

Abstract
Murine phosphatidyl choline (PtC)–specific B cells in normal mice belong exclusively to the B-1 subset. Analysis of anti-PtC (VH12 and VH12/Vκ4) transgenic (Tg) mice indicates that exclusion from B-0 (also known as B-2) occurs after immunoglobulin gene rearrangement. This predicts that PtC-specific B-0 cells are generated, but subsequently eliminated by either apoptosis or differentiation to B-1. To investigate the mechanism of exclusion, PtC-specific B cell differentiation was examined in mice expressing the X-linked immunodeficiency (xid) mutation. xid mice lack functional Bruton's tyrosine kinase (Btk), a component of the B cell receptor signal transduction pathway, and are deficient in B-1 cell development. We find in C57BL/ 6.xid mice that VH12 pre-BII cell selection is normal and that PtC-specific B cells undergo modest clonal expansion. However, the majority of splenic PtC-specific B cells in anti-PtC Tg/xid mice are B-0, rather than B-1 as in their non-xid counterparts. These data indicate that PtC-specific B-0 cell generation precedes segregation as predicted, and that Btk function is required for efficient segregation to B-1. Since xid mice exhibit defective B cell differentiation, not programmed cell death, these data are most consistent with an inability of PtC-specific B-0 cells to convert to B-1 and a single B cell lineage.
At least two B cell subsets, B-1 and B-2, are present in the mouse periphery (1–4). One of the most intriguing aspects of these subsets is that they exhibit different repertoires (5), presumably reflecting different functions in the immune system. B-2 cells appear to be responsible for T cell-dependent responses to exogenous antigens and for generating memory B cells (4). In contrast, the B-1 subset harbors a high frequency of cells with specificities to self-antigens such as phosphatidyl choline (PtC),1 immunoglobulin (rheumatoid factor), DNA, as well as specificities to common bacterial carbohydrate antigens like phosphorylcholine (6–10), and may be involved in T cell-independent responses to common environmental antigens.
The distinct B-1 and B-2 repertoires are the consequence of different selective pressures (11, 12), but the nature of these differences is not known. Critical to understanding how B-1 and B-2 repertoires arise is the relationship between the cells of these subsets. The more commonly held view (the lineage hypothesis) is that B-1 and B-2 cells derive from stem cells committed to one or the other subset before Ig gene rearrangement, thereby constituting two separate lineages (13–15). An alternative hypothesis (the induced differentiation hypothesis) is that they derive from a single lineage, and that an uncommitted B cell is induced to differentiate to a B-1 cell after Ig gene rearrangement by interaction with antigen, probably T cell-independent antigens in the absence of T cell help (16, 17). Since by this hypothesis the majority of splenic B cells are uncommitted, they are referred to as B-0 cells. Thus, the B-2 cells of the lineage hypothesis and the B-0 cells of the induced differentiation hypothesis are equivalent and referred to here as B-0. Each hypothesis predicts a different means of arriving at distinct B-1 and B-0 repertoires.
We have focused on the differentiation of B cells specific for the common membrane phospholipid, PtC, as a means to understand the bases for the repertoire differences between B-1 and B-0 cells. In normal mice, PtC-specific B cells appear to be exclusively B-1 (6, 12, 18, 19). They are driven to undergo considerable clonal expansion from birth (20, 21), and in normal adults eventually account for 2–10% of the peritoneal B-1 repertoire (6). Many anti-PtC B cells express VH12 and Vκ4 rearrangements (11, 22). The VH12 H chain is restricted to CDR3s of 10 amino acids with Gly in the fourth position and Tyr, encoded by the first codon of JH1, in the fifth position (referred to as 10/G4) (11). Selection for B cells of the appropriate gene rearrangements occurs at two stages during B cell development. The first results in the elimination of most non-10/ G4 VH12 pre-BII cells and in the enrichment of 10/G4 VH12 pre-B cells (23). This is probably due to positive selection of 10/G4 pre-BII cells, and an absence of positive selection of non-10/G4 pre-BII cells resulting in the death of the latter. The second stage of selection is at the B cell stage where 10/G4 VH12 B cells that express the appropriate Vκ4 L chain and bind PtC undergo antigen-driven clonal expansion in response to some ubiquitous environmental or self-antigen (24).
To understand the basis for the segregation of PtC-specific B cells to the B-1 subset, we have generated anti-PtC transgenic (Tg) mice using the VH12 and Vκ4 gene rearrangements of the anti-PtC lymphoma CH27 (21, 22). Mice with either the VH12 or the VH12 and Vκ4 Tgs continue to segregate PtC-specific B cells to the B-1 subset, indicating that the mechanism of segregation operates after Ig gene rearrangement (21). We predict that B-0 cells expressing the combination of VH12 and Vκ4 are generated in these Tg mice. Therefore, to achieve segregation, either PtC-specific B-0 cells are activated and induced to become B-1 cells (induced differentiation hypothesis), thereby depleting the B-0 subset of this specificity, or they are stimulated by antigen to undergo apoptosis (lineage hypothesis).
To distinguish between these possibilities, we have combined the VH12 and Vκ4 Tgs (21) with the xid mutation (25, 26). This mutation ablates function of Bruton's tyrosine kinase (Btk) (27–30), resulting in deficiencies in B cell differentiation (31–33) and responsiveness to T cell-independent type II antigens (26). This mutation also blocks development of a detectable peritoneal B-1 population in CBA/N mice (34). Analysis of the cellular defect indicates that signaling through surface IgM (sIgM) fails to drive xid B cells into cell cycle (35, 36). xid B cells do not appear to be deficient in induction of programmed cell death (36, 37). We demonstrate here that in xid mice, VH12 pre-B cell selection is normal and that PtC-specific B cells undergo modest clonal expansion. However, combining the VH12 and Vκ4 Tgs with xid, we demonstrate that the majority of splenic PtC-specific B cells fail to segregate to the B-1 subset and instead have a B-0 phenotype. These data argue that B-0 cells are an intermediate step in B-1 cell differentiation, consistent with the induced differentiation hypothesis and a single lineage of B cells.
Materials and Methods
Mice.
VH12 (6-1) and Vκ4 Tg mice have been described previously (21) and are maintained in our colony at the University of North Carolina by backcrossing to C.B17 mice. Offspring are identified by PCR analysis of tail genomic DNA as previously described (21). Double (dbl) Tg mice are generated by the intercrossing of 6-1 and Vκ4 Tg mice. C57BL/6.xid (B6/xid) mice were obtained from the National Institutes of Health (Bethesda, MD) and bred separately with the VH12 and Vκ4 Tg mice to obtain VH12 and Vκ4 Tg-xid/xid mice. These mice were intercrossed to obtain VH12/Vκ4 dbl Tg/xid mice.
PCR Analysis of VH12 CDR3 Sequences.
Extraction of genomic DNA, PCR, and cloning of VH12 rearrangements was performed as previously described (20). Clones of VH12 rearrangements were chosen randomly for sequence analysis.
Immunofluorescence and Flow Cytometry.
The antibodies used against IgMa (DS-1), IgMb (AF6-78), B220 (RA3-6B2), and CD5 (53-7.3) were obtained from PharMingen (San Diego, CA), and were fluoresceinated, biotinylated, or conjugated to PE. CD23 (B3B4) and CD43 (S7) were gifts of Dr. Tom Waldschmidt (University of Iowa, Iowa City, IA). In three-color experiments, directly fluoresceinated and PE-conjugated antibodies were combined with a biotinylated antibody revealed with streptavidin-RED670 (GIBCO BRL, Gaithersburg, MD). To detect PtC-binding B cells, liposomes encapsulating carboxyfluorescein were used as previously described (21).
To detect membrane expression of various molecules, single cell suspensions were prepared in HBSS (without Ca2+, Mg2+, or phenol red) containing 0.1% sodium azide and 1.0% FCS (buffer). Cells were incubated with previously determined optimal amounts of antibody in 25–50 μl buffer for 20 min, after which they were washed three times with buffer and incubated with second step reagents. After washing as before, the cells were analyzed using a FACScan (Becton Dickinson, Mountain View, CA) with acquisition and analysis software from Cytomation, Inc. (Ft. Collins, CO). All data represent cells that fall within the lymphocyte gate determined by forward and 90° light scatter. 1–5 × 104 cells were analyzed. All contour plots are 5% probability.
Results
The VH12 Repertoire in C57BL/6 xid/xid (B6/xid) Mice.
As a result of positive selection at the pre-B and B cell stages, mature VH12 B cells in the spleen and peritoneum are almost exclusively 10/G4 (24). To assess the ability of xid mice to select VH12 pre-B and B cells, we determined the ratio of the number of productive (P) VH12 rearrangements to the number of nonproductive (NP) VH12 rearrangements (P/NP). In the absence of selection, the P/NP for any V gene should be ∼2.3 (23, 38, 39), assuming that rearrangement is random. P/NP values <2.3 would indicate selection against cells expressing a P VH12 rearrangement, while a P/NP value >2.3 would indicate clonal expansion of B cells with P VH12 rearrangements. However, to independently assess selection at the pre-B and B cell stages, we calculated both 10/G4 and non-10/G4 P/NP values. Non-10/G4 P rearrangements include all VH12 rearrangements that are not 10/G4. Since most non-10/G4 rearrangements are eliminated at the pre-BII cell stage (23), the non-10/G4 P/NP is a measure of pre-BII cell selection. In the absence of selection, all but a small number of P rearrangements will be non-10/G4, and thus, the expected value for the non-10/G4 P/NP is essentially 2.3 (23). The 10/G4 P/NP is a measure of the extent of clonal expansion of PtC-specific B cells, since clonal expansion of PtC-specific B cells will increase the number of 10/G4 rearrangements in a given population with a negligible effect on the number of NP VH12 rearrangements. The 10/G4 P/NP is measured to be <0.05 in the absence of clonal expansion (23).
To measure the 10/G4 and non-10/G4 P/NPs in xid mice, VH12-D-JH1 rearrangements from genomic DNA of bone marrow, spleen, and peritoneal cells were PCR amplified. The amplified DNA was cloned, and clones were randomly selected for sequencing. As shown in Fig. 1 and Table 1, the majority of P rearrangements in the bone marrow are non-10/G4 (5 out of 8) and the non-10/G4 P/NP is 0.18, not different from that of wild-type mice, and significantly lower than the expected value of ∼2.3. This value is equally low in spleen and peritoneum. Thus, as in wild-type mice (24), non-10/G4 rearrangements contribute little to the central and peripheral repertoires, indicating that pre-B cell selection is unaffected by the xid mutation.
Figure 1.
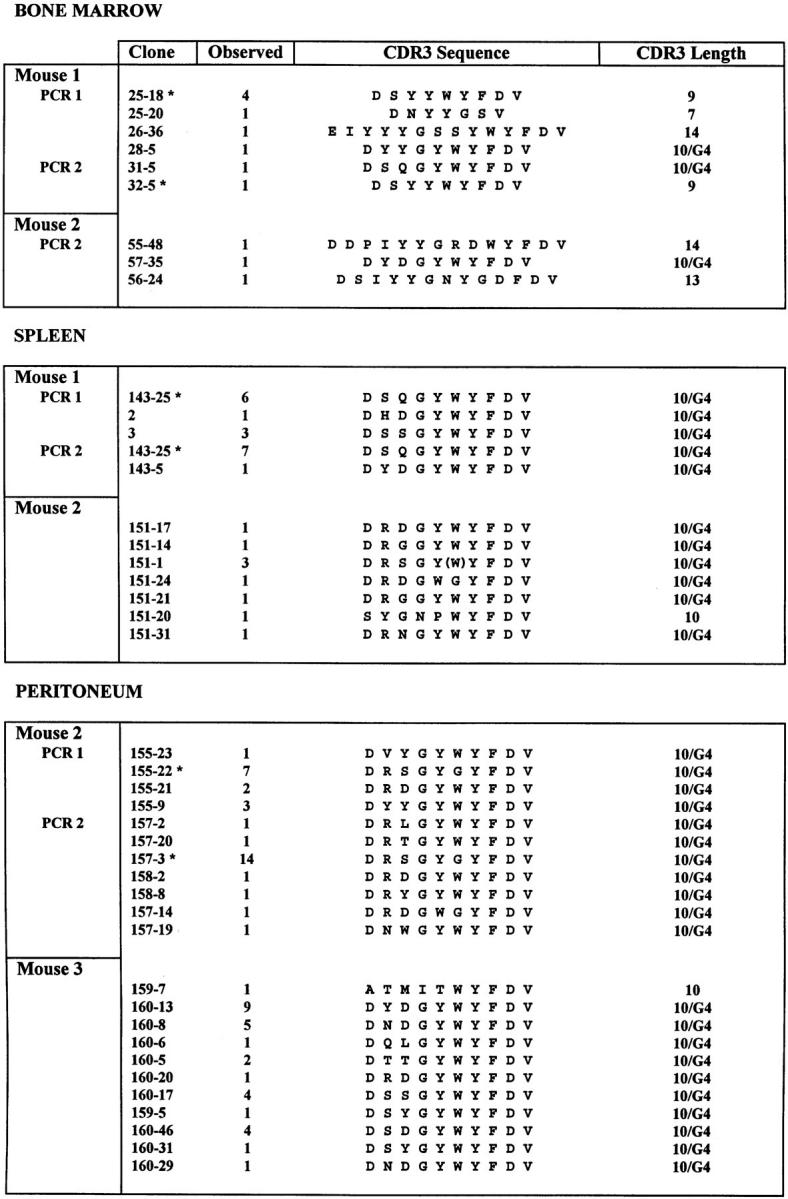
CDR3 amino acid sequences of VH12-D-JH1 rearrangements from bone marrow, spleen, and peritoneum of B6/xid mice. DNA was extracted from tissues of individual mice and either one or two separate PCR reactions were performed as indicated. PCR products were cloned and transfected, and individual colonies were chosen randomly for sequence analysis. The number of colonies that gave the identical nucleotide sequence is given in the second column. Asterisks indicate the clones originating from separate PCR that have identical nucleic acid sequences across the VH12-D-JH1 junction. The final column indicates the number of CDR3 amino acids encoded by each rearrangement. Whether or not a Gly is encoded at position 4 of CDR3 (G4) is indicated for clones with a length of 10 amino acids.
Table 1.
VH12 P/NP Values for B6/xid Bone Marrow, Spleen, and Peritoneum*
| P (10/G4)‡ | NP | P/NP | OVERALL P/NP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10/G4 | Non-10/G4 | 10/G4 | Non-10/G4 | |||||||||
| Bone marrow | ||||||||||||
| Mouse 1 | 5 (2) | 6 | 0.33 | 0.50 | 0.11 (0.33)§ | 0.18 (0.22) | ||||||
| Mouse 2 | 3 (1) | 22 | 0.05 | 0.091 | ||||||||
| Spleen | ||||||||||||
| Mouse 1 | 4 (4) | 19 | 0.21 | 0 | 0.29 (1.88) | 0.029 (0.13) | ||||||
| Mouse 2 | 7 (6) | 15 | 0.40 | 0.067 | ||||||||
| Peritoneum | ||||||||||||
| Mouse 2 | 10 (10) | 2 | 5 | 0 | 10 (32) | 0.50‖ | ||||||
| Mouse 3 | 11 (10) | 0 | ||||||||||
All P rearrangement data were taken from Fig. 1.
The total number of P rearrangements is followed in parentheses by the number of P rearrangements that are 10/G4.
The 10/G4 and non-10/G4 P/NP values for wild-type mice are given in parentheses and are taken from studies by Ye et al. (23, 24).
No non-10/G4 P rearrangements were observed in the peritonea of wild-type mice and therefore a non-10/G4 P/NP could not be calculated.
Clonal expansion of 10/G4 B cells occurs in xid mice, but to a lesser extent than in wild-type mice. Three out of eight P VH12 rearrangements in the bone marrow are 10/ G4, and the 10/G4 P/NP (0.11) is one-third that seen in wild-type mice (Fig. 1 and Table 1). However, the frequency of 10/G4 P rearrangements is higher than that observed in the absence of selection and clonal expansion (0 of 22 in μMT mice) (23), indicating that 10/G4 P rearrangements are enriched in xid bone marrow. Enrichment is greater in the spleen and particularly the peritoneum, where almost every P rearrangement is 10/G4. The 10/G4 P/NP is smaller than that observed in wild-type mice, reflecting a more modest clonal expansion. Nevertheless, clonal expansion of 10/G4 VH12 peripheral B cells, presumably because they are PtC-specific, occurs in B6/xid mice.
Ig Tg/xid Mice Can Develop B-1 Cells.
To determine the effect of the xid mutation on B-1 cell development, we combined the xid mutation with the VH12 and Vκ4 transgenes, since these transgenes exert a strong positive influence on B-1 cell development (21). We find that 6-1/xid mice have approximately one-fourth the number of splenic B cells found in 6-1 mice (Table 2). Thus, as expected (40), the xid mutation limits B cell development in this Tg model. B cells were stained for expression of a number of cell surface markers that distinguish B-1 and B-0 cells. B-1 cells are defined as CD43+, CD23−, B220lo, IgMhi, and they may or may not express CD5. B-0 cells, on the other hand, are defined as CD43−, CD23+, B220hi, and IgMlo, and they never express CD5. Shown in Fig. 2, A and B, and as we have previously published (21), a large fraction of the splenic B cells in 6-1 and dbl Tg mice are B-1 cells, i.e., CD5+, CD23−, B220lo, and IgMhi. A smaller number of B cells in these Tg mice are B-0, i.e., CD5−, CD23+, B220hi, and IgMlo. These cells in 6-1 mice are predominantly PtCneg (see below).
Table 2.
B Cell Subpopulations in the Spleen and Peritoneum of VH12 Tg Mice
| Genotype |
n
|
Total lymphs* | % IgM+ B cells | Total B cells‡ | Percentage of B cells that are | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % CD5+ | Total CD5+‡ | % CD23+ | Total CD23+‡ | % CD43+ | Total CD43+‡ | % Lipo+ | Total Lipo+‡ | |||||||||||||||||
| Spleen cells | ||||||||||||||||||||||||
| 6-1 | 9 | 4.8 ± | 24.9 ± | 11.6 ± | 46.2 ± | 5.2 ± | 35.4 ± | 4.1 ± | 35.9 ± | 4.6 ± | 40.1 ± | 5.4 ± | ||||||||||||
| 1.64 | 7.91 | 6.38 | 18.8 | 4.6 | 9.9 | 1.3 | 14.3 | 4.2 | 14.5 | 4.8 | ||||||||||||||
| 6-1/xid | 7 | 2.7 ± | 9.2 ± | 2.7 ± | 12.6 ± | 0.3 ± | 64.9 ± | 1.8 ± | 11.1 ± | 0.3 ± | 8.8 ± | 0.2 ± | ||||||||||||
| 0.6 | 3.2 | 0.9 | 7.9 | 0.2 | 4.4 | 0.5 | 3.8 | 0.1 | 2.0 | 0.1 | ||||||||||||||
| dbl Tg/xid | 5 | 4.3 ± | 25.3 ± | 11.1 ± | 11.3 ± | 1.1 ± | 60.9 ± | 6.5 ± | 8.4 ± | 0.8 ± | 84.3 ± | 9.3 ± | ||||||||||||
| 1.6 | 7.7 | 5.3 | 3.0 | 0.4 | 11.1 | 2.9 | 5.0 | 0.4 | 8.7 | 4.4 | ||||||||||||||
| Normal | 8 | 10.4 ± | 56.7 ± | 60.5 ± | 10.4 ± | 5.9 ± | 79.9 ± | 40.3 ± | 9.3 ± | 5.8 ± | 0.3 ± | 0.2 ± | ||||||||||||
| 5.7 | 5.5 | 35.3 | 4.4 | 3.8 | 10.0 | 24.7 | 2.2 | 4.3 | 0.3 | 0.39 | ||||||||||||||
| B6.xid | 7 | 5.8 ± | 48.7 ± | 31.2 ± | 13.3 ± | 2.9 ± | 73.4 ± | 23.6 ± | 13.5 ± | 3.4 ± | 0.6 ± | 0.1 ± | ||||||||||||
| 3.1 | 14.0 | 21.4 | 14.6 | 2.5 | 8.3 | 16.2 | 9.1 | 1.9 | 1.2 | 0.1 | ||||||||||||||
| Peritoneal cells | ||||||||||||||||||||||||
| 6-1 | 6 | 3.1 ± | 86.3 ± | 27.1 ± | 83.7 ± | 22.4 ± | 0.7 ± | 0.2 ± | 90.8 ± | 25.0 ± | 94.3 ± | 21.7 ± | ||||||||||||
| 0.9 | 5.0 | 8.9 | 5.9 | 6.4 | 0.4 | 0.1 | 6.4 | 9.7 | 7.3 | 8.0 | ||||||||||||||
| 6-1/xid | 5 | 0.6 ± | 31.2 ± | 2.1 ± | 54.7 ± | 0.9 ± | 10.1 ± | 0.3 ± | 73.4 ± | 1.9 ± | 91.3 ± | 1.7 ± | ||||||||||||
| 0.3 | 16.7 | 1.8 | 19.5 | 0.8 | 7.4 | 0.3 | 25.4 | 1.8 | 2.8 | 1.6 | ||||||||||||||
| dbl Tg/xid | 3 | 0.9 ± | 65.7 ± | 6.1 ± | 71.4 ± | 4.3 ± | 11.4 ± | 0.7 ± | 57.3 ± | 2.5 ± | 96.3 ± | 6.0 ± | ||||||||||||
| 0.4 | 11.9 | 3.2 | 10.5 | 2.3 | 6.3 | 0.7 | 27.2 | 0.9 | 5.4 | 3.3 | ||||||||||||||
| Normal | 6 | 2.2 ± | 78.0 ± | 17.1 ± | 43.4 ± | 7.7 ± | 12.6 ± | 2.3 ± | 69.9 ± | 10.0 ± | 4.3 ± | 0.8 ± | ||||||||||||
| 0.4 | 4.3 | 3.0 | 17.3 | 3.5 | 4.2 | 0.8 | 14.6 | 4.3 | 1.4 | 0.3 | ||||||||||||||
| B6.xid | 6 | 1.2 ± | 55.8 ± | 6.9 ± | 14.3 ± | 0.9 ± | 42.9 ± | 2.9 ± | 15.3 ± | 1.0 ± | 3.6 ± | 0.4 ± | ||||||||||||
| 0.5 | 10.6 | 3.7 | 13.5 | 0.6 | 12.3 | 1.9 | 3.8 | 0.5 | 1.6 | 0.2 | ||||||||||||||
×107 for spleen cells and ×106 for peritoneal cells.
×106 for spleen cells and ×105 for peritoneal cells.
Standard deviation.
Figure 2.
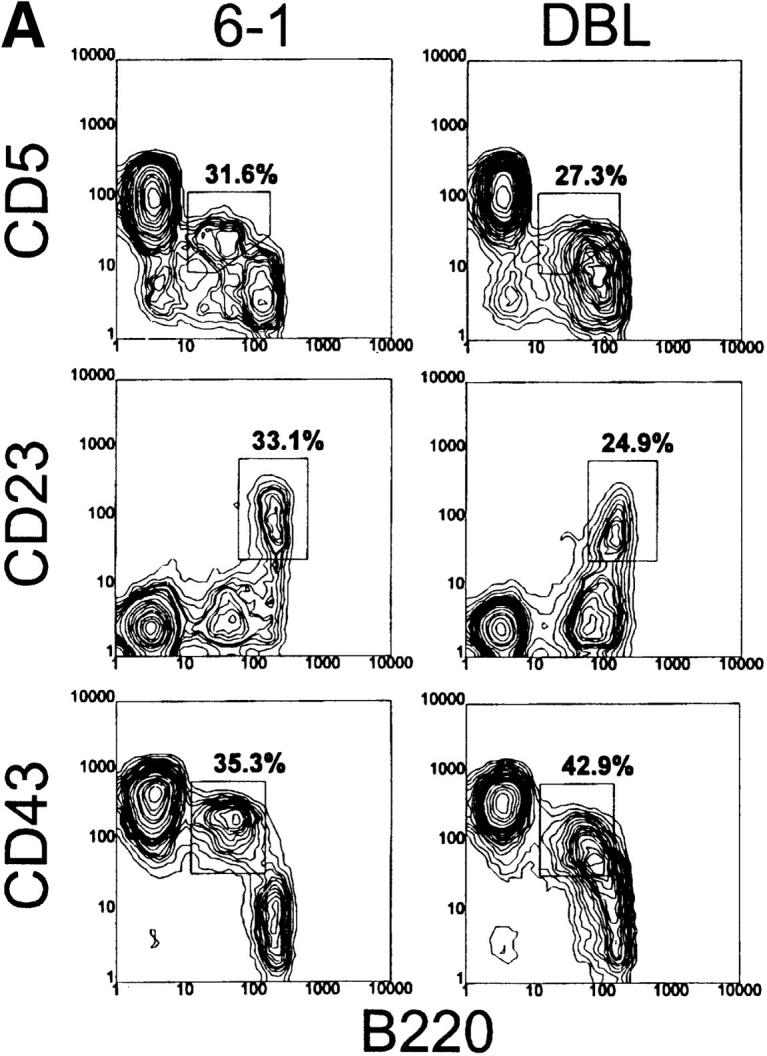


Analysis of spleen cells from 6-1 and dbl Tg mice. Spleen cells from 6-1 and dbl Tg mice were stained with FITC-conjugated anti-B220, PE-conjugated anti-IgM, and the biotinylated antibody indicated on the left (A and B). For C, cells were stained with carboxyfluorescein-encapsulating liposomes, PE-conjugated anti-B220, and the biotinylated reagent on the left. The biotinylated antibodies were visualized with PE-conjugated streptavidin. 10,000–20,000 cells were analyzed. Percentages are of the B220+ cells.
In contrast to their non-xid counterparts, B-1 cells in the spleens of Tg/xid mice are consistently a minority. Only 15–25% are B-1 cells with a CD43+, CD23−, B220lo, and IgMhi phenotype (Table 2 and Fig. 3). Most of these cells in 6-1/xid mice are CD5−, whereas most in dbl Tg/xid are CD5+ (follow the B220lo population in Fig. 3 A). The majority of the B cells in these Tg/xid mice have a B-0 phenotype, as they are CD5−, CD43−, CD23+, B220hi, and IgMlo. Cells with the B-1 phenotype are also evident in the peritoneum (CD43+, CD23−, B220lo, and IgMhi; Fig. 4, A and B), but their number is considerably lower than that in 6-1 and wild-type mice (Table 2). Thus, xid mice can generate B-1 cells, but the positive pressure exerted on B-1 cell development by the VH12 and Vκ4 transgenes is partially negated by the xid mutation.
Figure 3.



Analysis of spleen cells from VH12 Tg/xid mice. Spleen cells from 6-1/xid and dbl Tg/xid mice were stained as described in the legend to Fig. 2. As a control and for reference, analysis of 6-1 mice is included. Percentages are of B220+ cells.
Figure 4.



Analysis of peritoneal cells from VH12 Tg mice. Cells were stained as described in Fig. 2. Percentages are of B220+ cells.
Segregation of PtC-specific B Cells to the B-1 Subset in xid Spleens Is Impaired.
To determine the effect of xid on the segregation of PtC-specific B cells, we compared the phenotype of PtC-specific B cells from Tg/xid and Tg non-xid mice. PtC-specific cells can be detected by flow cytometry using as a probe fluorescein-encapsulating liposomes that contain PtC as a membrane constituent (6). As previously published (21), all of the B-1 cells of 6-1 and dbl Tg mice bind liposomes (CD23−, CD43+ cells in Fig. 2 C).
Dbl Tg mice have a liposome-binding population that is intermediate in staining (liposomeint) (Fig. 2 C). These cells have a B-0 phenotype since they are CD23+, IgMlo, and B220hi (Fig. 2 C, and follow the CD23+ population in Fig. 2, A and B). The liposomeint B-0 cells account for 10–30% of the B cells in a dbl Tg spleen. These cells were not previously detected probably because the liposome probe used in this study is much brighter than that used in our earlier study (21). Since these cells likely express the VH12 and Vκ4 Tgs, we conclude that they are liposomeint by virtue of the fact that they are IgMlo. We speculate that they constitute the PtC-specific B-0 cells predicted to undergo either programmed cell death or conversion to B-1 to achieve segregation of this specificity (21). A smaller number of these cells (5.6% in Fig. 2 C) appear to be present in 6-1 mice.
The xid mutation affects the relative proportions of the PtC-specific B-0 and B-1 subsets. The number of liposome binding cells in 6-1/xid is ∼10% of that in 6-1 mice. Although their number is small, the liposome-binding B cells appear to be equally divided between the liposomeint B-0 (CD23+, CD43−) and liposomebri B-1 (CD23−, CD43+) populations (Fig. 3 C). The presence of two subsets of PtC-specific B cells in xid mice is more apparent in dbl Tg/ xid mice, since the addition of the Vκ4 transgene increases the number of B cells present in the xid spleen fourfold (Table 2 and Fig. 3 C). In these mice, nearly two-thirds of the PtC-specific B cells are liposomeint B-0 cells, as they are CD43− and CD23+ (Fig. 3 C) and B220hi and IgMlo (follow the CD23+ population in Fig. 3, A and B). The remainder of the PtC-specific B cells are liposomebri B-1 cells that are CD43+ and CD23− (Fig. 3 C), and B220lo and IgMhi (follow the CD43+ population in Fig. 3, A and B). Since most IgMhi B cells in these mice are CD5+ (Fig. 3, A and B), most of the liposomebri B-1 cells are CD5+. This dramatic shift in the distribution of liposome binding cells from B-1 to B-0 in xid mice indicates that the xid mutation impairs the segregation of PtC-specific B cells to the B-1 subset.
Segregation of PtC-specific B Cells to the B-1 Subset Is Largely Intact in xid Peritonea.
The majority of B-1 cells in normal adult mice are located in the peritoneum. B6/xid mice have ∼40% of the number of B cells in the peritoneum, and on average 15% are CD5+ and CD43+, a much smaller percentage than in wild-type mice (Table 2). In addition, 2–4% of peritoneal B cells bind liposomes. Thus, xid mice are not devoid of peritoneal B-1 cells or of liposome-binding B cells. This provides an explanation for our observation of enrichment of 10/G4 rearrangements in the peritonea of B6/xid mice (Fig. 1 and Table 1). This is different from previous reports that xid mice lack peritoneal B-1 cells. This is likely due to background genetic differences between B6/xid mice and the CBA/N mice used in previous studies (34).
Essentially all peritoneal B cells in 6-1 mice are liposomebri B-1 cells (Fig. 4 C and Table 2), as previously reported (21). They are CD23−, CD43+, B220lo, and IgMhi (Fig. 4, A and B). Approximately 80% of liposome binding cells are CD5+. In contrast to the spleen, nearly all of the peritoneal B cells in 6-1/xid and dbl Tg/xid mice are liposomebri B-1 cells (Fig. 4 C). The number of PtC-specific B cells in 6-1/ xid mice is almost 10% of that in 6-1 mice, whereas that in dbl Tg/xid mice is nearly 30% (Table 2). Thus, the combination of these two transgenes does not increase the number of PtC-specific B cells in the peritoneum as dramatically as it does in the spleen (3.5× versus 46×). The liposomebri cells in 6-1/xid and dbl Tg/xid mice are mostly CD23− (Fig. 4 C) and B220lo and IgMhi (Fig. 4, A and B). About half express CD5 (Fig. 4, A and B), but only about one-third express CD43, giving them a somewhat unusual phenotype relative to those in 6-1 mice. Thus, the peritoneum contains PtC-specific B-1 cells and few, if any, of the PtC-specific B-0 cells that predominate in the spleen.
Discussion
We demonstrate here that xid mice can generate PtC-specific B cells. Although their number is small in non-Tg xid mice, when they are provided the VH12 and Vκ4 Tgs, which encode anti-PtC antibodies (11) and exert strong selective pressure on B-1 cell generation and clonal expansion (21), their numbers increase substantially. However, two-thirds of the splenic PtC-specific B cells in 6-1/xid and dbl Tg/xid mice are B-0. There appears to be an equivalent but smaller B-0 population in dbl Tg non-xid mice. Thus, the VH12 and Vκ4 transgenes reveal a deficit in the ability of xid mice to segregate PtC-specific B cells to the B-1 subset, indicating a role for Btk in this process. That some PtC-specific B cells are B-1 indicates that either the mutant Btk has residual function, or that its function is compensated for by other signals, as may occur for others of its functions (31, 41). These data establish that PtC-specific B-0 cells are generated, and that segregation to B-1 is achieved by their subsequent elimination, consistent with our previous conclusion that PtC-specific B cells segregate to the B-1 subset by a mechanism operating after Ig gene rearrangement (21).
xid mice are also deficient in B-1 cell clonal expansion. The large number of the PtC-specific B-1 cells in 6-1 mice is due to clonal expansion (21); 6-1 mice are free to use the endogenous Vκ repertoire and the majority of the developing B cells will be PtCneg. Indeed, at birth only 4% of 6-1 splenic IgM+ cells are liposome binding. But clonal expansion is so significant that by day 6 >80% of the splenic IgM+ cells are liposome-binding B-1 cells. 6-1/xid mice generate B-1 cells, but the fact that their numbers remain small in adult mice indicates that these cells are unable to undergo significant clonal expansion. This is corroborated by the analysis of 10/G4 rearrangements in B6/xid mice; the 10/G4 P/NP in the spleens and peritonea of B6/xid mice, although high, is lower than in wild-type mice, indicating modest clonal expansion of PtC-specific B cells (Table 1). Thus, B-1 cell clonal expansion is impaired in xid mice, consistent with the known function of Btk in IgM-mediated signaling (35, 36). This defect is probably a major contributor to the absence of detectable numbers of B-1 cells in xid mice. Larger numbers of PtC-specific B-1 cells are seen in dbl Tg/xid mice, but this would not require clonal expansion, since most newly generated B cells will express both Tgs and bind PtC.
Segregation of B cells to the B-1 subset must be antigen driven, as PtCneg VH12- or Vκ4-expressing B cells are B-0 in 6-1 and Vκ4-only Tg mice (21). Although the identity of the antigen that drives segregation is not known, it is not likely that the number of cells in Tg/xid mice exceeds its availability; dbl Tg/xid have only 1.5 times the number of PtC-specific B cells as 6-1 mice, where segregation is intact, and 6-1/xid mice have only 4% the number (Table 2). Thus, the defect in segregation is likely to be related to the responsiveness of PtC-specific B cells to antigen, consistent with the known function of Btk in IgM mediated signal transduction. This leaves two possible mechanisms; either PtC-specific B-0 cells are eliminated by apoptosis after stimulation by antigen, or they convert to B-1 cells.
Signaling of programmed cell death through sIgM in xid B cells appears to be intact. Anti-IgM stimulation induces xid B cells to undergo apoptosis as it does wild type B cells (36). In addition, xid B cells are less resistant to radiation induced apoptosis (37), indicating that programmed cell death proceeds normally, but that these cells are deficient at transducing environmental signals to turn off the cell death program. Moreover, xid mice are not autoimmune, arguing that negative selection of autoreactive B cells is normal in these mice. In fact, the xid mutation prevents the development of autoimmunity in New Zealand black mice (42, 43). These findings cast doubt on apoptosis as the mechanism of segregation to B-1.
On the other hand, it is well established that the xid mutation affects B cell differentiation. xid mice are unable to respond to TI-2 antigens and have impaired responses to some T cell-dependent antigens, particularly at suboptimal doses (26), and they are unable to generate a normal number of peritoneal B-1 cells (34). Even naive B-0 cells appear to not differentiate fully, as they do not downregulate sIgM to wild type levels (44, 45). xid B cells, in cotransfer experiments with non-xid B cells, are at a competitive disadvantage in survival (32), and are similarly disadvantaged in xid heterozygous female mice (33). Studies of the biochemical defect in xid B cells indicate that xid B cells fail to enter cell cycle and differentiate upon anti-IgM signaling due to a deficiency in cyclin induction (46). Therefore, we propose that PtC-specific B-0 cells in xid mice bind antigen, but are unable to differentiate in response to it, and consequently remain B-0. This would place B-0 as an intermediate stage in B-1 cell differentiation.
This mechanism is consistent with the induced differentiation hypothesis in which commitment to the B-1 lineage occurs after Ig gene rearrangement, is dependent on the specificity of the B cell, and follows antigen stimulation. It is incompatible with the two-lineage model of B cell development, which requires commitment to one cell lineage or the other before Ig gene rearrangement, and which does not accommodate movement of cells from one subset to the other. Thus, these data argue for a single B cell lineage that can give rise to B-1 cells upon activation. An implication of this conclusion is that the B-1 repertoire includes only antigen-selected B cells, and that at no time is it equivalent to the B-0 repertoire.
The induced differentiation hypothesis offers an explanation for the existence of the liposomeint B-0 population in non-xid dbl Tg mice. We suggest that these cells are newly generated B-0 cells that have not yet converted to B-1, either because the large number of PtC-specific B cells produced by the bone marrow reveals a population that is normally too small to detect, or because the number produced exceeds the available antigen, leaving PtC specific B-0 cells unconverted for an extended length of time. Consistent with these possibilities is that there are fewer liposomeint cells in 6-1 mice (Fig. 2 C) in which the production rate of PtC-specific B cells is lower than in dbl Tg mice because of the available use of multiple Vκ genes.
The proposal that B-1 cells derive from B-0 cells after antigen stimulation is consistent with the findings of Ying-zi et al. (47), demonstrating that stimulation of B-0 cells in vitro with anti-μ and IL-6 induces a B-1 cell phenotype (i.e., CD5+, CD23−, IgDlo, B220lo). It is also consistent with in vivo studies indicating that the formation of the B-1 subset is dependent on the action of coreceptors that deliver activation signals in concert with surface IgM, as well as on the presence of an intact signal transduction pathway from surface IgM. For example, mice deficient in the IgM coreceptor CD19 lack B-1 cells, whereas mice that overexpress CD19 have large numbers of B-1 cells (48–50). Likewise, mice deficient in the complement receptor CD21 lack B-1 cells (51). Both these receptors amplify the IgM-mediated signal delivered by antigen and play important costimulatory roles in responses to T cell-dependent antigens (48, 49, 51, 52). Deficiencies in the cytoplasmic protein kinases Vav and protein kinase C–βI/II (PKC-βI/II) also result in the absence of B-1 cells (53–55), and deficiency in phosphatase SHP-1 results in an excessive number of B-1 cells (56). Like Btk, these molecules are involved in signal transduction from IgM. Both Vav- and PKC–βI/II-deficient mice also exhibit impaired responses to antigen (53, 54), further evidence that signals initiated by IgM are essential for B-1 cell formation. Thus, the same signaling pathways used to respond to antigen are required for the generation of B-1 cells, consistent with an essential role for antigen in B-1 cell formation and the induced differentiation hypothesis. Based on our findings with xid mice, we predict that PtC-specific B cells in mice deficient for these receptor and signal transduction molecules will be similarly impaired in their ability to segregate PtC-specific B cells to B-1. Indeed, recent analysis indicates that PtC-specific B cells in dbl Tg, CD19-deficient mice are B-0 (Rickert, R., and S.H. Clarke, unpublished observation).
It is notable that PtC-specific B-0 cells are dominant in the spleen and lymph nodes (data not shown), but not in the peritoneum of dbl Tg/xid mice. This is most likely an indication that PtC-specific B cells home to the peritoneum only after differentiation to a B-1 cell. Since PtC-specific B cells in xid Tg mice are deficient in signal transduction, they remain in the spleen and lymph nodes as B-0 cells. The number of B cells in the peritoneum of dbl Tg/ xid mice is nearly as low as it is in 6-1/xid mice, suggesting that only differentiated B-1 cells are able to migrate to the peritoneum, consistent with this possibility. However, it cannot be excluded that peritoneal B cells are more able to overcome the deficit in signal transduction because of some unique aspect of the peritoneal microenvironment, such as antigen or cytokine availability. Such factors might overcome the absence of wild-type Btk, thereby permitting a greater degree of differentiation in the peritoneum than anywhere else. Regardless of the reason, the peritoneum is an environment in which segregation is largely intact in xid mice.
This analysis also indicates that pre-B cell selection resulting in the loss of most VH12 pre-BII cells and consequent enrichment for VH12 10/G4 rearrangements is normal in xid mice. This is in accord with the findings that pre-B cell proliferation and production is normal in xid mice (57), and that a defect is not observed until the IgM+ stage, as evidenced by the observation that xid B cells are at a survival disadvantage relative to non-xid B cells in xid/+ female mice (33). Thus, pre-B cell development is not measurably affected by the xid mutation, as others have noted previously (32, 33, 47), in contrast to Btk deficiencies in humans (58).
Acknowledgments
We are grateful to Suzanne McCray for her excellent technical assistance and to the members of our laboratory for their many helpful discussions and review of this manuscript. We also acknowledge the assistance of the Flow Cytometry Facility.
This work was supported by National Institutes of Health grants AI-29576 and AR-42573, by a research award from the Arthritis Foundation, and by the American Cancer Society grant IM-772.
Footnotes
1 Abbreviations used in this paper: Btk, Bruton's tyrosine kinase; dbl, double; NP, nonproductive; P, productive; PtC, phosphatidyl choline; sIgM, surface IgM; Tg, transgenic.
References
- 1.Herzenberg LA, Stall AM, Lalor PA, Sidman C, Moore WA, Parks D, Herzenberg LA. The Ly-1 B cell lineage. Immunol Rev. 1986;93:81–102. doi: 10.1111/j.1600-065x.1986.tb01503.x. [DOI] [PubMed] [Google Scholar]
- 2.Kipps TJ. The CD5 B cell. Adv Immunol. 1989;47:117–185. doi: 10.1016/s0065-2776(08)60663-x. [DOI] [PubMed] [Google Scholar]
- 3.Herzenberg, L.A., G. Haughton, and K. Rajewsky, editors. 1992. CD5 B cells in development and disease. Ann. NY Acad. Sci. Vol. 651:1–601.
- 4.Kantor AB. A new nomenclature for B cells. Immunol Today. 1991;12:388. doi: 10.1016/0167-5699(91)90135-G. [DOI] [PubMed] [Google Scholar]
- 5.Lalor PA, Morahan G. The peritoneal Ly-1 (CD5) B cell repertoire is unique among murine B cell repertoires. Eur J Immunol. 1990;20:485–492. doi: 10.1002/eji.1830200305. [DOI] [PubMed] [Google Scholar]
- 6.Mercolino TJ, Arnold LW, Hawkins LA, Haughton G. Normal mouse peritoneum contains a large number of Ly-1+(CD5) B cells that recognize phosphatidyl choline. Relationship to cells that secrete hemolytic antibody specific for autologous erythrocytes. J Exp Med. 1988;168:687–698. doi: 10.1084/jem.168.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masmoudi H, Mota-Santos S, Huetz F, Coutinho A, Casenave PA. All T15 Id-positive antibodies (but not the majority of VHT15+antibodies) are produced by peritoneal CD5 B lymphocytes. Int Immunol. 1990;2:515–520. doi: 10.1093/intimm/2.6.515. [DOI] [PubMed] [Google Scholar]
- 8.Hayakawa K, Hardy RR, Honda M, Herzenberg LA, Steinberg AD, Herzenberg LA. Ly-1 B cells: functionally distinct lymphocytes that secrete IgM autoantibodies. Proc Natl Acad Sci USA. 1984;81:2494–2498. doi: 10.1073/pnas.81.8.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardy RR, Hayakawa K, Shimizu M, Yamasaki K, Kishimoto T. Rheumatoid factor secretion from human Leu-1+ B cells. Science. 1987;236:81–83. doi: 10.1126/science.3105057. [DOI] [PubMed] [Google Scholar]
- 10.Forster I, Rajewsky K. Expansion and functional activity of Ly-1+ B cells upon transfer of peritoneal cells into allotype-congenic newborn mice. Eur J Immunol. 1987;17:521–528. doi: 10.1002/eji.1830170414. [DOI] [PubMed] [Google Scholar]
- 11.Pennell CA, Mercolino TJ, Grdina TA, Arnold LW, Haughton G, Clarke SH. Biased immunoglobulin variable region gene expression by Ly-1 B cells due to clonal selection. Eur J Immunol. 1989;19:1289–1295. doi: 10.1002/eji.1830190721. [DOI] [PubMed] [Google Scholar]
- 12.Carmack CE, Shinton SA, Hayakawa K, Hardy RR. Rearrangement and selection of VH11 in the Ly-1 B cell lineage. J Exp Med. 1990;172:371–374. doi: 10.1084/jem.172.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–1568. doi: 10.1084/jem.161.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy RR, Hayakawa K. A developmental switch in B lymphopoiesis. Proc Natl Acad Sci USA. 1991;88:11550–11554. doi: 10.1073/pnas.88.24.11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kantor AB, Herzenberg LA. Origin of murine B cell lineages. Annu Rev Immunol. 1993;11:501–538. doi: 10.1146/annurev.iy.11.040193.002441. [DOI] [PubMed] [Google Scholar]
- 16.Wortis HH. Surface markers, heavy chain sequences and B cell lineages. Int Rev Immunol. 1992;8:235–246. doi: 10.3109/08830189209055576. [DOI] [PubMed] [Google Scholar]
- 17.Haughton G, Arnold LW, Whitmore AC, Clarke SH. B1 cells are made, not born. Immunol Today. 1993;14:84–87. doi: 10.1016/0167-5699(93)90064-R. [DOI] [PubMed] [Google Scholar]
- 18.Hayakawa K, Hardy RR, Herzenberg LA. Peritoneal Ly-1 B cells: genetic control, autoantibody production, increased lambda light chain expression. Eur J Immunol. 1986;16:450–456. doi: 10.1002/eji.1830160423. [DOI] [PubMed] [Google Scholar]
- 19.Hardy RR, Carmack CE, Shinton SA, Riblet RJ, Hayakawa K. A single VH gene is utilized predominantly in anti-BrMRBC hybridomas derived from purified Ly-1 B cells. Definition of the VH11 family. J Immunol. 1989;142:3643–3651. [PubMed] [Google Scholar]
- 20.Clarke SH, McCray SK. VHCDR3-dependent positive selection of murine VH12-expressing B cells in the neonate. Eur J Immunol. 1993;23:3327–3334. doi: 10.1002/eji.1830231240. [DOI] [PubMed] [Google Scholar]
- 21.Arnold LW, Pennell CA, Suzanne, McCray K, Clarke SH. Development of B-1 cells: segregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J Exp Med. 1994;179:1585–1595. doi: 10.1084/jem.179.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pennell CA, Arnold LW, Haughton G, Clarke SH. Restricted Ig variable region gene expression among Ly-1+B cell lymphomas. J Immunol. 1988;141:2788–2796. [PubMed] [Google Scholar]
- 23.Ye J, McCray SK, Clarke SH. The transition of pre-BI to pre-BII cells is dependent on the structure of the m/surrogate L chain receptor. EMBO (Eur Mol Biol Organ) J. 1996;15:1524–1533. [PMC free article] [PubMed] [Google Scholar]
- 24.Ye J, McCray SK, Clarke SH. The majority of murine VH12-expressing B cells are excluded from the peripheral repertoire in adults. Eur J Immunol. 1995;25:2511–2521. doi: 10.1002/eji.1830250916. [DOI] [PubMed] [Google Scholar]
- 25.Scher I, Ahmed A, Steinberg DM, Steinberg AD, Paul WE. X-linked B lymphocyte defect in CBA/N mice. J Exp Med. 1975;141:788–803. [PMC free article] [PubMed] [Google Scholar]
- 26.Scher I. The CBA/N mouse strain: an experimental model illustrating the influence of the X-chromosome on immunity. Adv Immunol. 1982;33:1–71. doi: 10.1016/s0065-2776(08)60834-2. [DOI] [PubMed] [Google Scholar]
- 27.Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 28.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi J, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 29.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Muller S, Kantor AB, Herzenberg LA, Rosen FS, Sideras P. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 30.Kerner JD, Appleby MW, Mohr RN, Chien S, Rawlings DJ, Maliszewski CR, Witte ON, Perlmutter RM. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:301–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 31.Wortis HH, Burkly L, Hughes D, Roschelle S, Waneck G. Lack of mature B cells in nude mice with X-linked immune deficiency. J Exp Med. 1982;155:903–913. doi: 10.1084/jem.155.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sprent J, Bruce J. Physiology of B cells in mice with X-linked immunodeficiency (xid). III. Disappearance of xidB cells in double bone marrow chimeras. J Exp Med. 1984;160:711–723. doi: 10.1084/jem.160.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrester LM, Ansell JD, Micklem HS. Development of B lymphocytes in mice heterozygous for the X-linked immunodeficiency (xid) mutation. xidinhibits development of all splenic and lymph node B cells at a stage subsequent to their initial formation in bone marrow. J Exp Med. 1987;165:949–958. doi: 10.1084/jem.165.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayakawa K, Hardy RR, Parks DR, Herzenberg LA. The Ly-1 B cell subpopulation in normal, immunodeficient, and autoimmune mice. J Exp Med. 1983;157:202–218. doi: 10.1084/jem.157.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sieckmann DG, Scher I, Asofsky R, Mosier DE, Paul WE. Activation of mouse lymphocytes by anti-immunoglobulin. II. A thymus-independent response by a mature subset of B lymphocytes. J Exp Med. 1978;148:1628–1643. doi: 10.1084/jem.148.6.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson JS, Teutsch, Dong Z, Wortis HH. An essential role for tyrosine kinase in the regulation of Bruton's B-cell apoptosis. Proc Natl Acad Sci USA. 1996;93:10966–10971. doi: 10.1073/pnas.93.20.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodland RT, Schmidt MR, Riggs JE, Korsmeyer SJ, Lussier AM, Gravel KA. Radiation-induced apoptosis is differentially regulated in primary B cells from normal mice and mice with the CBA/N X-linked immunodeficiency. J Immunol. 1995;155:3453–3463. [PubMed] [Google Scholar]
- 38.Alt FW, Yancopoulos GD, Blackwell TK, Wood C, Thomas E, Boss M, Coffman R, Rosenberg N, Tonegawa S, Baltimore D. Ordered rearrangement of immunoglobulin heavy chain variable region segments. EMBO (Eur Mol Biol Organ) J. 1984;3:1209–1219. doi: 10.1002/j.1460-2075.1984.tb01955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loffert D, Ehlich A, Hardy RR, Zou YR, Muller W, Rajewsky K. Early B-cell development in mouse: insights from mutations introduced by gene targeting. Immunol Rev. 1994;137:135–153. doi: 10.1111/j.1600-065x.1994.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 40.Sprent J, Bruce J, Ron Y, Webb SR. Physiology of B cells in mice with X-linked immunodeficiency. I. Size, migratory properties, and turnover of the B cell pool. J Immunol. 1985;134:1442–1448. [PubMed] [Google Scholar]
- 41.Karagogeos D, Rosenberg N, Wortis HH. Early arrest of B cell development in nude, X-linked immune-deficient mice. Eur J Immunol. 1986;16:1125–1130. doi: 10.1002/eji.1830160916. [DOI] [PubMed] [Google Scholar]
- 42.Taurog JD, Raveche ES, Smathers PA, Glimcher LH, Huston DP, Hansen CT, Steinberg AD. T cell abnormalities in NZB mice occur independently of autoantibody production. J Exp Med. 1981;153:221–234. doi: 10.1084/jem.153.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinberg BJ, Smathers PA, Frederiksen K, Steinberg AD. Ability of the xid gene to prevent autoimmunity in (NZB × NZW)F1mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J Clin Invest. 1982;70:587–597. doi: 10.1172/JCI110651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scher I, Sharrow SO, Paul WE. X-linked B-lymphocyte defect in CBA/N mice. III. Abnormal development of B-lymphocyte populations defined by their density of surface immunoglobulin. J Exp Med. 1976;144:507–519. doi: 10.1084/jem.144.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardy RR, Hayakawa K, Haaijman J, Herzenberg LA. B cell subpopulations identified by two-color fluorescence analysis. Nature. 1982;297:589–591. doi: 10.1038/297589a0. [DOI] [PubMed] [Google Scholar]
- 46.Brorson K, Brunswick M, Ezhevsky S, Wei DG, Berg R, Scott D, Stein KE. xidaffects events leading to B cell cycle entry. J Immunol. 1997;159:135–143. [PubMed] [Google Scholar]
- 47.Ying-zi C, Rabin E, Wortis HH. Treatment of murine CD5−B cells with anti-Ig, but not LPS, induces surface CD5: two B-cell activation pathways. Int Immunol. 1991;3:467–476. doi: 10.1093/intimm/3.5.467. [DOI] [PubMed] [Google Scholar]
- 48.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell–dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 49.Engel P, Zhou L-J, Ord DC, Sato S, Koller B, Tedder TF. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 50.Sato S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B lymphocyte signaling thresholds critical for the development of B-1 lineage cells and autoimmunity. J Immunol. 1996;157:4371–4378. [PubMed] [Google Scholar]
- 51.Ahearn JM, Fischer MB, Croix D, Goerg S, Ma M, Xia J, Zhou X, Howard RG, Rothstein TL, Carroll MC. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4:251–262. doi: 10.1016/s1074-7613(00)80433-1. [DOI] [PubMed] [Google Scholar]
- 52.Sato S, Steeber DA, Tedder TF. The CD19 signal transduction molecule is a response regulator of B-lymphocyte differentiation. Proc Natl Acad Sci USA. 1995;92:11558–11562. doi: 10.1073/pnas.92.25.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang R, Alt FW, Davidson L, Orkin SH, Swat W. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the vav proto-oncogene. Nature. 1995;374:470–473. doi: 10.1038/374470a0. [DOI] [PubMed] [Google Scholar]
- 54.Fisher K-D, Zmuidzinas A, Gardner S, Barbacid M, Berstein A, Guidos C. Defective T-cell receptor signalling and positive selection of Vav-deficient CD4+ CD8+thymocytes. Nature. 1995;374:474–477. doi: 10.1038/374474a0. [DOI] [PubMed] [Google Scholar]
- 55.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Immunodeficiency in protein kinase Cβ-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- 56.Sidman CL, Shultz LD, Hardy RR, Hayakawa K, Herzenberg LA. Production of immunoglobulin isotypes by Ly-1+B cells in viable motheaten and normal mice. Science. 1986;232:1423–1425. doi: 10.1126/science.3487115. [DOI] [PubMed] [Google Scholar]
- 57.Reid GK, Osmond DG. B lymphocyte production in the bone marrow of mice with x-linked immunodeficiency (xid) . J Immunol. 1985;135:2299–2302. [PubMed] [Google Scholar]
- 58.Rosen FS, Cooper MD, Wedgwood RJP. The primary immunodeficiencies. N Engl J Med. 1984;311:235–242. doi: 10.1056/NEJM198407263110406. [DOI] [PubMed] [Google Scholar]