

Abstract
We have previously shown that a tyrosine to leucine replacement in the transmembrane region of T cell receptor (TCR)-β results in a deficient induction of CD95-L and apoptosis upon TCR triggering in a transfected T cell line. By contrast, interleukin (IL)-2 production and the expression of CD25 and CD69 were normally induced. Since the mutation in TCR-β also resulted in impaired association of CD3-ζ, it was proposed that this chain is specifically required for the induction of apoptosis. We now show that the deficient induction of CD95-L and apoptosis does not derive from a general lower production of second messengers, since intracellular Ca2+ fluxes and tyrosine phosphorylation of total proteins were elicited at wild-type levels. Unlike in T cell clones stimulated with partial agonists, both p21 and p18 forms of tyrosine-phosphorylated CD3-ζ were detected, although the overall level of tyrosine-phosphorylated CD3-ζ was low. More strikingly, inducible association of ZAP70 to CD3-ζ was strongly inhibited, despite a normal induction of ZAP70 tyrosine phosphorylation. Finally, ZAP70 was not concentrated near the plasma membrane in the apoptosis-deficient cells. These results suggest that CD3-ζ is necessary for engagement of a specific signaling pathway leading to CD95-L expression that also needs the recruitment of ZAP70.
The TCR complex is composed of two functionally distinct modules. Whereas the TCR-α/β heterodimer is responsible for recognition of the antigen/MHC ligand, the cytoplasmic tails of the CD3 components (CD3-γ, CD3-δ, CD3-ε, CD3-ζ) are responsible for signal transduction. Thus, engagement of the TCR initiates a cascade of signal transduction events that trigger T cell proliferation and differentiation. It seems that the earliest activation event measurable is the recruitment and activation of nonreceptor tyrosine kinases of the Src family that in turn phosphorylate the tyrosine residues of the immunoreceptor tyrosine-based activation motifs (ITAMs)1 present in the cytoplasmic tails of the CD3 chains (1–4). CD3-γ, CD3-δ, and CD3-ε each contain one ITAM, whereas CD3-ζ contains three. It has been proposed that the multiplicity of ITAMs in the TCR complex may serve primarily to amplify TCR activation signals. Once phosphorylated, the ITAMs become sites for high-affinity binding of tyrosine kinases of the Syk family, mainly of ZAP70 in T cells, through their tandem src homology 2 (SH2) domains (5– 8). After binding to the phosphorylated ITAMs, ZAP70 becomes tyrosine phosphorylated and activated by a src kinase, which is thought to be primarily Lck (9). Once activated, ZAP70 probably autophosphorylates on multiple tyrosine residues (2), thus generating docking sites for SH2 domain–containing proteins, including Lck and Vav (10, 11). Subsequently, downstream effector functions are triggered, including the mobilization of intracellular Ca2+ and the transport to the nucleus of an array of transcription factors that drive, among others, cytokine gene expression and programmed cell death.
Programmed cell death and its accompanying morphological changes, called apoptosis, are active processes by which unnecessary or harmful cells are self-eliminated in multicellular organisms (12, 13). Evidence has accumulated that signaling through the TCR complex can elicit apoptosis in immature thymocytes, human leukemic T cells, and mature peripheral T cells (14). This mechanism contributes to the downregulation of ongoing peripheral immune responses and to the establishment of tolerance to self-antigens. Engagement of the TCR frequently triggers both proliferation and death of mature cells, raising the question of how these two outcomes are differentially regulated. Several members of the growing families of the TNF and TNFR have been shown to be involved in mediation of the final stages of programmed cell death (for recent reviews see references 15–17). Of these members, it seems that the fas-ligand (CD95-L), TNF, and their receptors (fas or CD95, TNFR1, and TNFR2) are the most important mediators of apoptosis in peripheral T cells (18, 19). Stimulation of the TCR complex results in upregulation of CD95 and CD95-L, and subsequent binding of CD95 to its ligand results in the direct activation of a cascade of proteases that finally lead to apoptosis (for review see references 16, 17). Although this process has been and is still under intense scrutiny, the activation pathways that lead to CD95 and CD95-L expression are mostly unknown. A recently cloned gene, TDAG51, seems necessary for TCR induction of CD95 in T cell hybridomas (20). On the other hand, it is known that the induction of CD95-L gene transcription is nuclear factor of activated cells (NFAT) dependent (21–24). Indeed, the recent characterization of the CD95-L promoter has shown the presence of an inducible NFAT-binding site that could be responsible for the regulation of CD95-L expression in T cells (25).
It was recently described that cross-linking of a Tac–ζ chimera results in induction of apoptosis in a transfected murine T hybridoma, suggesting that CD3-ζ is capable of inducing CD95-L expression on its own (26). Furthermore, Combadière et al. (27) showed in transgenic mice that the CD3-ζ chain, through one of its ITAMs, might play unique roles in TCR responses leading to apoptosis by engaging specific signaling pathways. In addition, we have previously described that transfection of a transmembrane tyrosine to leucine mutant of TCR-β into TCR-β–negative Jurkat cells results in a defective association of CD3-ζ to the other subunits of the TCR complex (28). Compared with cells transfected with wild-type TCR-β, the mutant cells responded normally to TCR cross-linking by IL-2 secretion, TCR downregulation, and expression of the activation-induced receptors CD25 and CD69. By contrast, compared with wild-type cells, mutant cells were resistant to programmed death after TCR cross-linking. This defective induction of apoptosis was found to be based on a defective induction of CD95-L. On the basis of this evidence, we suggested that CD3-ζ is needed for activation of a signaling pathway leading to expression of CD95-L and apoptosis. We now show that the defective signaling in the mutant cells is not derived from a generalized lower production of second messengers, including intracellular Ca2+ increases and total protein tyrosine phosphorylation, but must be derived from a specific defect in a signaling pathway. In this regard, we have found that both a defective association of ZAP70 to CD3-ζ and a defective recruitment of ZAP70 to a near-plasma membrane location might be responsible for the apoptosis-defective phenotype.
Materials and Methods
Cell Lines.
The wild-type and mutant cell lines were obtained by reconstitution of the TCR-β–negative Jurkat variant 31.13 (generously provided by Dr. A. Alcover, Institut Pasteur, Paris, France), with either a wild-type Vβ3+ TCR-β cDNA derived from the T cell clone HA1.7, or with a mutant TCR-β cDNA that contains a transmembrane tyrosine to leucine mutation (28).
DNA Constructs and Antibodies.
The plasmid encoding for the chimeric protein consisting of the extracellular and transmembrane domain of human IL-2R α chain and the intracytoplasmic domain of CD3-ζ was generously given by Dr. F. Letourneur (29). The ZAP70–green fluorescent protein (GFP) construct was prepared by PCR using as template the HA-tagged ZAP70 cDNA construct generously provided by Dr. A. Weiss (University of California at San Francisco, CA). The PCR primers used added a EcoRI site in 5′ and a BamHI site in 3′. The PCR product was digested and subcloned in the plasmid pEGFP-N1 (CLONTECH Laboratories, Inc., Palo Alto, CA) previously digested with the same restriction enzymes.
The antibody 448, specific for CD3-ζ, was generated by immunization of a New Zealand rabbit with a synthetic peptide corresponding to the 109–132-amino acid sequence of human CD3-ζ coupled to KLH, as previously described (30). The anti-ZAP70–specific rabbit antiserum ZAP4, as well as the antiserum specific for human Vav, were generously donated by Dr. S. Ley (Medical Research Council, Mill Hill, London, UK) (31). The rabbit anti–human PLC-γ1 was kindly given by Dr. Peter Parker (Imperial Cancer Research Fund, London, UK). The anti-Vβ3 antibody Jovi.3 was a generous gift of Dr. M. Owen (Imperial Cancer Research Fund, London, UK). The anti-Tac antibody MAR108 was generously provided by Dr. Miguel López Botet (Hospital de la Princesa, Madrid, Spain). The anti-CD3-ε and anti-CD3-δ antibodies, APA1/1 and APA1/2, respectively, have been previously described (32). The anti-CD3 antibodies OKT3 and UCHT1 were purchased from Ortho Diagnostics (Raritan, NJ) and Immunokontact (Bioggio, Switzerland), respectively. The anti-Vβ8 and anti-CD95 antibodies were from PharMingen (San Diego, CA). The anti-phosphotyrosine antibody 4G10 was acquired from Upstate Biotechnology Inc. (Lake Placid, NY) and the anti-phosphotyrosine antibody PY20 from Transduction Laboratories (Lexington, KY).
Cytokine Assays.
A total of 5 × 105 cells/ml were stimulated with soluble Staphylococcal enterotoxin B (SEB; 10 μg/ml) for 48 h or with soluble UCHT1 (10 μg/ml) plus PMA (10 ng/ml) for 24 h. The IL-2 content in the supernatants of these cultures was determined by a commercial ELISA, following the manufacturer's instructions (Genzyme Corp., Boston, MA). IFN-γ was also detected in the same supernatants by ELISA using the mAbs 43.11 and 45.11 (kindly donated by Dr. Sefik Alkan, Novaitis, Basel, Switzerland) as previously described (33). In both cases, supernatants were tested in duplicate and OD values were converted to U/ml by comparison with standard curves determined with recombinant human IL-2 (Hoffman-La Roche, Nutley, NY) and IFN-γ (Genzyme Corp.). The culture supernatants were diluted to make the OD values fall in the linear range of the curves.
Analysis of Intracellular Ca2+ Fluxes.
A total of 107 cells/ml were washed three times with serum-free RPMI and loaded with 5 μg/ml fura-2AM (Sigma Chemical Co., St. Louis, MO) for 1 h at 37°C in the dark. They were then washed twice with RPMI, resuspended at 5 × 106 cells/ml in PSS-Ca2+ (140 mM NaCl, 4.6 mM KCl, 2 mM CaCl2, 10 mM MgCl2, 10 mM d-glucose, 10 mM Hepes, pH 7.4), placed in a cuvette, and allowed to equilibrate in a Perkin-Elmer fluorimeter at 37°C under gentle stirring until the baseline was stable. The excitation wavelength was of 339 nm and emission was measured at 510 nm. Primary and secondary fluorimetric responses were recorded after stimulation with 10 μg/ml OKT3 and subsequent cross-linking with 30 μg/ml polyclonal rabbit anti–mouse Ig, respectively. Triton X-100 (Sigma Chemical Co.) was added to 1% to establish a maximum signal (Fmax), followed by EGTA to 100 mM to establish a minimum (Fmin). Absolute Ca2+ concentration increases were calculated as described (34).
Activation, Immunoprecipitation and Immunoblotting.
For activation 3 × 107 cells per time point were collected, washed, and resuspended in 1 ml of RPMI supplemented with 10 mM Hepes, pH 7.4 and prewarmed at 37°C for 15 min. Afterwards, 10 μg/ml OKT3 or SEB (Sigma Chemical Co.) was added and the cells were collected at different time intervals by brief centrifugation in an Eppendorf centrifuge. Each cell pellet was subsequently resuspended in 1 ml of 1% Brij96 lysis buffer (1% Brij96, 140 mM NaCl, 10 mM Tris-HCl, pH 7.8, 10 mM iodoacetamide, 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mM sodium orthovanadate, 20 mM sodium fluoride) and maintained for 30 min on ice. The cell lysates were centrifuged at 12,000 g for 15 min and the supernatants were subjected to immunoprecipitation with protein A– or G–Sepharose beads precoated with specific antibodies as previously described (32). The immunoprecipitates were subjected to SDS-PAGE and the electrophoresed proteins were transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA) by standard procedures. The membrane was blocked in a solution of 10% nonfat dry milk in PBS for 1 h at room temperature. The membrane was rinsed three times with 100 ml of 0.1% Tween 20 in PBS and incubated with the appropriate dilutions of the specific antibodies in PBS-Tween for 1 h at room temperature. The membrane was then washed five times with 100 ml PBS-Tween and subsequently incubated with a peroxidase-labeled sheep anti–mouse Ig (Amersham International, Little Chalfont, UK) in PBS-Tween for 1 h. After six washes with 100 ml PBS-Tween the membrane was processed by the enhanced chemiluminescence (ECL) method according to the manufacturer's specifications (Amersham International).
To analyze tyrosine phosphorylation of total proteins, an aliquot of the 1% Brij96 cell lysate containing the equivalent of 300,000 cells was mixed with an equal volume of ice-cold acetone and kept on ice for 10 min. The samples were centrifuged at 12,000 g for 10 min, air-dried and resuspended in sample buffer for SDS-PAGE.
In Vitro Kinase Assay.
For the in vitro kinase assay, the anti-ZAP70 immunoprecipitates were washed six times with lysis buffer and once with kinase buffer (100 mM NaCl, 50 mM Hepes, pH 7.5, 5 mM MgCl2, 5 mM MnCl2, 1 mM sodium orthovanadate). The beads were resuspended in 50 μl kinase buffer plus 5 μM cold ATP and 10 μCi γ-[32P]ATP (5,000 Ci/ mmol; Amersham International) and incubated for 20 min at room temperature. The beads were then washed three times with lysis buffer containing 20 mM EDTA, and finally were boiled in Laemmli sample buffer and the samples subjected to SDS-PAGE. To measure the phosphorylation of an exogenous substrate, 8 μg of purified bovine brain tubulins (generously given by Dr. J. Avila, Centro de Biología Molecular, Madrid, Spain) were added to the kinase reaction.
Confocal Microscopy.
Wild-type and mutant cells at 106/ml in RPMI were stimulated with 10 μg/ml OKT3 for 1 min at 37°C and then diluted in 10 ml ice-cold PBS and centrifuged onto coverslips. The cells were fixed in 2% paraformaldehyde in PBS for 20 min at room temperature and subsequently blocked and permeabilized in PBS containing 1% BSA and 0.1% saponin. The cells were then stained with a 1:1,000 dilution of ZAP4 antibody in blocking buffer for 30 min at room temperature, followed by staining with a mixture of a Texas red–labeled goat anti–rabbit Ig and fluoresceinated goat anti–mouse Ig antibodies (Southern Biotechnology Associates Inc., Birmingham, AL). The coverslips were subsequently washed and mounted in mowiol as described (35). The preparations were examined in a Zeiss confocal microscope.
Transient Transfections.
For transient transfection, 5 × 106 cells were collected, resuspended in 500 μl of RPMI plus 20% FCS and transferred to a 0.4-mm electroporation cuvette (Bio-Rad Laboratories). A total of 50 μg of plasmid DNA was added and the cells were incubated for 15 min at room temperature before electroporation was performed at 260 V and 960 μF in a Bio-Rad Gene Pulser. After electroporation, the cells were allowed to stand for 2 min and then centrifuged and put in culture. Expression of the transfected protein was usually examined 24 h after transfection.
Apoptosis Assays.
A total of 105 transfected cells/well were plated 24 h after electroporation on 96-well plates (Costar Corp., Cambridge, MA) precoated with variable concentrations of anti-CD3 or anti-Tac antibodies. To estimate the number of cells in apoptosis, 20 h later they were stained with biotinylated annexin V (Boehringer Mannheim GmbH, Mannheim, Germany) followed by staining with PE-labeled streptavidin (Southern Biotechnology Associates Inc.). To distinguish the transfected from the nontransfected cells, the samples were double stained with anti-Tac antibody, followed by a fluoresceinated goat anti–mouse Ig antibody (Southern Biotechnology Associates Inc.) and analyzed in an EPICS-XL flow cytometer (Coulter Immunology, Hialeah, FL). ZAP-GFP transfected cells were single stained with biotinylated annexin V– and PE-labeled streptavidin and subjected to two-color analysis. The level of apoptosis within the transfected population was estimated as the percentage of annexin V–positive cells.
Results
Apoptosis but Not Cytokine Release Is Defective in Jurkat T Cell Clones Expressing a TCR-β Mutant That Impairs CD3-ζ Association.
The ability of the TCR complex to elicit different activation events in several Jurkat cell clones that express either wild-type TCR-β or a transmembrane tyrosine to leucine mutant have been studied. Mutant clones that expressed similar amounts of the TCR complex to those of wild-type transfectants (Fig. 1 A) were chosen for comparative purposes. A comparison of the ability of wild-type and mutant clones to secrete cytokines and to die by apoptosis upon stimulation of the TCR complex with SEB is shown in Fig. 1 B. Strikingly, although the mutant clone C2 released similar amounts of IL-2 and IFN-γ to the wild-type clone B7, it was, however, refractory to activation-induced cell death. Indeed, as shown in Fig. 1 B, the mutant cell clone was defective in the induction of CD95-ligand, although it did maintain an intact apoptosis machinery that could be triggered by direct stimulation of CD95 (CH11 stimulation). These data suggest that, as previously shown (28), the mutation in the transmembrane domain of TCR-β results in a selective defect in the induction of CD95-L, but not of IL-2 and IFN-γ.
Figure 1.


TCR-β mutant clones release normal levels of cytokines but are resistant to TCR-mediated apoptosis. (A) Flow cytometry analysis of clones expressing either wild-type (B7) or mutant TCR-β (C2) chains. B7 and C2 cells were analyzed for the cell surface expression of the CD3 complex (antibody UCHT1), the transfected TCR-β chain (Vβ3, continuous line), the Jurkat endogenous TCR-β (Vβ8, broken line) and CD95. (B) Wild-type B7 (filled bars) and mutant C2 cells (open bars) were stimulated with 10 μg/ml SEB for 48 h at 37°C. The percentages of cells with sub-G1 amounts of DNA were quantified by flow cytometry after staining the cells with propidium iodide. Secretion of IL-2 and IFN-γ was measured by ELISA in the supernatants of stimulated cells. CD95-L mRNA expression of cells stimulated for 3 h was determined by Northern blotting and quantified by densitometry. Cells were also stimulated with the anti-CD95 antibody CH11 to demonstrate that the apoptotic machinery works well in both types of cell. Values are expressed in arbitrary units as a result of the normalization of the wild-type (WT) values to 1.
Despite the high cell surface expression of the TCR– CD3 complex in the wild-type and mutant cell clones studied (Fig. 1 A and reference 28) we found that the association of CD3-ζ to the remaining chains of the complex is impaired. Thus, immunoprecipitation with an anti–TCR-β antibody from 1% Brij96 lysates of the mutant cell clone (C2) resulted in the coprecipitation of very low amounts of CD3-ζ compared with those coprecipitated from a wild-type clone (G7, Fig. 2, A and B). However, normal amounts of CD3-δ and CD3-ε chains were coprecipitated (Fig. 2 A), suggesting that the TCR–CD3 complex in the mutant clone is assembled without CD3-ζ. Thus, these data confirm those obtained by surface iodination (28), where an otherwise complete TCR–CD3 complex was shown to be expressed at the cell surface in the almost complete absence of CD3-ζ. As a control for the expression of CD3-ζ in the mutant clone, aliquots of the total lysates from the wild-type and the mutant clones were subjected to SDS-PAGE and to immunoblotting with anti-ζ antibody. The result showed that, despite the low association of CD3-ζ to the TCR complex in the mutant clone, the total levels of CD3-ζ were normal (Fig. 2 B). The defective association of CD3-ζ was not reverted in the mutant clone upon TCR cross-linking with an anti-CD3 antibody while, in the wild-type clone, the amount of CD3-ζ associated to the TCR complex seems to decrease upon activation (Fig. 2 B), suggesting this chain may be inducibly internalized and degraded, as recently proposed (36, 37).
Figure 2.


CD3-ζ is loosely associated to the TCR–CD3 complex in the apoptosis-defective mutant cells. (A) Wild-type G7 and mutant C2 cell clones were lysed in 1% Brij96 immunoprecipitation buffer and the cell lysates were immunoprecipitated (Ip) with either anti–TCR-β antibody Jovi.3 or anti-CD3-ζ antibody 448. The immunoprecipitates were subjected to SDS-PAGE, transferred to nitrocellulose and immunoblotted with anti-CD3-ζ antibody 448, anti-CD3-ε antibody APA1/1 and anti-CD3-δ antibody APA1/2. (B) Wild-type G7 and mutant C2 cells were stimulated with 10 μg/ml OKT3 for the indicated times (in min), and then lysed in 1% Brij96. Immunoprecipitation was performed with anti-Vβ3 antibody Jovi.3. The immunoprecipitates were resolved by SDS-PAGE under nonreducing conditions and the proteins were transferred to a nitrocellulose membrane. Immunoblotting was then performed with anti-ζ antibody 448. A portion of the total cell lysate was run in parallel to verify the level of CD3-ζ expression. The position of the CD3-ζ homodimer is indicated by an arrow. The running positions of the molecular mass standards are indicated with arrowheads.
According to these data, the defective induction of CD95-L upon stimulation of the TCR–CD3 complex could be the result of weak association of the CD3-ζ chain. By contrast, a high level of CD3-ζ association would not be required for activation of the signaling mechanisms leading to secretion of IL-2 and other cytokines, as was previously established in murine T cell hybridomas (38).
Defective CD95-L Expression Does Not Result from a Generalized Decrease of Second Messengers.
Since the CD3-ζ homodimer contributes with six ITAMs to the TCR complex, it could be hypothesized that a stimulation of a complex with low levels of associated CD3-ζ would elicit lower levels of intracellular second messengers. Thus, if the threshold of second messengers required for CD95-L were higher than for IL-2 induction, then a defective recruitment of CD3-ζ to the TCR complex should result in a selective defect on CD95-L induction. Alternatively, CD3-ζ could trigger a special activation pathway required for the induction of CD95-L but not for cytokines. To investigate whether the ability of mutant cells to produce second messengers is diminished, intracytosolic Ca2+ increases were recorded in two wild-type and two mutant clones. As shown in Fig. 3, all four clones responded similarly to anti-CD3 stimulation before and after cross-linking with a second anti-mouse Ig antibody. Although there were some differences in the intensity of the Ca2+ signal and in the response to the cross-linking antibody, these differences could not be ascribed to wild-type or mutant phenotypes. Similar results were obtained upon stimulation with SEB (data not shown).
Figure 3.

TCR triggering elicits normal intracellular Ca2+ increases in apoptosis-deficient cells. Wild-type clones G7 and C3 and mutant clones C2 and B5 loaded with the Ca2+ sensitive dye fura-2 were stimulated with 10 μg/ml OKT3 (first arrow), and when the peaks of intracytosolic Ca2+ began to decrease, a second stimulation was produced by adding 30 μg/ml of a rabbit anti–mouse Ig cross-linking antibody (second arrow).
Despite the normal Ca2+ fluxes induced upon TCR cross-linking, it was possible that the level of tyrosine phosphorylation in total proteins was diminished in the clones that have a low level of CD3-ζ associated to the TCR complex. This was not the case and, indeed, stimulation of the TCR complex with antibodies (Fig. 4) or with superantigen (not shown) did not result in lower tyrosine phosphorylation. On the contrary, activation-induced tyrosine phosphorylation reached consistently higher levels in mutant clones. Interestingly, tyrosine phosphorylation of total proteins was considerably higher in nonactivated mutant than in wild-type cells. At present, we do not know what could be the cause of this effect. Some protein bands were readily identified after reprobing the membrane with specific antibodies. Thus, it was clear that PLC-γ1 became tyrosine-phosphorylated upon activation of wild-type and mutant clones. Since tyrosine phosphorylation activates PLC-γ1 (39–42), this result is consistent with the normal intracellular Ca2+ fluxes observed in mutant clones. Tyrosine phosphorylation of ZAP70 was also clearly induced to wild-type levels in mutant clones, as well as tyrosine phosphorylation of Vav, one of the ZAP70 binding proteins (31, 43). A major difference between the wild-type clone G7 and the mutant clone C2 was that a 50-kD protein was tyrosine phosphorylated at much higher levels in the mutant clone. This protein had the mobility of β-tubulin, as demonstrated after reprobing the membrane with a specific antibody (data not shown). However, in other mutant clones β-tubulin was not as heavily phosphorylated as in C2, suggesting that it is not relevant to the apoptosis-resistant phenotype. In summary, the Ca2+ flux measurements and the total protein tyrosine phosphorylation results suggest that TCR triggering in mutant cells does not result in a generalized lower production of second messengers, and favors the existence of a specific signaling defect for CD95-L induction.
Figure 4.

TCR activation in apoptosis-deficient cells results in high level of total tyrosine phosphorylation. Wild-type G7 and mutant C2 cells were stimulated with 10 μg/ml OKT3 for the indicated times and total cytoplasmic proteins were obtained by acetone precipitation from detergent cell lysates. The samples (equivalent to 3 × 105 cells per lane) were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. Immunoblotting was performed with antiphosphotyrosine antibody 4G10. Afterwards, the membrane was stripped and sequentially reprobed with antibodies specific for PLC-γ1, Vav and ZAP70. The position of β-tubulin was identified in a separate experiment. Molecular mass standards are indicated by arrowheads.
Both Tyrosine-phosphorylated Forms of CD3-ζ Are Detected in Apoptosis-deficient Cell Clones.
Further experiments to assess the level of tyrosine phosphorylation in wild-type and mutant clones were performed by immunoprecipitation with anti-phosphotyrosine antibodies from anti-CD3 stimulated cell lysates. As shown in Fig. 5 A, the immunoprecipitation and immunoblotting with the antiphosphotyrosine antibody showed higher levels of total tyrosine-phosphorylated proteins in mutant than in wild-type cells (Fig. 5, left). Again, as in Fig. 4, a considerable level of constitutive tyrosine phosphorylation was detected in nonstimulated mutant cells. After reprobing the membrane with an anti-ζ antibody it was found that the amount of CD3-ζ homodimer that had been immunoprecipitated with the anti-phosphotyrosine antibody was considerably reduced in mutant cells (Fig. 5 A, right). This effect was specially clear when the intensity of the CD3-ζ bands was compared with those of the tyrosine-phosphorylated proteins. Note that CD3-ζ is not one of the major tyrosine-phosphorylated proteins and is not visualized in the film exposure shown in Fig. 5 A.
Figure 5.



Both tyrosine phosphorylation forms of CD3-ζ are found at low levels in apoptosis-deficient cells. (A) After activation of wild-type and mutant cells with OKT3 for the indicated times, the cells were lysed and immunoprecipitation was performed with anti-phosphotyrosine antibody PY20. The samples were subjected to SDS-PAGE under nonreducing conditions. Immunoblotting was afterwards carried out with anti-phosphotyrosine (P-Tyr) antibody 4G10 (left). The nitrocellulose membrane was stripped and reprobed with anti-ζ antibody 448 (right). (B) After activation with 10 μg/ml OKT3 or 10 μg/ml SEB, the cells were lysed and immunoprecipitation was carried out with anti-ζ antibody 448. The samples were subjected to SDS-PAGE under reducing conditions to resolve the high (p21) and low (p18) molecular mass forms of CD3-ζ. Immunoblotting was performed with antiphosphotyrosine antibody 4G10. NMS, non-immune serum used for control immunoprecipitation. (C) Wild-type G7 and mutant C2 cells were stimulated with OKT3, lysed in 1% Brij96 and immunoprecipitation was performed with anti-Vβ3 antibody Jovi.3. The immunoprecipitates were resolved by SDS-PAGE under non-reducing conditions and immunoblotting was performed with 4G10.
There are marked differences in the patterns of tyrosine-phosphorylated proteins obtained either after direct immunoblotting of a total lysate with the antiphosphotyrosine antibody (Fig. 4) or by immunoblotting of an antiphosphotyrosine immunoprecipitate (Fig. 5 A). The differences could be attributed to nonspecific binding of either the anti-phosphotyrosine or the peroxidase-conjugated second antibody to nontyrosine-phosphorylated bands; this could hold true for the low molecular mass noninducible proteins bands in Fig. 4. In addition, the immunoprecipitation with the antiphosphotyrosine antibody could introduce a bias by selecting the most heavily phosphorylated proteins or the tyrosine-phosphorylated sequences for which the antibody has higher affinity.
Since the mutation in the transmembrane domain of TCR-β resulted in impaired association of CD3-ζ, it was compelling to examine whether it became phosphorylated to its low and high molecular mass forms. Therefore, a direct immunoprecipitation under reducing conditions with the anti-ζ antibody was performed from lysates of cells stimulated with superantigen or with anti-CD3 antibody (Fig. 5 B). As shown in this figure, although the levels of tyrosine-phosphorylated CD3-ζ were consistently lower in the mutant clone, both p18 and p21 phosphorylated forms were detected. Furthermore, the ratio of p21 to p18 was never lower in mutant than in wild-type cells. A control for gel loading was established after reprobing the membrane with the anti-ζ antibody. This demonstrated that the amounts of immunoprecipitated CD3-ζ were equivalent in the wild-type and mutant clones (data not shown). According to these experiments, and unlike in altered peptide ligands (APL)-stimulated T cell clones (44–46), CD3-ζ is tyrosine-phosphorylated to its high molecular mass form. Thus, in regard to the tyrosine phosphorylation of CD3-ζ, the only difference observed between wild-type and mutant clones was a decreased level of total phosphorylation in the latter. Notwithstanding, it was surprising that despite its low stoichiometry of association to the TCR complex (Fig. 2) CD3-ζ became tyrosine phosphorylated to relatively high levels in the mutant clone (Fig. 5, A and B). A possible explanation for this discrepancy is that tyrosine-phosphorylated CD3-ζ could associate in the mutant clone to the TCR complex with higher stoichiometry than nonphosphorylated one. To control for the amount of tyrosine-phosphorylated CD3-ζ associated to the TCR complex, an immunoprecipitation with anti–TCR-β antibody was performed from wild-type and mutant clones stimulated with anti-CD3 antibodies. As shown in Fig. 5 C, high levels of the tyrosine-phosphorylated CD3-ζ dimer were detected in the wild-type clone samples after activation whereas in the mutant clone it was practically undetectable. Thus, this result excludes the possibility that the TCR complex preferably associates with phospho-ζ in the mutant clone.
ZAP70 Becomes Tyrosine Phosphorylated in Apoptosis-deficient Clones but Does Not Stably Associate to CD3-ζ.
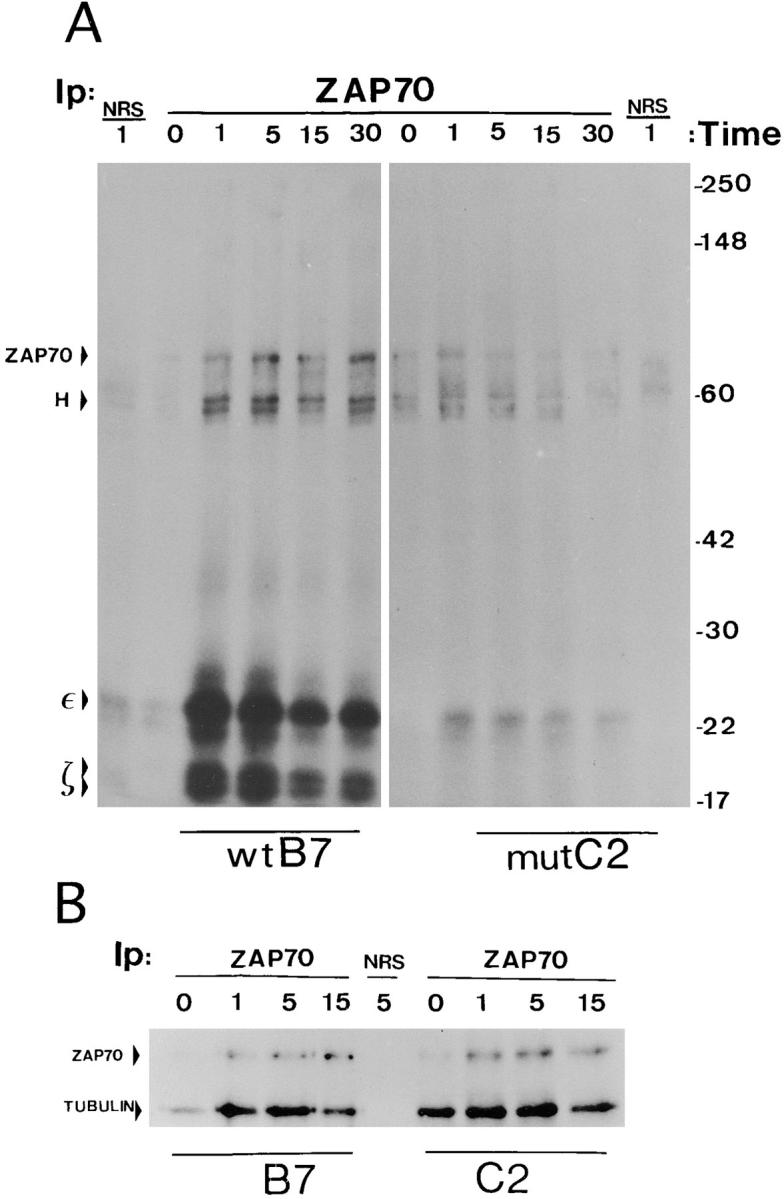
Although ZAP70 appeared to become tyrosine-phosphorylated in mutant cells (Fig. 4), it was compelling to test the level of ZAP70 phosphorylation by direct immunoprecipitation with a specific antibody. As shown in Fig. 6, after immunoprecipitation with an anti-ZAP70 antibody and immunoblotting with anti-phosphotyrosine, tyrosine-phosphorylated proteins of 35–38 and 70 kD were detected in samples from the wild-type clone G7 (Fig. 6, left). These proteins were inducibly phosphorylated or inducibly associated to ZAP70, although at different times after stimulation. To identify these protein bands, the membrane was stripped and reprobed sequentially with anti-ZAP70 and anti-ζ antibodies (Fig. 6, right). Thus, the 70-kD band was identified as ZAP70, whereas the mobility of the CD3-ζ band did not correspond to any of the major tyrosine-phosphorylated proteins.
Figure 6.

ZAP70 becomes tyrosine phosphorylated but does not associate to CD3-ζ in apoptosis-deficient cells. Wild-type and mutant cell clones were stimulated with 10 μg/ml OKT3 for the indicated times and lysed in 1% Brij96-containing buffer. The cell lysates were subjected to immunoprecipitation with anti-ZAP70 antiserum ZAP4, the immunoprecipitates were resolved by SDS-PAGE under nonreducing conditions, and the proteins were transferred to a nitrocellulose membrane. The membrane was subsequently incubated with antiphosphotyrosine antibody 4G10 (PTyr, left), and later stripped and sequentially reprobed with ZAP4 and anti-ζ antibody 448 (right). The running positions of molecular mass standards, as well as of ZAP70, the CD3-ζ homodimer (ζ2) and the immunoglobulin heavy chain (H), are indicated.
Several differences were observed with the samples from the mutant clone C2. Interestingly, higher levels of tyrosine-phosphorylated ZAP70 were detected in the mutant clones (Fig. 6, left), even though the amount of immunoprecipitated ZAP70 was equivalent (Fig. 6, right). Nevertheless, ZAP70 was tyrosine phosphorylated with similar kinetics in each type of cell. These results show that ZAP70 becomes tyrosine phosphorylated in the mutant T cell clones defective in induction of CD95-L. Interestingly, despite the fact that ZAP70 becomes tyrosine phosphorylated, its association to CD3-ζ in the mutant clone is negligible (Fig. 6, right). Accordingly, the 35–38-kD tyrosine-phosphorylated protein that is found associated to ZAP70 both in the wild-type and mutant clones must be a different protein. This protein may be pp36, a membrane-bound tyrosine-phosphorylated protein that has been described to associate to the adapter Grb2, to PLC-γ1, and to other proteins (47–49). Nevertheless, the 35–38-kD phosphoprotein band runs fuzzier in the wild-type than in the mutant samples, suggesting that the lowest part of the band could correspond to tyrosine-phosphorylated CD3-ζ.
The anti-ZAP70 immunoprecipitations were repeated, with similar results, using another wild-type and another mutant clone obtained from an independent transfection (data not shown). Although it has been established that ZAP70 must be bound to tyrosine-phosphorylated ITAMs, mainly of CD3-ζ, in order to become tyrosine-phosphorylated by one of the Src-family kinases associated to the TCR complex or its coreceptors (5), the results shown in Fig. 6 demonstrate that ZAP70 becomes tyrosine phosphorylated in the mutant clone in the almost total absence of CD3-ζ binding. It is possible that, in the mutant clones, ZAP70 becomes tyrosine-phosphorylated after binding to the ITAMs of other CD3 subunits or, alternatively, that ZAP70 becomes tyrosine-phosphorylated after a transient association with CD3-ζ.
To examine the kinase activities associated with ZAP70 in the mutant cell clones, an in vitro kinase assay was performed with anti-ZAP70 immunoprecipitates obtained from anti-CD3 stimulated cell samples. As shown in Fig. 7 A, a phosphorylated 70-kD protein band was observed both in wild-type and mutant clones, although the intensities of the bands were somewhat lower in the immunoprecipitates from the mutant clone samples. The 70-kD band, which corresponded to ZAP70, as demonstrated after reprobing with a specific antiserum (data not shown), was already phosphorylated at low levels in nonactivated wild-type and mutant cells, but its intensity increased after 1 min of activation in both types of cell. Most striking was the intensity of two series of ZAP70-associated protein bands with molecular masses in the range of 18-21 and 22-24 kD that became strongly phosphorylated in samples from stimulated wild-type cells. After reprobing with specific antibodies, it was demonstrated that the two bands just above the 17-kD molecular mass marker corresponded to the two forms of tyrosine-phosphorylated CD3-ζ, p18 and p21, and that the strong phosphorylated protein band of 23 kD corresponded to CD3-ε (data not shown). However, although the sizes of the 22- and 24-kD protein bands that run just below and above CD3-ε are reminiscent of the CD3-δ and CD3-γ subunits, their identity could not be confirmed by probing with specific antibodies, due to technical difficulties.
Figure 7.
In vitro kinase assays. ZAP70 is not associated to CD3 subunits in apoptosis-deficient cells. (A) Wild-type and mutant clones were stimulated with OKT3, lysed in 1% Brij96 and immunoprecipitation was performed with anti-ZAP70 antiserum. The immunoprecipitates were incubated with γ-[32P]ATP in kinase buffer, washed, resolved by SDS-PAGE under reducing conditions and transferred to a nitrocellulose membrane. The membrane was dried and exposed to x-ray film. The positions of ZAP70, CD3-ε and the p18 and p21 forms of CD3-ζ (arrowheads) were located by probing the membrane with specific antibodies. NRS, nonimmune rabbit serum used as a control; H, immunoglobulin heavy chain. (B) An in vitro kinase experiment was performed as in A but 8 μg per time point of purified bovine tubulin were added to the reaction mixture. The positions of the phosphorylated ZAP70 and tubulin are indicated.
Since it has been shown that purified ZAP70 does not phosphorylate CD3-ζ (50), the phosphorylation of this chain in the in vitro kinase assay (Fig. 7 A) suggests the participation of an inducible ZAP70-associated kinase different to ZAP70. On the other hand, the fact that the CD3 ITAMs are not substrates for ZAP70 makes it difficult to estimate the kinase activity of ZAP70 in the mutant clone in the experiment shown in Fig. 7 A. To solve this problem, an exogenous substrate specific for ZAP70 (50) was added to the in vitro kinase assay. As shown in Fig. 7 B, tubulin was phosphorylated with similar efficiency in ZAP70 immunoprecipitates from wild-type and mutant cells, suggesting that ZAP70 was equally active in both types of cell. Interestingly, ZAP70 was constitutively more active in samples from nonstimulated mutant cells (time 0) than from the wild type, suggesting that ZAP70 could contribute to the higher level of constitutive general tyrosine phosphorylation observed in mutant cells (Figs. 4 and 5 A).
Although there was an inducible kinase activity that phosphorylated ZAP70 in mutant cell samples, the phosphorylation of CD3-ε was strongly diminished, and both forms of phosphorylated CD3-ζ were undetectable (Fig. 7 A). In principle, the low level of phosphorylation in CD3-ε and CD3-ζ could be due either to low kinase activity in the samples or to low concentration of these substrates. This latter possibility seems to be correct since, after reprobing the membrane, CD3-ζ and CD3-ε were not detected in the mutant clone samples (data not shown). Thus, the results shown in Fig. 7 suggest that ZAP70 is active and becomes tyrosine phosphorylated in mutant cells despite its low-level association to CD3-ζ and other CD3 subunits.
ZAP70 Is Not Recruited to a Juxtamembrane Localization in Apoptosis-deficient Cell Clones.
Despite being mostly present as a cytosolic protein, ZAP70 has been described to be intracellularly located in a juxtamembrane position in activated lymphoblasts (51), Jurkat cells, and transfected fibroblasts (52). Although it could have been expected that ZAP70 location in a juxtamembrane position depends on binding to the phosphorylated ITAMs of the TCR–CD3 complex, it was demonstrated that a ZAP70 mutant deprived of the SH2 domains was still located near the membrane (52). By contrast, a kinase dead mutant was not located near the membrane, suggesting that this domain is necessary for the localization of ZAP70 by a yet unknown mechanism. To examine whether the low association of ZAP70 to CD3-ζ in the mutant clones caused a mislocation of ZAP70, wild-type and mutant cells were stimulated for 1 min with anti-CD3 antibody and subsequently fixed, permeabilized with saponin and stained with the anti-ZAP70 antiserum. ZAP70 and CD3 stainings were observed after incubation with fluoresceinated and Texas red– labeled second antibodies, respectively. As shown in Fig. 8 A, ZAP70 was concentrated in a near-plasma membrane location in most of the wild-type clone cells, whereas it remained more evenly distributed throughout the cytosol in most of the mutant clone cells and was not specially concentrated near the membrane. On the other hand, CD3 staining corresponded to a plasma membrane pattern, as was expected. A higher magnification of single wild-type and mutant cells is shown in Fig. 8 B. The observed patterns obtained with the anti-peptide ZAP4 antibody were absent in cells that had been stained in the presence of the competing peptide, suggesting that they were specific (data not shown). In addition, the ZAP70 staining patterns characteristic of wild-type and mutant clones were reproduced by a transfected HA-tagged ZAP70 construct (data not shown), adding further support to their specificity.
Figure 8.


ZAP70 is not located near the plasma membrane in apoptosis-deficient cells. Wild-type and mutant cells were stimulated for 1 min with 10 μg/ml OKT3, fixed, permeabilized, and stained with anti-ZAP70 antiserum. The cells were then incubated with a Texas red–labeled goat anti–mouse Ig antibody and with a fluoresceinated goat anti–rabbit Ig antibody. A general field is shown in A, whereas magnifications of single wild-type and mutant cells are shown in B. ZAP70 appears in green and CD3 in red.
Although the immunofluorescence data indicated that ZAP70 was located near the plasma membrane in wild-type cells, in a cell fractionation experiment it was mostly found in a nonmembrane-bound cytosolic fraction (data not shown), such as has been previously described (52). As these authors proposed, the localization of ZAP70 near the plasma membrane seems to be the result of a dynamic process of association and dissociation to a yet unidentified receptor in the plasma membrane, rather than the result of permanent binding. Accordingly, a ZAP70 gradient is generated. On the other hand, our results show that ZAP70 is not concentrated near the plasma membrane in TCR-activated mutant cells where the association of ZAP70 to CD3-ζ is impaired, suggesting that this binding might be required for its localization near the plasma membrane in Jurkat cells. In addition, these results suggest that the defective induction of CD95-L in mutant cells may derive from the mislocation of ZAP70.
Direct Stimulation of a CD3-ζ Chimera, but Not ZAP70 Overexpression, Reestablishes the Sensitivity of Mutant Cells to Apoptosis.
Since the defective induction of CD95-L and apoptosis in Jurkat cells transfected with a plasma membrane TCR-β point mutant may derive from the concomitant impaired association of CD3-ζ to the remaining subunits of the TCR complex, it was compelling to test whether direct stimulation of CD3-ζ resulted in induction of apoptosis. To this end, wild-type B7 and mutant C2 cells were transiently transfected with a Tac–ζ chimera that contains the intracytoplasmic tail of CD3-ζ bound to the extracellular and transmembrane domains of CD25. The transfected cells were plated on anti-Tac–coated plastic wells and the induction of apoptosis was measured 24 h later by annexin V staining. As shown in Fig. 9 A, the percentage of annexin V and Tac double positive cells increased to similar extents in wild-type and mutant cells after stimulation with anti-Tac antibody. These results suggest that the poor induction of apoptosis in mutant cells can be overcome if CD3-ζ is directly stimulated, and reinforces the idea that this poor induction results from a defective transmission of the activation signal from the TCR to the CD3-ζ chain. On the other hand, since ZAP70 is poorly associated to CD3-ζ in apoptosis-resistant mutant cells, we tried to determine whether overexpression of ZAP70 could overcome the apoptosis induction defect. To this end, a ZAP70–GFP chimera was constructed, in order to distinguish transfected from untransfected cells by flow cytofluorimetry in a transient transfection assay. Wild-type B7 and mutant C2 cells were transfected with either ZAP70-GFP or GFP alone, and 24 h later the cells were plated on anti-CD3 antibody-coated plastic wells. The percentage of apoptotic cells within the transfected population was evaluated 20 h later by annexin V staining. As shown in Fig. 9 B, the overexpression of ZAP70 resulted in enhanced induction of apoptosis in the wild type, but not in the mutant clone. Interestingly, ZAP70 overexpression also increased the level of basal apoptosis in nonstimulated wild-type cells, suggesting that it is sufficient to trigger the apoptosis pathway. The results shown in Fig. 9 could be interpreted in terms of the impaired association of ZAP70 to CD3-ζ in the mutant clones; the overexpression of ZAP70 did not result in an increased percentage of apoptotic cells because the underlying association defect was not overcome.
Figure 9.

Direct stimulation of a Tac–ζ chimera, but not ZAP70 overexpression, results in apoptosis of the apoptosis-deficient cells. (A) Apoptosis through a Tac–ζ chimera. Wild-type and mutant cell clones were transiently transfected with a Tac–ζ chimera and 24 h later were stimulated with plastic-bound anti-Tac antibody (striped bars) or left unstimulated (filled bars). 20 h later the percentage of cells in apoptosis within the Tac– ζ-transfected population was measured by annexin V staining. (B) Effect of ZAP70 overexpression on apoptosis induction. Wild-type B7 (open symbols) and mutant C2 (filled symbols) cell clones were transiently transfected with plasmids expressing either a ZAP70-GFP fusion protein (continuous lines) or GFP (broken lines). 24 h after electroporation the cells were plated on wells precoated with the indicated concentrations of OKT3, and 20 h later, the percentage of apoptotic cells within the transfected populations was measured by annexin V staining.
Discussion
In this paper we have analyzed early biochemical events in an apoptosis-resistant variant of Jurkat cells as a tool for dissecting possible pathways specific for the induction of CD95-L. Since the variant was derived from transfection of an amino acid substituted TCR-β cDNA that resulted in impaired association of CD3-ζ, and because the CD3-ζ homodimer contributes with six of the ITAMs of the TCR–CD3 complex, it could have been expected that the defective induction of CD95-L was the result of a generalized lower production of second messengers. In other words, a different requirement for second messenger levels could be the basis for a defective CD95-L induction versus IL-2 production. This was not the case, and similar levels of intracellular Ca2+ fluxes were recorded in wild-type and mutant clones. These results are consistent with those describing the production of normal levels of IL-2 and Ca2+ fluxes in murine T hybridomas that express a CD3-ζ chain deprived of the cytoplasmic tail (38). Furthermore, the levels of total protein tyrosine phosphorylation were never lower in the mutant than in the wild-type clones, thus suggesting that impaired CD95-L production results from alteration of a specific signaling pathway. Although the putative specific pathway, necessary for CD95-L induction but not for cytokine production nor for expression of activation antigens, has not yet been characterized, we have found two specific alterations in mutant cells: first, a lower level of tyrosine phosphorylation of CD3-ζ; second, and more important, a lower level of ZAP70/CD3-ζ association.
The fact that CD3-ζ becomes phosphorylated, albeit at low level, despite its low stoichiometry of association to the other chains of the complex, suggests that CD3-ζ becomes transiently associated to the complex and then phosphorylated. Alternatively, either isolated CD3-ζ or CD3-ζ associated to other membrane receptors could become tyrosine phosphorylated by a trans-acting mechanism. Although we do not yet know which of these possibilities is correct, we favor the idea of the existence of an equilibrium of association of CD3-ζ on the cell surface that is displaced towards dissociation in cells expressing the TCR-β mutant. It would be in the TCR-associated form when CD3-ζ becomes phosphorylated. Interestingly, CD3-ζ was found in its two tyrosine-phosphorylated forms of 18 and 21 kD in the apoptosis-deficient clones, contrarily to the findings on T cell clones stimulated with partial agonists, where only the lower molecular mass form of tyrosine-phosphorylated CD3-ζ was detected (44–46).
The involvement of CD3-ζ in the induction of apoptosis through the TCR complex has been shown earlier (27, 28). Although the components of the TCR–CD3 complex may be in part functionally redundant, we believe our data and those of Combadière et al. (27) suggest the existence of a specialized role for CD3-ζ in the induction of CD95-L. Furthermore, the data of Combadière et al. (27) showed that the membrane-proximal ITAM of CD3-ζ (ITAM1), and not the others, was effective in the induction of apoptosis. These results suggest that the functional specialization within the TCR–CD3 complex can be narrowed down not only to individual subunits but to particular regions of these subunits as well. In line with this idea, previous works in vitro have shown that different ITAMs of the TCR– CD3 complex have different affinities for components of the transduction machinery, including ZAP70 and the p85 subunit of PI 3-kinase (6, 8, 53). A further degree of specialization was made evident by Sunder-Plassman et al. (54), who showed that the NH2-terminal and COOH-terminal YxxL/I sequences within ITAM1 of CD3-ζ may each trigger different activation events.
In addition to a different pattern of CD3-ζ tyrosine phosphorylation, in APL-stimulated T cell clones ZAP70 was not tyrosine phosphorylated (44, 45). By contrast, in the apoptosis-deficient clones ZAP70 became tyrosine phosphorylated to similar or even higher levels than in wild-type clones. Although the impaired induction of CD95-L and apoptosis were not paralleled by a defective tyrosine phosphorylation of ZAP70, two results seem to link this tyrosine kinase to the underlying apoptosis induction defect. First, the stoichiometry of ZAP70/CD3-ζ association was very low in apoptosis-defective mutants. Second, in these mutants, ZAP70 is not found in a near-plasma membrane location. Tyrosine phosphorylation of ZAP70 is believed to be performed by Src family tyrosine kinases once ZAP70 is bound to tyrosine-phosphorylated CD3-ζ (reviewed in references 2, 3). Our findings in the apoptosis-deficient mutants are apparently contradictory to this idea, since ZAP70 becomes inducibly tyrosine phosphorylated even at higher levels than in wild-type cells, despite the fact that ZAP70 association to CD3-ζ and to the other subunits of the TCR complex is very poor. It may be that in the mutant cells ZAP70 associates transiently to the TCR complex, and that this association is sufficient for ZAP70 to become tyrosine phosphorylated and activated. It could also be speculated that the higher levels of tyrosine-phosphorylated ZAP70 detected in the mutant cells could be due to mislocation of ZAP70 that would make it less accessible to tyrosine phosphatases such as SHP-1, which binds and dephosphorylates ZAP70 (55). The differential induction of cytokines versus CD95-L in the apoptosis-resistant cells could thus derive from a quantitative difference in the levels of membrane-bound ZAP70. Alternatively, the differential signaling may derive from a different orientation adopted by ZAP70, since it has recently been suggested that ZAP70 may “sense” in this way different TCR ligands (56).
The tyrosine kinase activity of ZAP70 has recently been shown to be essential for TCR triggering of apoptosis via CD95-L expression (57) and, according to our data, the localization of ZAP70 near the plasma membrane is also necessary for the induction of CD95-L ligand. In this regard, it has been shown that the permanent association of ZAP70 to the plasma membrane by transfection of a CD2/ZAP70 chimera results in constitutive activation of the NFAT transcription factor and apoptosis (58). Thus, we could hypothesize the existence of a ZAP70 substrate, located at or near the plasma membrane, necessary for the induction of CD95-L expression. The transient recruitment of ZAP70 to the plasma membrane in the mutant cells would cause a deficit in the recruitment or in the phosphorylation of that substrate. So far, we do not know the identity of this possible substrate, since the pattern of total tyrosine-phosphorylated proteins is very similar to that of wild-type cells. It has been described that ZAP70 recruitment to the plasma membrane does not involve its SH2 domains and needs its kinase activity intact (52). These results imply the existence of a membrane-bound ZAP70 substrate that, once phosphorylated, may promote the localization of ZAP70 near the plasma membrane. The translocation of ZAP70 to the plasma membrane in fibroblasts, in the complete absence of T cell–specific components, argues strongly against an involvement of the TCR–CD3 complex. However, we have found in our mutant cells a correlation between impaired ZAP70 association to CD3-ζ and mislocation of ZAP70. Thus, it is likely that the association to CD3-ζ is indirectly involved, at least in T cells, in the localization of ZAP70 near the plasma membrane; perhaps as a requisite for the tyrosine phosphorylation of the putative membrane-bound ZAP70 substrate. Nevertheless, it is still possible that the strongly diminished binding of ZAP70 to CD3-ζ and the mislocation of ZAP70 observed in the mutant cells are independent processes. Overexpression of ZAP70 in the mutant clones, unlike in the wild-type ones, does not result in increased apoptosis, perhaps because the defective recruitment of ZAP70 to the cell surface is not bypassed. By contrast, the transfection and direct stimulation of a Tac–ζ chimera results in an induction of apoptosis comparable to that found in transfected wild-type cells. This result supports the hypothesis that the defective induction of apoptosis in the mutant clones results from an impaired recruitment of CD3-ζ to the TCR complex.
The expression of CD95-L is inhibited by cyclosporin A treatment (21–23) and is not induced in NFATp-deficient mice (24), strongly suggesting that it is dependent on NFAT transcription factors. Indeed, an inducible NFAT-binding site has been demonstrated in the CD95-L promoter (25). Thus, the regulation of the CD95-L gene expression seems to be very similar to that of cytokine genes. Furthermore, the induction of CD95-L expression is dependent on early signals that involve Lck (59, 60) and CD45 (25), as well as the participation of the ras pathway (25). Nevertheless, the inducible NFAT-binding site in the CD95-L promoter is more reminiscent of that of the TNF-α promoter than the IL-2 promoter (25). For instance, the CD95-L promoter does not contain a predicted AP-1–binding site in the proximity of the NFAT site. Against this background, it seems clear that the fine regulation of CD95-L versus IL-2 expression is dependent on rather subtle differences. The results shown in this paper and Rodríguez-Tarduchy et al. (28) show that it is possible to distinguish between the two pathways, and that the signaling pathways diverge very early in the signaling cascade, involving the ZAP70/CD3-ζ interaction. The unstable interaction of ZAP70 with CD3-ζ may cause an unbalanced cascade of phosphorylations, which may be detrimental for CD95-L expression but not for induction of IL-2 expression and for other activation events. Interestingly, it has recently been proposed that Lck, but not Fyn, may mediate a signaling activation pathway that results in CD95-L upregulation and apoptosis (60). Since it has been shown that tyrosine-phosphorylated ZAP70 recruits Lck through its SH2 domain (11, 51), an intriguing possibility is that CD95-L is not induced in the apoptosis-deficient cells analyzed here because Lck is not recruited by ZAP70. In support of this idea, it has been shown that Lck lies downstream of ZAP70 when apoptosis is triggered by permanent localization of ZAP70 near the plasma membrane (58).
Our results suggest that a point mutation in the transmembrane domain of TCR-β impairs the association of CD3-ζ, and that this is translated into transient association of ZAP70. This association is sufficient for activation of ZAP70 and to elicit the gene expression of IL-2, IFN-γ, CD25, and CD69, but not for induction of CD95-L. We are at present trying to characterize the pathway that links ZAP70 with CD95-L induction, and which must be inactive in the apoptosis-resistant cells.
Acknowledgments
We are indebted to Drs. Steven Ley, Arthur Weiss, Michael Owen, Sefik Alkan, Jesús Avila, and Peter Parker for providing reagents, and to Drs. Oreste Acuto, Andrés Alcover, and Gary Koretzky for critically reading the manuscript and for helpful discussions.
This work was supported by grants from Comisión Interministerial de Ciencia y Tecnologia (PM95.0005 and PM95.0047), Fondo de Investigaciones Samitarias (97/0597), and Comunidad de Madrid (07/047/96 and 07/055/96). The institutional support of the Fundación Ramón Areces is also acknowledged. A.G. Sahuquillo and E. Teixeiro are fellows of the Fundación Conchita Rábago and Universidad Complutense de Madrid, respectively.
Footnotes
Abbreviations used in this paper: APL, altered peptide ligands; ECL, enhanced chemiluminescence; GFP, green fluorescent protein; ITAM, immunoreceptor tyrosine-based activation motif; NFAT, nuclear factor of activated T cells; SEB, Staphylococcal enterotoxin B; SH2, src homology 2.
The first two authors contributed equally to this work.
References
- 1.Reth M. Antigen receptor tail clue. Nature. 1989;338:383–384. [PubMed] [Google Scholar]
- 2.Qian D, Weiss A. T cell antigen receptor transduction. Curr Opin Cell Biol. 1997;9:205–212. doi: 10.1016/s0955-0674(97)80064-6. [DOI] [PubMed] [Google Scholar]
- 3.Wange RL, Samelson LE. Complex complexes: signaling at the TCR. Immunity. 1996;5:197–205. doi: 10.1016/s1074-7613(00)80315-5. [DOI] [PubMed] [Google Scholar]
- 4.Chan AC, Shaw AS. Regulation of antigen receptor signal transduction by protein tyrosine kinases. Curr Opin Immunol. 1995;8:394–401. doi: 10.1016/s0952-7915(96)80130-0. [DOI] [PubMed] [Google Scholar]
- 5.Iwashima M, Irving BA, Van Oers NSC, Chan AC, Weiss A. Sequential interactions of the TCR with distinct cytoplasmic tyrosine kinases. Science. 1994;263:1136–1139. doi: 10.1126/science.7509083. [DOI] [PubMed] [Google Scholar]
- 6.Isakov N, Wange RL, Burgess WH, Watts JD, Aebersold R, Samelson LE. ZAP-70 binding specificity to T cell receptor tyrosine-based activation motifs: the tandem SH2 domains of ZAP-70 bind distinct tyrosine-based activation motifs with varying affinity. J Exp Med. 1995;181:375–380. doi: 10.1084/jem.181.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bu JY, Shaw AS, Chan AC. Analysis of the interaction of ZAP-70 and syk protein-tyrosine kinases with the T-cell antigen receptor by plasmon resonance. Proc Natl Acad Sci USA. 1995;92:5106–5110. doi: 10.1073/pnas.92.11.5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osman N, Lucas SC, Turner H, Cantrell D. A comparison of the interaction of Shc and the tyrosine kinase ZAP-70 with the T cell antigen receptor zeta chain tyrosine-based activation motif. J Biol Chem. 1995;270:13981–13986. doi: 10.1074/jbc.270.23.13981. [DOI] [PubMed] [Google Scholar]
- 9.Van Oers NSC, Killeen N, Weiss A. Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J Exp Med. 1996;183:1053–1062. doi: 10.1084/jem.183.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J, Zhao Q, Kurosaki T, Weiss A. The Vav binding site (Y315) in ZAP-70 is critical for antigen receptor-mediated signal transduction. J Exp Med. 1997;10:1877–1882. doi: 10.1084/jem.185.10.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duplay P, Thome M, Hervé F, Acuto O. p56lck interacts via its src homology 2 domain with the ZAP-70 kinase. J Exp Med. 1994;179:1163–1172. doi: 10.1084/jem.179.4.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–1462. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 13.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 14.Strasser A. Death of a T cell. Nature. 1995;373:385–386. doi: 10.1038/373385a0. [DOI] [PubMed] [Google Scholar]
- 15.Van Parijs L, Abbas AK. Role of Fas-mediated cell death in the regulation of immune responses. Curr Opin Immunol. 1996;8:355–361. doi: 10.1016/s0952-7915(96)80125-7. [DOI] [PubMed] [Google Scholar]
- 16.Wong B, Choi Y. Pathways leading to cell death in T cells. Curr Opin Immunol. 1997;9:358–364. doi: 10.1016/s0952-7915(97)80082-9. [DOI] [PubMed] [Google Scholar]
- 17.Winoto A. Cell death in the regulation of immune responses. Curr Opin Immunol. 1997;9:365–370. doi: 10.1016/s0952-7915(97)80083-0. [DOI] [PubMed] [Google Scholar]
- 18.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumor necrosis factor. Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 19.Sytwu HK, Libau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5:17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 20.Park CG, Lee SY, Kandala G, Lee SY, Choi Y. A novel gene product that couples TCR signaling to Fas (CD95) expression in activation-induced cell death. Immunity. 1996;4:583–591. doi: 10.1016/s1074-7613(00)80484-7. [DOI] [PubMed] [Google Scholar]
- 21.Anel A, Buferne M, Boyer C, Schmitt-Verhulst AM, Golstein P. T cell receptor-induced Fas ligand expression in cytotoxic T lymphocyte clones is blocked by protein tyrosine kinase inhibitors and cyclosporin A. Eur J Immunol. 1994;24:2469–2476. doi: 10.1002/eji.1830241032. [DOI] [PubMed] [Google Scholar]
- 22.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 23.Brunner T, Nam JY, LaFace D, Ware CF. Activation-induced cell death in murine T cell hybridomas: differential regulation of Fas (CD95) versus Fas ligand expression by cyclosporin A and FK506. Int Immunol. 1996;8:1017–1026. doi: 10.1093/intimm/8.7.1017. [DOI] [PubMed] [Google Scholar]
- 24.Hodge MR, Ranger AM, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity. 1996;4:397–405. doi: 10.1016/s1074-7613(00)80253-8. [DOI] [PubMed] [Google Scholar]
- 25.Latinis KM, Carr LL, Peterson EJ, Norian LA, Eliason SL, Koretzky GA. Regulation of CD95 (Fas) ligand expression by TCR-mediated signaling events. J Immunol. 1997;158:4602–4611. [PubMed] [Google Scholar]
- 26.Vignaux F, Vivier E, Malissen B, Depraetere V, Nagata S, Golstein P. TCR/CD3 coupling to Fas-based cytotoxicity. J Exp Med. 1995;181:781–786. doi: 10.1084/jem.181.2.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Combadière B, Freedman M, Chen L, Shores EW, Love P, Lenardo MJ. Qualitative and quantitative contributions of the T cell receptor ζ chain to mature T cell apoptosis. J Exp Med. 1996;183:2109–2117. doi: 10.1084/jem.183.5.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodríguez-Tarduchy G, Sahuquillo AG, Alarcón B, Bragado R. Apoptosis but not other activation events is inhibited by a mutation in the transmembrane domain of T cell receptor β that impairs CD3ζ association. J Biol Chem. 1996;271:30417–30425. doi: 10.1074/jbc.271.48.30417. [DOI] [PubMed] [Google Scholar]
- 29.Letourneur F, Klausner RD. T-cell and basophil activation through the cytoplasmic tail of T-cell receptor ζ family proteins. Proc Natl Acad Sci USA. 1991;88:8905–8909. doi: 10.1073/pnas.88.20.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sancho J, Ledbetter J, Choi MS, Kanner SB, Deans JP, Terhorst C. CD3-ζ surface expression is required for CD4-p56lck-mediated up-regulation of T cell antigen receptor-CD3 signaling in T cells. J Biol Chem. 1992;267:7871–7879. [PubMed] [Google Scholar]
- 31.Huby RDJ, Carlile GW, Ley SC. Interactions between the protein-tyrosine kinase ZAP-70, the proto-oncoprotein Vav, and tubulin in Jurkat T cells. J Biol Chem. 1995;270:30241–30244. doi: 10.1074/jbc.270.51.30241. [DOI] [PubMed] [Google Scholar]
- 32.Alarcón B, Ley SC, Sánchez-Madrid F, Blumberg RS, Ju ST, Fresno M, Terhorst C. The CD3-γ and CD3-δ subunits of the T cell antigen receptor can be expressed within distinct functional TCR/CD3 complexes. EMBO (Eur Mol Biol Organ) J. 1991;10:903–912. doi: 10.1002/j.1460-2075.1991.tb08023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alkan S, Akdis C, Towbin H. Chemiluminescent and enzyme-linked immunoassays for sensitive detection of human IFN-γ. J Immunoassay. 1994;15:217–238. doi: 10.1080/15321819408009574. [DOI] [PubMed] [Google Scholar]
- 34.Shaw AC, Mitchell RN, Wever YK, Campos-Torres J, Abbas AK, Leder P. Mutations of immunoglobulin transmembrane and cytoplasmic domains: effects on intracellular signaling and antigen presentation. Cell. 1990;63:381–392. doi: 10.1016/0092-8674(90)90171-a. [DOI] [PubMed] [Google Scholar]
- 35.Bonay P, Munro S, Fresno M, Alarcón B. Intra-Golgi transport inhibition by megalomicin. J Biol Chem. 1996;271:3719–3726. doi: 10.1074/jbc.271.7.3719. [DOI] [PubMed] [Google Scholar]
- 36.Valitutti, S., Müller, S., M. Salio, and A. Lanzavecchia. 1997. Degradation of T cell receptor (TCR)-CD3-ζ complexes after antigenic stimulation. J. Exp. Med. 185:1859–1864. [DOI] [PMC free article] [PubMed]
- 37.Kishimoto H, Kubo RT, Yorifuji H, Nakayama T, Asano Y, Tada T. Physical dissociation of the TCR/ CD3 complex accompanies receptor ligation. J Exp Med. 1995;182:1997–2006. doi: 10.1084/jem.182.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wegener AMK, Letourneur F, Hoeveler A, Brocker T, Luton F, Malissen B. The T cell receptor/CD3 complex is composed of at least two autonomous transduction modules. Cell. 1992;68:83–95. doi: 10.1016/0092-8674(92)90208-t. [DOI] [PubMed] [Google Scholar]
- 39.Granja C, Lin LL, Yunis EJ, Relias V, Dasgupta JD. PLCγ1: a possible mediator of T cell receptor function. J Biol Chem. 1991;266:16277–16280. [PubMed] [Google Scholar]
- 40.Secrist JP, Karnitz L, Abraham RT. T cell antigen receptor ligation induces tyrosine phosphorylation of phospholipase C-γ1. J Biol Chem. 1991;266:12135–12139. [PubMed] [Google Scholar]
- 41.Weiss A, Koretzky G, Schatzman RC, Kadlecek T. Functional activation of the T cell antigen receptor induces tyrosine phosphorylation of phospholipase C-γ1. Proc Natl Acad Sci USA. 1991;88:5484–5488. doi: 10.1073/pnas.88.13.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park DJ, Rho HW, Rhee SG. CD3 stimulation causes phosphorylation of phospholipase C-γ1 on serine and tyrosine residues in a human T cell line. Proc Natl Acad Sci USA. 1991;88:5453–5456. doi: 10.1073/pnas.88.12.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katzav S, Sutherland M, Packham G, Yi T, Weiss A. The protein tyrosine kinase ZAP-70 can associate with the SH2 domain of proto-vav. J Biol Chem. 1994;269:32579–32585. [PubMed] [Google Scholar]
- 44.Sloan-Lancaster J, Shaw AS, Rothbard JB, Allen PM. Partial T cell signaling: Altered phospho-ζ and lack of ZAP70 recruitment in APL-induced T cell anergy. Cell. 1994;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 45.Madrenas J, Wange RL, Wang JL, Isakov N, Samelson LE, Germain RN. ζ phosphorylation without ZAP70 activation induced by TCR antagonists or partial agonists. Science. 1995;267:515–518. doi: 10.1126/science.7824949. [DOI] [PubMed] [Google Scholar]
- 46.Smith JA, Tso JY, Clark MR, Cole MS, Bluestone JA. Nonmitogenic anti-CD3 monoclonal antibodies deliver partial T cell receptor signal and induce clonal anergy. J Exp Med. 1997;185:1413–1422. doi: 10.1084/jem.185.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sieh M, Batzer A, Schlessinger J, Weiss A. Grb2 and phospholipase Cγ1 associate with a 36- to 38-kilodalton phosphotyrosine protein after T cell receptor stimulation. Mol Cell Biol. 1994;14:4435–4442. doi: 10.1128/mcb.14.7.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buday L, Egan SE, Rodríguez-Viciana P, Cantrell DA, Downward J. A complex of Grb2-adapter protein, SOS exchange factor and a 36 kD membrane bound tyrosine phosphoprotein is implicated in Ras activation in T cells. J Biol Chem. 1994;269:9019–9023. [PubMed] [Google Scholar]
- 49.Motto D, Ross S, Jackman J, Sun Q, Olson AL, Findell P, Koretzky G. In vivo association of Grb2 with pp116, a substrate of the T cell antigen receptor activated protein tyrosine kinase. J Biol Chem. 1994;269:21608–21613. [PubMed] [Google Scholar]
- 50.Isakov N, Wange RL, Watts JD, Aebersold R, Samelson LE. Purification and characterization of human ZAP70 protein-tyrosine kinase from a baculovirus expression system. J Biol Chem. 1996;271:15753–15761. doi: 10.1074/jbc.271.26.15753. [DOI] [PubMed] [Google Scholar]
- 51.Thome M, Duplay P, Guttinger M, Acuto O. Syk and ZAP-70 mediate recruitment of p56lck/CD4 to the activated T cell receptor/CD3/ζ complex. J Exp Med. 1995;181:1997–2006. doi: 10.1084/jem.181.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huby RDJ, Iwashima M, Weiss A, Ley SC. ZAP-70 protein tyrosine kinase is constitutively targeted to the T cell cortex independently of its SH2 domains. J Cell Biol. 1997;137:1639–1649. doi: 10.1083/jcb.137.7.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Exley M, Varticovski L, Peter M, Sancho J, Terhorst C. Association of phosphatidylinositol 3-kinase with a specific sequence of the T cell receptor ζ chain is dependent on T cell activation. J Biol Chem. 1994;269:15140–15146. [PubMed] [Google Scholar]
- 54.Sunder-Plassmann R, Lialios F, Madsen M, Koyasu S, Reinherz EL. Functional analysis of immunoreceptor tyrosine-based activation motif (ITAM)-mediated signal transduction: the two YxxL segments within a single CD3ζ-ITAM are functionally distinct. Eur J Immunol. 1997;27:2001–2009. doi: 10.1002/eji.1830270826. [DOI] [PubMed] [Google Scholar]
- 55.Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, Chan AC, Thomas ML. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science. 1996;272:1173–1176. doi: 10.1126/science.272.5265.1173. [DOI] [PubMed] [Google Scholar]
- 56.Graef IA, Holsinger LJ, Diver S, Schreiber SL, Crabtree GR. Proximity and orientation underlie signaling by the non-receptor tyrosine kinase ZAP70. EMBO (Eur Mol Biol Organ) J. 1997;16:5618–5628. doi: 10.1093/emboj/16.18.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eischen CM, Williams BL, Zhang W, Samelson LE, Lynch DH, Abraham RT, Leibson PJ. ZAP-70 tyrosine kinase is required for the up-regulation of fas ligand in activation-induced T cell apoptosis. J Immunol. 1997;159:1135–1139. [PubMed] [Google Scholar]
- 58.Yamasaki S, Takamatsu M, Iwashima M. The kinase, SH3 and SH2 domains of Lck play critical roles in T-cell activation after ZAP-70 membrane localization. Mol Cell Biol. 1996;13:7408–7417. doi: 10.1128/mcb.16.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oyaizu N, Than S, McCloskey TW, Pahwa S. Requirement of p56lckin T-cell receptor/CD3 mediated apoptosis and Fas-ligand induction in Jurkat cells. Biochem Biophys Res Commun. 1995;213:994–1001. doi: 10.1006/bbrc.1995.2227. [DOI] [PubMed] [Google Scholar]
- 60.González-García, A., L.R. Borlado, E. Leonardo, I. Mérida, C. Martínez-A, and A.C. Carrera. 1997. Lck is necessary and sufficient for Fas-ligand expression and apoptotic cell death in mature cycling T cells. J. Immunol. 158:4104–4112. [PubMed]