looks very similar to the biphasic TCR-triggered response (Figs. 2 a and 6 a). In comparison, the maximal thapsigargin [Ca2+]i response peaks at ∼800 nM and is maintained at >700 nM (Fig. 6 b). At lower concentrations, between 10 and 200 nM, the ionomycin [Ca2+]i response is quantitatively lower, but qualitatively similar, retaining a biphasic profile (Fig. 6 a). Thapsigargin, on the other hand, induces a quantitatively and qualitatively different response at lower concentrations than did ionomycin (Fig. 6 b). Between 1 and 20 nM, thapsigargin induces a Ca2+ response that did not have a fast initial increase, but induces a slow rise in [Ca2+]i (Fig. 6 b). Interestingly, this Ca2+ response looks remarkably like the TCR-triggered Ca2+ response in 14-7FD (Figs. 2 b and 6 b). These results demonstrate that ionomycin and thapsigargin can induce quantitatively and qualitatively different Ca2+ signals.
Figure 6.

Thapsigargin or ionomycin induction of perforin versus FasL/Fas killing. 14-7 was stimulated with 2,000, 1,000, 200, 100, 20, or 10 nM of ionomycin (a) or thapsigargin (b) and [Ca2+]i was measured by indo-1 fluorescence. 14-7 was stimulated with PMA (25 ng/ml) plus different doses of ionomycin or thapsigargin, and granzyme A granule exocytosis was determined (c and d, ▪), or 14-7 was stimulated with ionomycin or thapsigargin in the absence of PMA and percentage of specific killing of the L1210Fas+ target was determined (c and d, ○).
We next wanted to determine whether these different Ca2+ signals that had been generated with ionomycin and thapsigargin have functional consequences in regulating perforin or FasL/Fas killing. In other words, can we use ionomycin or thapsigargin to make 14-7 act like 14-7FD with regard to CTL effector function? Using ionomycin or thapsigargin at various concentrations, we assayed for granule exocytosis and lysis of Fas+ targets. At concentrations up to 100 nM, thapsigargin induces FasL/Fas killing in the absence of granule exocytosis, but ionomycin does not (Fig. 6, c and d). The concentrations of thapsigargin that selectively induced FasL/Fas killing corresponds exactly with those that give a [Ca2+]i response that resembles the 14-7FD TCR-triggered Ca2+ response (Figs. 2, b, and 6, b and d). At higher concentrations of thapsigargin, both perforin/ granule exocytosis and FasL/Fas killing are induced. In contrast, ionomycin always induced both types of killing (Fig. 6 d). These data demonstrated that we could make 14-7 behave like 14-7FD by stimulation with low amounts of thapsigargin.
Fig. 7, a and b, demonstrates that the selective blocking of perforin cytolysis using nifedipine seen for clone 14-7 (Fig. 4, a and c) can be extended to additional CD8+ CTL clones, 11-1 and 14-13. The inability of low concentrations of ionomycin to trigger both granule exocytosis (Fig. 7 c) and FasL/Fas cytolysis (Fig. 7 e) also holds for clones 11-1 and 14-13. Low concentrations of thapsigargin trigger cytolysis (Fig. 7 f ) in the absence of significant granule exocytosis (Fig. 7 d). That this cytolysis is FasL/Fas mediated is demonstrated by inhibition seen in the presence of anti-FasL antibody (Fig. 7 f ). In summary, these data demonstrate that quantitatively and qualitatively distinct Ca2+ signals can differentially activate FasL/Fas versus perforin cytotoxicity.
Figure 7.
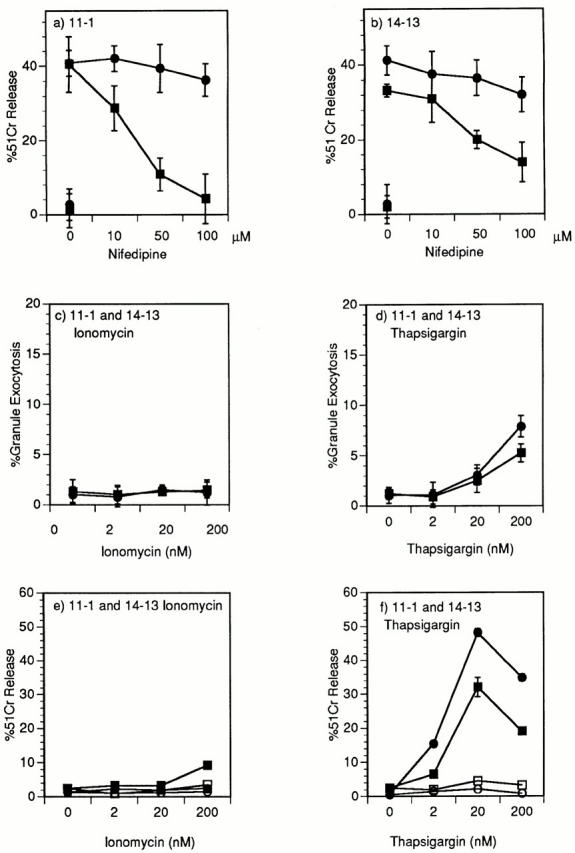
Demonstration in two additional CD8+ CTL clones of nifidipene inhibition of perforin cytolysis and thapsigargin induction of cytolysis that is inhibited by anti-FasL. Clone 11-1 (a) and 14-13 (b) were pretreated for 40 min with nifedipine at various concentrations before the start of a 51Cr–release assay. The CTLs were incubated with 0.10 μM HA204–212 (a) or 0.01 μM nucleoprotein 147–155 (b) peptide–pulsed L1210Fas− targets (▪) or L1210Fas+ targets (•) in a 51Cr–release assay (a and b). Clones 11-1(▪) and 14-13(•) were stimulated with PMA (25 ng/ ml) plus different doses of ionomycin (c) or thapsigargin (d) and granzyme A granule exocytosis was determined. Clones 11-1(▪) and 14-13(•) were stimulated with 200, 20, or 2 nM of ionomycin (e) or thapsigargin (f) and percentage of specific 51Cr–release of the L1210Fas+ target determined in the absence (▪, •) or presence (□, ○) of anti-FasL antibody.
Discussion
We have previously reported that distinct TCR signal transduction pathways activate perforin versus FasL/Fas killing (45, 46). This study demonstrated that soluble anti-CD3ε worked as effectively as Ag/MHC to trigger perforin killing by 14-7 (Fig. 1 a). This is most likely due to the fact that L1210 expresses an FcR (64), which allows a close physical proximity between the CTL and the target. Cell to cell contact is required for the lethal hit as plate-bound anti-CD3ε was ineffective in triggering killing (Fig. 1), although it efficiently initiated granule exocytosis (Fig. 1 b). In comparison, soluble or plate-bound anti-CD3ε or Ag/MHC effectively induced FasL/Fas killing (Fig. 1 c), leading us to believe that the physical interaction between the CTL and the target is not as critical for FasL/Fas killing as it is for perforin killing. In either case, anti-CD3ε mimicked the effect of Ag/MHC in our 14-7/14-7FD system.
To further dissect the TCR signaling differences between 14-7 and 14-7FD, we examined Ca2+ signaling in the two clones. It was apparent that the TCR-triggered Ca2+ signal in 14-7FD was qualitatively and quantitatively different from that of 14-7 (Fig. 2, a and b). 14-7 had a typical biphasic response, characterized by a large transient increase followed by a sustained plateau phase (Fig. 2 a). In contrast, 14-7FD did not have a biphasic response, but had a small (50–100 nM), sustained increase (Fig. 2 b). Additionally the resting level of [Ca2+]i in 14-7FD was consistently higher than that in 14-7, ranging anywhere from 80 to 300 nM (Figs. 2 and 3). To determine if the slow increase in [Ca2+]i in 14-7FD was coming from intracellular stores, extracellular influx, or both we examined the effect of treating the CTL with EGTA before or after TCR triggering. Adding EGTA before TCR engagement in 14-7 revealed that the large transient [Ca2+]i increase is made up of both intracellular (20%) and extracellular (80%) Ca2+ (Fig. 2 c), whereas the plateau phase is made up of almost entirely extracellular Ca2+ influx (Fig. 2 e). Adding EGTA before TCR stimulation almost completely inhibited the Ca2+ response in 14-7FD (Fig. 2 d), whereas adding EGTA after stimulation revealed that a large component of the Ca2+ response was mediated by extracellular Ca2+ influx (Fig. 2 f ). The 50–100-nM Ca2+ response in 14-7FD is near the limits of sensitivity of our instrument, and a small but functional intracellular response such as a “Ca2+ puff ” from the ER as has been described by M. Berridge (65) can not be ruled out, especially considering that the Ca2+ response in 14-7 stimulated with low doses of thapsigargin looks very similar to the TCR response in 14-7FD (Fig. 6 b). Nevertheless, the qualitatively and quantitatively distinct Ca2+ signaling patterns in 14-7FD make it a unique CTL for examining the role Ca2+ plays in activating and regulating CTL effector functions.
We next wanted to determine if the Ca2+ signaling defect in 14-7FD was at the level of activating intracellular Ca2+ release or if there was a defect in the Ca2+ release–activated Ca2+ influx pathway as has been described in mutant Jurkat cells (66, 67). Using the Ca2+ ATPase ER pump inhibitor thapsigargin, in the presence of EGTA demonstrated that 14-7FD had a functional intracellular store of Ca2+ (Fig. 3 a). Additionally, it could be demonstrated that release of Ca2+ from this pool activated the Ca2+ influx pathway (Fig. 3 b). These results suggest that the signaling defect lies upstream of ER Ca2+ release along the pathway between TCR stimulation, phospholipase C activation, and inositol triphosphate production and binding.
Although it is apparent that differential TCR signaling is occurring in 14-7FD, as of yet we do not know the reason why. 14-7FD has equivalent levels of TCR, CD45, CD8α/β, CD3ε, zeta, Lck, Fyn, ZAP70, PLCγ1, and MAPK (Esser, M.T., unpublished observations), but we have not yet tested the TCR-triggered activity of these known signaling intermediates. One possible explanation for the signaling defect in 14-7FD is that the higher resting level of [Ca2+]i modulates the activity of early TCR signaling kinases and phosphatases, preventing “full activation”. Another explanation is that a key signaling event such as zeta chain phosphorylation or Fyn kinase activation is not occurring in 14-7FD. From other studies, we know that differential tyrosine kinase activity occurs after TCR engagement in 14-7FD as compared with 14-7 (Esser, M.T., unpublished observations). How these early tyrosine kinase signaling events regulate perforin versus FasL/Fas killing is not known.
Since FasL/Fas killing had originally been reported to be Ca2+ independent, we used our 14-7/14-7FD CTL to test the intracellular Ca2+ release and Ca2+ influx requirements for perforin/granule exocytosis and FasL/Fas killing. The L type Ca2+ channel antagonist verapamil had no effect on perforin killing as had been seen by Gray et al. (68) or FasL/Fas killing (data not shown). Interestingly, the L type antagonist nifedipine inhibited perforin killing but not FasL/Fas killing (Fig. 4). Our results confirm the findings by Sitkovsky et al. that extracellular Ca2+ is required for granule exocytosis (41) and extend these observations by characterizing the T type Ca2+ channel antagonists NDGA and SK&F-96365, both of which inhibited perforin killing. The extracellular Ca2+ requirement for granule exocytosis is most likely due to the fact that the large initial increase in [Ca2+]i mediates the cytoskeletal changes required for granule exocytosis is comprised of 70–80% extracellular Ca2+ (Fig. 2 c). These cytoskeletal changes occur at ∼200 nM, can occur in the presence of CsA (69), and may explain why perforin killing is CsA insensitive (46). We also found that extracellular Ca2+ is required for the induction of FasL expression (Fig. 4 d ) as well as FasL/Fas killing (Fig. 4 c). These results extend the observations of Vignaux, who first showed that Ca2+ is required for the induction of FasL, but once expressed, FasL/Fas killing is Ca2+ independent (32). From Fig. 4 d it appears that higher concentrations of the antagonists are required to inhibit killing than are required to inhibit expression of FasL. We do not find this surprising in light of our findings and those of others, that neither the level of FasL or Fas correlate with the levels of killing (20, 70, 71). Our data suggests that the initial peak of TCR-stimulated increase in [Ca2+]i is required for cytoskeletal changes and perforin/granule exocytosis, whereas the sustained influx is sufficient for calcineurin activation, NFATc translocation to the nucleus, and FasL gene transcription.
Since PMA and ionomycin induced granzyme A exocytosis in 14-7FD (Fig. 5), we wanted to determine if replacing the increase in [Ca2+]i (via ionomycin or thapsigargin) in the presence of a TCR signal could induce perforin/ granule exocytosis. Using Ag/MHC or anti-CD3ε plus ionomycin or thapsigargin did not restored perforin/granule exocytosis killing in 14-7FD, suggesting that in addition to a strong Ca2+ signal other signaling events lacking in 14-7FD are required to induce perforin/granule exocytosis killing (Fig. 5). Since we could restore perforin-mediated cytotoxicity with PMA and ionomycin or thapsigargin we asked if we could selectively induce FasL/Fas killing without triggering perforin killing. Low doses of thapsigargin induced FasL/Fas killing without triggering perforin/ granule exocytosis in 14-7 (Fig. 6 d). Concentrations of thapsigargin which selectively activated FasL/Fas killing were qualitatively different in the time course and extent to which they increased [Ca2+]i than were higher concentrations that activated perforin killing. At lower concentrations, thapsigargin induced a slow sustained increase in [Ca2+]i that looked very similar to the TCR-stimulated changes in [Ca2+]i in 14-7FD (Figs. 2 b and 6 b). These results suggest that qualitatively and quantitatively distinct Ca2+ signals can differentially activate CTL effector functions.
In addition to the quantitative and qualitative Ca2+ signaling differences in 14-7FD, there could also be spatial and temporal differences in Ca2+ signaling. Studies performed by Gray et al. revealed that following Ag/MHC engagement, Ca2+ mobilization from intracellular stores occurred at a site distal to target cell contact and was transient (39), whereas extracellular Ca2+ influx occurred proximal to the T cell–target interface, was prolonged, and oscillated with a periodicity of 3–4 min (39). These early studies demonstrate the need to examine the magnitude, duration, and subcellular localization of the Ca2+ response in 14-7 and 14-7FD. These studies should provide new understanding into the mechanisms of Ca2+ regulated cytotoxicity and gene transcription.
In summary, our present findings demonstrate that signaling via the TCR is a dynamic process, allowing perforin/granule exocytosis and FasL/Fas killing to be independently activated. A large transient increase in [Ca2+]i is required for perforin killing, whereas sustained Ca2+ influx appears to be both necessary and sufficient for induction of FasL/Fas killing. This differential Ca2+ signaling allows selective activation of FasL/Fas versus perforin killing and may represent a mechanism that lymphocytes use to selectively activate different effector functions, proliferation, or death. By modulating these TCR signal transduction pathways, the immune system could selectively execute (perforin) or downregulate (FasL/Fas) a cellular immune response.
Acknowledgments
We thank Dr. David Lynch (Immunex, Seattle, WA) for the generous gift of Fas.Fc fusion protein.
This work was supported in part by the Beirne B. Carter Center for Immunology Research and grants from the U.S. Public Health Service (to T.J. Braciale), by funds from U.S. Public Health Service Training Grant T32 CA-09109 (M.T. Esser).
Footnotes
1 Abbreviations used in this paper: BLT, N-α-benzyloxycarbonyl-l-lysine thiobenzyl; [Ca2+]i, intracellular Ca2+ concentration; CsA, cyclosporin A; CTL, cytolytic T cell; ER, endoplasmic reticulum; FasL, fas ligand; FD, factor dependent; HA, hemagglutinin; NBCS, newborn calf serum; NDGA, nordihydroguaiaretic acid; NFAT, nuclear factor of activated T cells.
References
- 1.Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell–mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 2.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 3.Kagi D, Hengartner H. Different roles for cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr Opin Immunol. 1996;8:472–477. doi: 10.1016/s0952-7915(96)80033-1. [DOI] [PubMed] [Google Scholar]
- 4.Podack ER. Execution and suicide: cytotoxic lymphocytes enforce Draconian laws through separate molecular pathways. Curr Opin Immunol. 1995;7:11–16. doi: 10.1016/0952-7915(95)80023-9. [DOI] [PubMed] [Google Scholar]
- 5.Clark WR, Walsh CM, Glass AA, Huang MT, Ahmed R, Matloubian M. Cell-mediated cytotoxicity in perforin-less mice. Int Rev Immunol. 1995;13:1–14. doi: 10.3109/08830189509061734. [DOI] [PubMed] [Google Scholar]
- 6.Berke G. The CTL's kiss of death. Cell. 1995;81:9–12. doi: 10.1016/0092-8674(95)90365-8. [DOI] [PubMed] [Google Scholar]
- 7.Liu CC, Persechini PM, Young JD. Perforin and lymphocyte-mediated cytolysis. Immunol Rev. 1995;146:145–175. doi: 10.1111/j.1600-065x.1995.tb00688.x. [DOI] [PubMed] [Google Scholar]
- 8.Henkart PA. Lymphocyte-mediated cytotoxicity: two pathways and multiple effector molecules. Immunity. 1994;1:343–346. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 9.Zychlinsky A, Zheng LM, Liu CC, Young JD. Cytolytic lymphocytes induce both apoptosis and necrosis in target cells. J Immunol. 1991;146:393–400. [PubMed] [Google Scholar]
- 10.Yagita H, Nakata M, Kawasaki A, Shinkai Y, Okumura K. Role of perforin in lymphocyte-mediated cytolysis. Adv Immunol. 1992;51:215–242. doi: 10.1016/s0065-2776(08)60488-5. [DOI] [PubMed] [Google Scholar]
- 11.Martz, E. 1993. Overview of CTL-target adhesion and other critical events in the cytotoxic mechanism. In Cytotoxic Cells: Recognition, Effector Function, Generation, and Methods. M. Sitkovsky and P. Henkart, editors. Birkhauser, Boston, MA. 9–48.
- 12.Russell JH. Activation-induced death of mature T cells in the regulation of immune responses. Curr Opin Immunol. 1995;7:382–388. doi: 10.1016/0952-7915(95)80114-6. [DOI] [PubMed] [Google Scholar]
- 13.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 14.Lynch DH, Ramsdell F, Alderson MR. Fas and FasL in the homeostatic regulation of immune responses. Immunol Today. 1995;16:569–574. doi: 10.1016/0167-5699(95)80079-4. [DOI] [PubMed] [Google Scholar]
- 15.van Parijs L, Abbas AK. Role of Fas-mediated cell death in the regulation of immune responses. Curr Opin Immunol. 1996;8:355–361. doi: 10.1016/s0952-7915(96)80125-7. [DOI] [PubMed] [Google Scholar]
- 16.Rieux-Laucat F. Mutations in Fas associated with a human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–1349. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 17.Rouvier E, Luciani MF, Golstein P. Fas involvement in Ca2+-independent T cell–mediated cytotoxicity. J Exp Med. 1993;177:195–200. doi: 10.1084/jem.177.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kojima H, Shinohara N, Hanaoka S, Someya-Shirota Y, Takagaki Y, Ohno H, Saito T, Katayama T, Yagita H, Okumura K, et al. Two distinct pathways of specific killing revealed by perforin mutant cytotoxic T lymphocytes. Immunity. 1994;1:357–364. doi: 10.1016/1074-7613(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 19.Ramsdell F, Seaman MS, Miller RE, Picha KS, Kennedy MK, Lynch DH. Differential ability of Th1 and Th2 T cells to express Fas ligand and to undergo activation-induced cell death. Int Immunol. 1994;6:1545–1553. doi: 10.1093/intimm/6.10.1545. [DOI] [PubMed] [Google Scholar]
- 20.Esser M, Dinglasan R, Krishnamurthy B, Gullo C, Graham M, Braciale V. IL-2 induces Fas ligand/Fas (CD95L/CD95) cytotoxicity in CD8+ and CD4+T lymphocyte clones. J Immunol. 1997;158:5612–5618. [PubMed] [Google Scholar]
- 21.Qian D, Weiss A. T cell antigen receptor signal transduction. Curr Opin Cell Biol. 1997;9:205–212. doi: 10.1016/s0955-0674(97)80064-6. [DOI] [PubMed] [Google Scholar]
- 22.Shaw AS, Dustin ML. Making the T cell receptor go the distance: a topological view of T cell activation. Immunity. 1997;6:361–369. doi: 10.1016/s1074-7613(00)80279-4. [DOI] [PubMed] [Google Scholar]
- 23.Ward SG, June CH, Olive D. PI 3-kinase: a pivotal pathway in T-cell activation? . Immunol Today. 1996;17:187–197. doi: 10.1016/0167-5699(96)80618-9. [DOI] [PubMed] [Google Scholar]
- 24.Izquierdo Pastor, M., K. Reif, and D. Cantrell. The regulation and function of p21ras during T-cell activation and growth. Immunol Today. 1995;16:159–164. doi: 10.1016/0167-5699(95)80134-0. [DOI] [PubMed] [Google Scholar]
- 25.Cardenas ME, Heitman J. Role of calcium in T-lymphocyte activation. Adv Second Messenger Phosphoprotein Res. 1995;30:281–298. doi: 10.1016/s1040-7952(05)80011-4. [DOI] [PubMed] [Google Scholar]
- 26.Lewis RS, Cahalan MD. Potassium and calcium channels in lymphocytes. Annu Rev Immunol. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- 27.Premack BA, Gardner P. Signal transduction by T-cell receptors: mobilization of Ca and regulation of Ca-dependent effector molecules. Am J Physiol. 1992;263:C1119–C1140. doi: 10.1152/ajpcell.1992.263.6.C1119. [DOI] [PubMed] [Google Scholar]
- 28.Crabtree GR, Clipstone NA. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- 29.Baeuerle PA, Henkel T. Function and activation of NF-κ B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 30.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 31.Rao A. NF-ATp: a transcription factor required for the co-ordinate induction of several cytokine genes. Immunol Today. 1994;15:274–281. doi: 10.1016/0167-5699(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 32.Vignaux F, Vivier E, Malissen B, Depraetere V, Nagata S, Golstein P. TCR/CD3 coupling to Fas-based cytotoxicity. J Exp Med. 1995;181:781–786. doi: 10.1084/jem.181.2.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anel A, Simon AK, Auphan N, Buferne M, Boyer C, Golstein P, Schmitt-Verhulst AM. Two signaling pathways can lead to Fas ligand expression in CD8+cytotoxic T lymphocyte clones. Eur J Immunol. 1995;25:3381–3387. doi: 10.1002/eji.1830251227. [DOI] [PubMed] [Google Scholar]
- 34.Eischen CM, Williams BL, Zhang WG, Samelson LE, Lynch DH, Abraham RT, Leibson PJ. Zap-70 tyrosine kinase is required for the up-regulation of Fas ligand in activation-induced T cell apoptosis. J Immunol. 1997;159:1135–1139. [PubMed] [Google Scholar]
- 35.Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- 37.Gray LS, Gnarra JR, Engelhard VH. Demonstration of a calcium influx in cytolytic T lymphocytes in response to target cell binding. J Immunol. 1987;138:63–69. [PubMed] [Google Scholar]
- 38.Engelhard VH, Gnarra JR, Sullivan J, Mandell GL, Gray LS. Early events in target-cell lysis by cytotoxic T cells. Ann NY Acad Sci. 1988;532:303–313. doi: 10.1111/j.1749-6632.1988.tb36348.x. [DOI] [PubMed] [Google Scholar]
- 39.Gray LS, Gnarra JR, Sullivan JA, Mandell GL, Engelhard VH. Spatial and temporal characteristics of the increase in intracellular Ca2+induced in cytotoxic T lymphocytes by cellular antigen. J Immunol. 1988;141:2424–2430. [PubMed] [Google Scholar]
- 40.Haverstick DM, Engelhard VH, Gray LS. Three intracellular signals for cytotoxic T lymphocyte–mediated killing. Independent roles for protein kinase C, Ca2+ influx, and Ca2+release from internal stores. J Immunol. 1991;146:3306–3313. [PubMed] [Google Scholar]
- 41.Takayama H, Sitkovsky MV. Antigen receptor-regulated exocytosis in cytotoxic T lymphocytes. J Exp Med. 1987;166:725–743. doi: 10.1084/jem.166.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romeo C, Amiot M, Seed B. Sequence requirements for induction of cytolysis by the T cell antigen/Fc receptor zeta chain. Cell. 1992;68:889–897. doi: 10.1016/0092-8674(92)90032-8. [DOI] [PubMed] [Google Scholar]
- 43.Ostergaard H, Clark WR. The role of Ca2+in activation of mature cytotoxic T lymphocytes for lysis. J Immunol. 1987;139:3573–3579. [PubMed] [Google Scholar]
- 44.Young JD, Clark WR, Liu CC, Cohn ZA. A calcium- and perforin-independent pathway of killing mediated by murine cytolytic lymphocytes. J Exp Med. 1987;166:1894–1899. doi: 10.1084/jem.166.6.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao W, Tykodi SS, Esser MT, Braciale VL, Braciale TJ. Partial activation of CD8+T cells by a self-derived peptide. Nature. 1995;378:295–298. doi: 10.1038/378295a0. [DOI] [PubMed] [Google Scholar]
- 46.Esser MT, Krishnamurthy B, Braciale VL. Distinct T cell receptor signaling requirements for perforin- or FasL-mediated cytotoxicity. J Exp Med. 1996;183:1697–1706. doi: 10.1084/jem.183.4.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walsh CM, Glass AA, Chiu V, Clark WR. The role of the Fas lytic pathway in a perforin-less CTL hybridoma. J Immunol. 1994;153:2506–2514. [PubMed] [Google Scholar]
- 48.Braciale TJ, Andrew ME, Braciale VL. Heterogeneity and specificity of cloned lines of influenza-virus specific cytotoxic T lymphocytes. J Exp Med. 1981;153:910–923. doi: 10.1084/jem.153.4.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thastrup O, Dawson A. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions. 1989;27:17–23. doi: 10.1007/BF02222186. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Estacion M, Mordan LJ. Ca2+influx via T-type channels modulates PDGF-induced replication of mouse fibroblasts. Am J Physiol. 1993;265:C1239–C1246. doi: 10.1152/ajpcell.1993.265.5.C1239. [DOI] [PubMed] [Google Scholar]
- 51.Chung SC, McDonald TV, Gardner P. Inhibition by SK&F 96365 of Ca2+current, IL-2 production and activation in T lymphocytes. Br J Pharmacol. 1994;113:861–868. doi: 10.1111/j.1476-5381.1994.tb17072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature. 1983;302:790–794. doi: 10.1038/302790a0. [DOI] [PubMed] [Google Scholar]
- 53.Leo O, Foo M, Sachs D, Samelson L, Bluestone J. Identification of a monoclonal antibody specific for a mutine T3 polypeptide. Proc Natl Acad Sci USA. 1987;84:1374–1378. doi: 10.1073/pnas.84.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Braciale TJ, Sweetser MT, Morrison LA, Kittlesen DJ, Braciale VL. Class I major histocompatibility complex–restricted cytolytic T lymphocytes recognize a limited number of sites on the influenza hemagglutinin. Proc Natl Acad Sci USA. 1989;86:277–281. doi: 10.1073/pnas.86.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ju ST, Cui H, Panka DJ, Ettinger R, Marshak-Rothstein A. Participation of target Fas protein in apoptosis pathway induced by CD4+ Th1 and CD8+cytotoxic T cells. Proc Natl Acad Sci USA. 1994;91:4185–4189. doi: 10.1073/pnas.91.10.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Isaaz S, Baetz K, Olsen K, Podack E, Griffiths G. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a nongranule pathway. Eur J Immunol. 1995;25:1071–1079. doi: 10.1002/eji.1830250432. [DOI] [PubMed] [Google Scholar]
- 57.Nisbet-Brown E, Cheung RK, Lee JW, Gelfand EW. Antigen-dependent increase in cytosolic free calcium in specific human T-lymphocyte clones. Nature. 1985;316:545–547. doi: 10.1038/316545a0. [DOI] [PubMed] [Google Scholar]
- 58.Imboden JB, Weiss A, Stobo JD. The antigen receptor on a human T cell line initiates activation by increasing cytoplasmic free calcium. J Immunol. 1985;134:663–665. [PubMed] [Google Scholar]
- 59.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haverstick DM, Gray LS. Increased intracellular Ca2+ induces Ca2+influx in human T lymphocytes. Mol Biol Cell. 1993;4:173–184. doi: 10.1091/mbc.4.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Premack BA, McDonald TV, Gardner P. Activation of Ca2+ current in Jurkat T cells following the depletion of Ca2+ stores by microsomal Ca(2+)-ATPase inhibitors. J Immunol. 1994;152:5226–5240. [PubMed] [Google Scholar]
- 62.Gouy H, Cefai D. Ca2+ influx in human T lymphocytes is induced independently of inositol phosphate production by mobilization of intracellular Ca2+ stores: a study with the endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin. Eur J Immunol. 1990;20:2269–2275. doi: 10.1002/eji.1830201016. [DOI] [PubMed] [Google Scholar]
- 63.Liu J. FK506 and cyclosporin, molecular probes for studying intracellular signal transduction. Immunol Today. 1993;14:290–295. doi: 10.1016/0167-5699(93)90048-P. [DOI] [PubMed] [Google Scholar]
- 64.Anel A, Gamen S, Alava MA, Schmitt-Verhulst AM, Pineiro A, Naval J. Inhibition of CPP32-like proteases prevents granzyme B- and Fas-, but not granzyme A-based cytotoxicity exerted by CTL clones. J Immunol. 1997;158:1999–2006. [PubMed] [Google Scholar]
- 65.Berridge M. The AM and FM of calcium signalling. Nature. 1997;386:759–760. doi: 10.1038/386759a0. [DOI] [PubMed] [Google Scholar]
- 66.Serafini AT, Lewis RS, Clipstone NA, Bram RJ, Fanger C, Fiering S, Herzenberg LA, Crabtree GR. Isolation of mutant T lymphocytes with defects in capacitative calcium entry. Immunity. 1995;3:239–250. doi: 10.1016/1074-7613(95)90093-4. [DOI] [PubMed] [Google Scholar]
- 67.Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gray LS, Gnarra JR, Russell JH, Engelhard VH. The role of K+ in the regulation of the increase in intracellular Ca2+mediated by the T lymphocyte antigen receptor. Cell. 1987;50:119–127. doi: 10.1016/0092-8674(87)90668-4. [DOI] [PubMed] [Google Scholar]
- 69.Negulescu PA, Krasieva TB, Khan A, Kerschbaum HH, Cahalan MD. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity. 1996;4:421–430. doi: 10.1016/s1074-7613(00)80409-4. [DOI] [PubMed] [Google Scholar]
- 70.Glass A, Walsh CM, Lynch DH, Clark WR. Regulation of the Fas lytic pathway in cloned CTL. J Immunol. 1996;156:3638–3644. [PubMed] [Google Scholar]
- 71.Wang R, Rogers AM, Rush BJ, Russell JH. Induction of sensitivity to activation-induced death in primary CD4+ cells: a role for interleukin-2 in the negative regulation of responses by mature CD4+T cells. Eur J Immunol. 1996;26:2263–2270. doi: 10.1002/eji.1830260944. [DOI] [PubMed] [Google Scholar]