

Abstract
Signal transducers and activators of transcription 6 (STAT6) is essential for interleukin 4–mediated responses, including class switching to IgE and induction of type 2 T helper cells. To investigate the role of STAT6 in allergic asthma in vivo, we developed a murine model of allergen-induced airway inflammation. Repeated exposure of actively immunized C57BL/6 mice to ovalbumin (OVA) aerosol increased the level of serum IgE, the number of eosinophils in bronchoalveolar lavage (BAL) fluid, and airway reactivity. Histological analysis revealed peribronchial inflammation with pulmonary eosinophilia in OVA-treated mice. In STAT6-deficient (STAT6−/−) C57BL/6 mice treated in the same fashion, there were no eosinophilia in BAL and significantly less peribronchial inflammation than in wild-type mice. Moreover STAT6−/− mice had much less airway reactivity than wild-type mice. These findings suggest that STAT6 plays a crucial role in the pathogenesis of allergen-induced airway inflammation.
Atopic bronchial asthma is characterized by an increased serum IgE level and an eosinophilic inflammation of airway, which are accompanied by increased airway reactivity (1–6). The cytokine IL-4 has pleiotropic effects and appears to play a key role in the pathogenesis of atopic diseases (7, 8). IL-4–deficient (IL-4−/−) mice fail to develop both increase in the level of IgE in serum (9–12) and eosinophil recruitment into airways (11, 12). Moreover, the airway hyperreactivity normally resulting from allergen challenge is abolished in IL-4−/− mice (12) and mice treated with anti–IL-4 antibody (13). These data show that IL-4 is a central mediator in the pathogenesis of allergic asthma.
Signal transducers and activators of transcription (STAT) proteins are family of transcription factors that mediate many cytokine-induced responses (14). STAT6 is tyrosine phosphorylated and activated in response to IL-4 (15, 16). Similar to IL-4−/− mice (9, 10), STAT6-deficient (STAT6−/−) mice also abrogate IL-4–mediated functions including Th2 differentiation, expression of cell surface markers, and Ig class switching to IgE (17–19). These findings demonstrate that STAT6 is required for IL-4–specific functions, despite the existence of multiple signaling pathways activated by IL-4 (20, 21). However, it is still unclear whether a STAT6-mediated signal is also in the pathogenesis of both the peribronchial inflammation and the airway hyperreactivity.
In this study, we examined the roles played by STAT6 in a murine model of allergen-induced airway inflammation. Our findings show that STAT6 may play a critical role in the development of the pathophysiology of allergic asthma.
Materials and Methods
Animals.
C57/BL6 mice with targeted disruption of the gene encoding STAT6 (STAT6−/− mice) were generated in the Department of Biochemistry, Hyogo College of Medicine (Hyogo, Japan), as previously reported (17), and inbred in Experimental Technology Research Center, Daiichi Pharmaceutical Co., Ltd. (Tokyo, Japan). Age-matched C57/BL6 female mice were purchased from SLC (Shizuoka, Japan). All animals were housed under specific pathogen-free conditions, and had free access to commercial diet and water.
Immunization and Exposure of Mice.
On the first day of the experiment (day 0) and day 12, 5- or 6-wk-old female mice were actively immunized by intraperitoneal injection of 50 μg of OVA (Sigma Chemical Co., St. Louis, MO), adsorbed to 1 mg of alum (Kyowa Kagaku, Kagawa, Japan) in 0.9% sterile saline. On day 23, mice were exposed to an aerosol of OVA in 0.9% sterile saline for 30 min three times at 1-h intervals, and then every second day thereafter, for 8 d. The aerosol was generated with an ultrasonic nebulizer (NE-U12; Omron, Mie, Japan) driven by a plastic filling box (length: 21.5 cm; width: 28.5 cm; height: 21.5 cm). According to the specifications supplied by the manufacturer, the output of the nebulizer is 0.5 ml/min, and the particles produced have a mean diameter of 3.5 μm. The concentration of OVA in the nebulizer was 10 mg/ml. 24 h after the last aerosol exposure, the mice were anesthetized by intraperitoneal injection of pentobarbital 80 mg/kg body weight (Tokyo Kasei, Tokyo, Japan) and used for the following experiments.
Measurement of Serum Concentrations of IgE and IgM.
Blood was drawn from the sinus cavernosus, and serum was obtained by centrifugation at 1,500 g for 10 min. Serum IgE level was determined with a commercial ELISA kit (Yamasa, Chiba, Japan). Concentrations of IgM were determined by ELISA using goat anti–mouse IgM (Southern Biochemistry Assoc., Birmingham, AL) as capture antibody and goat anti–mouse IgM labeled with biotin (Southern Biochemistry Assoc.) as detection antibody (17).
Bronchoalveolar Lavage.
The trachea was cannulated and the airway lumina were washed twice with 0.5 ml of phosphate-buffered saline (free of ionized calcium and magnesium) supplemented with 0.05 mM sodium EDTA (Sigma Chemical Co.) and 0.1% bovine serum albumin (Sigma Chemical Co.). The bronchoalveolar lavage (BAL) fluid was immediately centrifuged (10 min, 4°C, 700 g). The BAL cells were resuspended in 0.2 ml of rat serum and counted using a hemocytometer.
For differential cell counts, 2–4 × 103 cells were spun onto glass slides (Cytospin2; Cytospin, Shandon, UK), air dried, fixed with ethanol, and stained with Giemsa solution. The number of eosinophils, neutrophils, and macrophages/monocytes in 200 cells were counted based on morphology and staining characteristics.
Lung Histology.
The lungs were resected, fixed in 10% phosphate-buffered formalin, embedded into paraffin, sectioned, stained with hematoxylin-eosin solution, and examined by light microscopy for histologic changes.
Measurement of Airway Hyperreactivity.
The trachea was cannulated and connected to a rodent ventilator (683; Harvard Apparatus, South Natick, MA) with an in-line pressure transducer (DP45-28; Validyne Engineering Corp., Northridge, CA) that was coupled to a pulmonary mechanics analyzer (6; Buxco Electronics, Inc., Sharon, CT). Flows were determined by measuring the differential pressure (DP45-14; Validyne Engineering Corp.) across six layers of 400-mesh wire cloth covering a 1.3-cm hole in a plethysmograph box (Plyan-M; Buxco Electronics, Inc.). Mice were placed in the plethysmograph box and were then ventilated at 120 strokes/min with a stroke volume of 0.3 ml after neuromuscular blockade with 10 mg/kg succinylcholine (Tokyo Kasei). After establishing a stable baseline of lung resistance (RL), acetylcholine (Ach; Daiichi Pharmaceutical Co., Ltd., Tokyo, Japan) was cumulatively administered (31.5–1,000 μg/kg) via the tail vein, and changes in RL were monitored.
Statistical Analysis.
All results are expressed as the mean ±SD. Lung resistances in various groups were compared with a Dunnett's test.
Results
No Elevation of Serum IgE in STAT6− /− Mice.
C57BL/6 wild-type and STAT6−/− mice immunized with OVA were treated with aerosol of OVA. At the 24-h time point after OVA challenge, mice were bled, and serum concentration of Ig was measured by ELISA. C57BL/6 wild-type mice displayed the expected increase in IgE concentration after the treatment of OVA (Fig. 1 a). In contrast, the IgE level was below 10 ng/ml in OVA-challenged STAT6−/− mice of C57BL/6 background. Serum concentration of IgM was equally increased after OVA challenge in both wild-type and STAT6−/− mice, indicating that both mice were sensitized with OVA (Fig. 1 b). Thus, STAT6−/− mice displayed no IgE response to OVA challenge.
Figure 1.

Changes in serum concentrations of IgE and IgM induced by OVA treatment. The columns represent the concentration of serum IgE (a) and IgM (b) in wild-type and STAT6−/− mice after treatment with (OVA) or without (Control) OVA. Results shown are from a single experiment representative of three separate experiments. N.D., not detectable (<10 ng/ml).
No Eosinophilia in BAL Fluid of STAT6−/− Mice.
To determine whether STAT6 deficiency influences airway eosinophilia, we analyzed the cells in BAL fluid 24 h later than the last exposure to OVA aerosol (Fig. 2). In actively immunized wild-type mice, the total cell number of BAL cells was dramatically increased after repeated exposure to OVA. Cell differentiation analysis revealed the marked elevation of eosinophil population and slight increase in macrophages and neutrophils in wild-type mice. In contrast, the total cell number of BAL cells was not increased after OVA exposure in STAT6−/− mice. Furthermore, eosinophils were scarcely detected in BAL fluid of STAT6−/− mice, even after OVA exposure, demonstrating that STAT6−/− mice exhibited no eosinophilia in the airway, even after aerosolization with OVA.
Figure 2.

Effect of STAT6 deficiency on total BAL cell number and cellular distribution of BAL fluid. BAL fluid was obtained from wild-type and STAT6−/− mice after treatment with (OVA) or without (Control) OVA. The columns represent the absolute numbers of total BAL cells, eosinophils, neutrophils, macrophages, and other cells (mostly lymphocytes) per animal. Results shown are from a single experiment representative of three separate experiments.
No Inflammatory Response of Lungs in STAT6−/− Mice.
In wild-type mice, exposure to OVA induced patchy inflammatory infiltrates in the lungs (Fig. 3 A). These infiltrates were mainly located around the small bronchi, bronchioles, and blood vessels. Most of peribronchial inflammatory cells consisted of lymphocytes and eosinophils. In contrast, any inflammatory changes, including infiltration of eosinophils, were not observed in the lungs of STAT6−/− mice after aerosolization with OVA (Fig. 3 B). Examination of the lungs taken from OVA-treated STAT6−/− mice revealed normal lung histology similar to that in the lungs from untreated wild-type mice (Fig. 3 C). Thus, STAT6−/− mice displayed no inflammatory responses to OVA.
Figure 3.
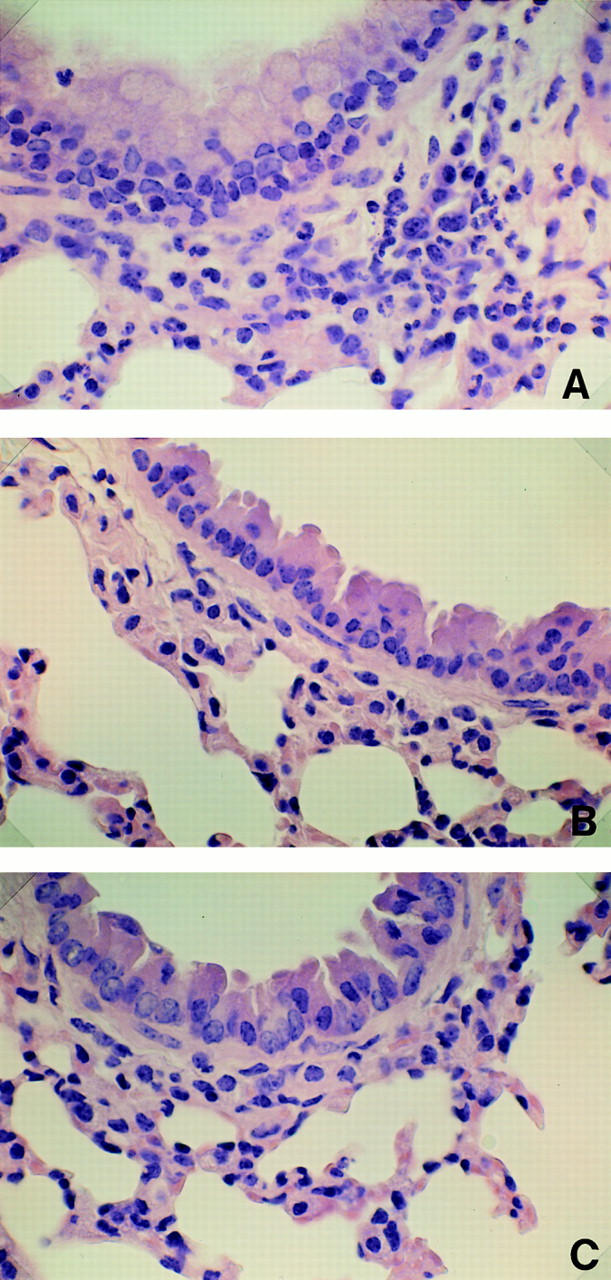
Microscopic examination of lung tissues from wild-type and STAT6−/− mice. Lung preparations from both wild-type and STAT6−/− mice treated with or without OVA were stained with hematoxylin-eosin. Photomicrographs of these preparations were evaluated infiltration of eosinophils and other lymphocytes in bronchial mucosa and perivascular sheaths in lung. Noticeable cell accumulation of eosinophils appeared in lungs from OVA-treated wild-type mice (A). In contrast, the inflammatory cells were not shown in lungs from OVA-treated STAT6−/− mice (B). The lungs taken from untreated wild-type mice represent normal lung histology (C). Original magnification: 200.
No Increased Airway Reactivity in STAT6−/− Mice.
Mice immunized and subsequently aerosolized with OVA were analyzed for airway reactivity after intravenous administration of Ach (Fig. 4). In wild-type mice, OVA treatment significantly increased the airway response to Ach, as compared with untreated mice. However, the airway response to Ach in OVA-treated STAT6−/− mice was same as that in nontreated STAT6−/− and wild-type mice, demonstrating that STAT6−/− mice did not exhibit airway hyperreactivity after OVA sensitization.
Figure 4.

Effect of STAT6 deficiency on airway reactivity in OVA-treated mice. Airway reactivity assessed by measurement of RL after intravenous Ach challenge in wild-type mice (circles) and STAT6−/− mice (squares) treated with (closed symbols) or without (open symbols) OVA. Only in wild-type mice, OVA treatment significantly increased airway reactivity compared with that in the untreated group (*P <0.05). Results shown are from a single experiment representative of three separate experiments.
Discussion
In this study, we demonstrate that pulmonary eosinophilia, airway hyperreactivity, and lung damage usually seen in mice immunized and challenged with antigen are not observed in STAT6−/− mice, suggesting that STAT6 activation plays an essential role in the pathogenesis of allergic airway inflammation.
Bronchial asthma is a chronic airway disease with reversible airway obstruction and airway inflammation. The pathophysiological changes in asthma are characterized by increased serum IgE level, eosinophil infiltration around airways, bronchial mucosal injury, and airway hyperreactivity (1–6). This pathophysiologic process of asthma is thought to involve T helper cells with a Th2 cytokine phenotype. It has been reported that depletion of cluster of differentiation (CD)4-positive lymphocytes prevents antigen-induced airway reactivity and recruitment of eosinophils to the airways (22). Bronchoalveolar lymphocytes and T cell clones from airway mucosa with allergic respiratory disorders synthesized and released IL-3, -4, -5, and GM-CSF, indicating predominant differentiation of Th2 (23, 24). Moreover, either IL-4 (12) or -5 (25) deficiency abolishes airway hyperreactivity in mouse asthma models. Thus, there is no doubt that IL-4 and -5 are key cytokines participating in the various aspects of manifestations of asthma.
However, there are apparently discrepant reports on their relative importance in airway hyperreactivity. Corry et al. showed that neutralization of IL-4 using monoclonal antibodies abrogated airway hyperreactivity but had little effect on the influx of eosinophils, and that administration of anti–IL-5 antibodies suppressed eosinophil recruitment but had no effect on the subsequent airway response (13). These data are similar to what was found in another report in which bronchial responses to carbachol and serotonin were substantially less in IL-4−/− mice than in wild-type mice (12), implicating IL-4 as the key cytokine and indicating that airway hyperreactivity can occur independently of IL-5 and recruitment of eosinophils. In contrast, Foster et al. provide equally convincing evidence that IL-5 is the critical cytokine (25). They showed that eosinophilia, lung damage, and airway hyperreactivity were abolished in IL-5–deficient mice, and that reconstitution of IL-5 production with recombinant vaccinia viruses completely restored aeroallergen-induced eosinophilia and airway dysfunction. Drazen et al. discussed two conflicting studies and speculated that there are two distinct mechanisms for induction of airway hyperreactivity, that is, IgE-stimulated activation of mast cells that occurred in the model used by Corry et al. and eosinophil-dependent pathway demonstrated in the experiment by Foster et al. (26). Recently, Foster et al. published a report showing that the recruitment of eosinophils to airways was not abolished or the characteristic airway damage and hyperreactivity were not attenuated in IL-4−/− mice (27). Moreover, they showed that treatment of IL-4−/− mice with anti–IL-5 antibodies before aeroallergen challenge abolished blood and airway eosinophilia, lung damage, and airway hyperreactivity. These results indicate that IL-4 is not essential for the development of IL-5–producing CD4-positive T cells or for the induction of eosinophilic inflammation, airway damage, and hyperreactivity. This finding contrasts with our present finding that STAT6−/− mice did not develop airway eosinophilia, lung damage, or airway hyperreactivity.
Although STAT6 is clearly shown to be essential for IL-4 signaling, IL-4 is not the only cytokine that activates STAT6. It has been reported that IL-13 also activates STAT6 (28). Moreover, we have demonstrated that IL-13 signaling is completely abolished in STAT6−/− mice (29). IL-4 and -13 are related cytokines with many common biological properties (30). Overlapping biological properties of IL-4 and -13 is now explained by the sharing of IL-4 receptor α chain as a common receptor component between two cytokines (30). The IL-13 signaling pathway appears to be an alternative pathway for IL-4 signaling in humans (31–33). In patients with chronic allergic disease, repeated stimulation of allergen-specific memory CD4 T cells appears to induce IgE production largely via IL-13 (34). More importantly, like IL-4, IL-13 has the capacity to make naive T cells differentiate into Th2s (19). Besides IL-4 and -13, STAT6 is activated through a leptin receptor (35), surface Ig (36), CD40 (36), or may be activated through as yet unknown receptors. Some of these signalings may also participate in Th2 response. Although IL-4 plays a central role in the differentiation of Th0s into Th2s, there is the possibility that factors other than IL-4 mediate Th2 differentiation in the absence of IL-4. Indeed, the residual Th2 response has been clearly observed in IL-4−/− mice (37), in contrast to the almost complete abolishment of Th2 response in STAT6−/− mice (17–19). This may explain the marked difference in the morphological and functional changes of the airways in response to aeroallergen challenge between IL-4−/− mice and STAT6−/− mice.
In conclusion, this study demonstrates the critical role for STAT6 in the development of murine aeroallergen-induced airway inflammation, and suggests that blocking the action of STAT6 may be a therapeutic approach worth testing in the treatment of asthma.
Acknowledgments
We thank Drs. Shin-etsu Ono, Shunzo Aibara, Yasuo Kita, and Katsuhiko Nawa for valuable discussions in the course of this study. We also thank Dr. Kotaro Ishibashi for breeding STAT6−/− mice, and Dr. Yutaka Iigo for histologic assessment of lung tissues.
This work has been supported in part by Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Corporation (JST).
References
- 1.Burrow B, Martines F, Halonen M, Barbee R, Cline M. Association of asthma with serum IgE levels and skin test reactivity to allergens. N Engl J Med. 1989;320:271–277. doi: 10.1056/NEJM198902023200502. [DOI] [PubMed] [Google Scholar]
- 2.Sears MR, Burrows B, Flannery EM, Herbinson GP, Hewitt CJ, Holdaway MD. Relationship between airway hyperresponsiveness and serum IgE in child with asthma and in apparently normal children. N Engl J Med. 1991;325:1067–1071. doi: 10.1056/NEJM199110103251504. [DOI] [PubMed] [Google Scholar]
- 3.Bousquet J, Chanez P, Lacote JY, Barneon G, Ghavanian N, Enander I, Venge P, Ahlstedt S, Lafontaine JS, Godard P, Michel FB. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 4.Djukanovic R, Roche WR, Wilson JM, Beasley CRW, Twentyman OP, Howarth PH, Holgate ST. Mucosal inflammation in asthma. Am Rev Respir Dis. 1990;141:263–275. doi: 10.1164/ajrccm/142.2.434. [DOI] [PubMed] [Google Scholar]
- 5.Gleich G. The eosinophil and bronchial asthma. Current understanding. J Allergy Clin Immunol. 1990;85:422–436. doi: 10.1016/0091-6749(90)90151-s. [DOI] [PubMed] [Google Scholar]
- 6.Kay AB. Asthma and inflammation. J Allergy Clin Immunol. 1991;87:893–910. doi: 10.1016/0091-6749(91)90408-g. [DOI] [PubMed] [Google Scholar]
- 7.Paul WE, Ohara J. B cell stimulatory factor–1/ interleukin-4. Annu Rev Immunol. 1987;5:429–459. doi: 10.1146/annurev.iy.05.040187.002241. [DOI] [PubMed] [Google Scholar]
- 8.Paul WE. Interleukin-4: a prototypic immunoregulatory lymphokine. Blood. 1991;77:1859–1870. [PubMed] [Google Scholar]
- 9.Kühn R, Rajewsky K, Müller W. Generation and analysis of interleukin-4 deficient mice. Science. 1991;254:707–710. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- 10.Kopf M, LeGros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokines. Nature. 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 11.Brusselle GG, Kips JC, Tavernier JH, Van Der Heyden JG, Cuvelier CA, Pauwels RA, Bluethmann H. Attenuation of allergic airway inflammation in IL-4 deficient mice. Clin Exp Allergy. 1994;24:73–80. doi: 10.1111/j.1365-2222.1994.tb00920.x. [DOI] [PubMed] [Google Scholar]
- 12.Kips JC, Brussele GG, Joos GF, Peleman RA, Devos RR, Tavernier JH, Pauwels RA. Importance of interleukin-4 and interleukin-12 in allergen-induced airway changes in mice. Int Arch Allergy Immunol. 1995;107:115–118. doi: 10.1159/000236947. [DOI] [PubMed] [Google Scholar]
- 13.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ihle JN. Cytokine receptor signaling. Nature. 1995;377:591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 15.Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL. An interleukin-4–induced transcription factor: IL-4 stat. Science. 1994;265:1701–1706. doi: 10.1126/science.8085155. [DOI] [PubMed] [Google Scholar]
- 16.Quelle FW, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben SM, Cleveland JL, Pierce JH, Keegan AH, Nelms K, et al. Cloning of murine stat6 and human stat6, stat proteins that are tyrosine phosphorylated in response to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol. 1995;15:3336–3343. doi: 10.1128/mcb.15.6.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signaling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 18.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DAA, et al. Lack of IL-4–induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 20.Keegan AD, Nelms K, Wang L-M, Pierce JH, Paul WE. Interleukin 4 receptor: signaling mechanisms. Immunol Today. 1994;15:423–432. doi: 10.1016/0167-5699(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 21.Ryan JJ. Interleukin-4 and its receptor: essential mediators of the allergic response. J Allergy Clin Immunol. 1997;99:1–5. doi: 10.1016/s0091-6749(97)70293-8. [DOI] [PubMed] [Google Scholar]
- 22.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+ T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 23.Robinson DR, Hamid Q, Ying S, Tsicopoulus A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant Th2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 24.Del Prete GF, De Carli M, D'Elios MM, Maestrelli P, Ricci M, Fabbri L, Romagnani S. Allergen exposure induces the activation of allergen-specific Th2 cells in airway mucosa of patients with allergic respiratory disorders. Eur J Immunol. 1993;23:1445–1449. doi: 10.1002/eji.1830230707. [DOI] [PubMed] [Google Scholar]
- 25.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med. 1996;183:1–5. doi: 10.1084/jem.183.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horgan SP, Mould A, Kikutani H, Ramsay AJ, Foster PS. Aeroallergen-induced eosinophilic inflammation, lung damage, and airways hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J Clin Invest. 1997;99:1329–1339. doi: 10.1172/JCI119292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Köhler I, Alliger P, Minty A, Caput D, Ferrara P, Holl-Neugebauer B, Rank G, Rieber EP. Human interleukin-13 activates the interleukin-4–dependent transcription factor NF–IL-4 sharing a DNA binding motif with an interferon-γ–induced nuclear binding factor. FEBS Lett. 1994;345:187–192. doi: 10.1016/0014-5793(94)00438-2. [DOI] [PubMed] [Google Scholar]
- 29.Takeda K, Kamanaka M, Tanaka T, Kishimoto T, Akira S. Impaired IL-13–mediated functions of macrophages in STAT6-deficient mice. J Immunol. 1996;157:3220–3222. [PubMed] [Google Scholar]
- 30.Callard RE, Matthews DJ, Hibbert LM. Interleukin 4 and interleukin 13: same response, different receptors. Biochem Soc Trans. 1997;25:451–455. doi: 10.1042/bst0250451. [DOI] [PubMed] [Google Scholar]
- 31.Matthew DJ, Clark PA, Herbert J, Morgan G, Armitage RJ, Kinnon C, Minty A, Grabstein KH, Caput D, Ferrara P, Callard R. Function of the interleukin-2 (IL-2) receptor γ-chain in biologic responses of X-linked severe combined immunodeficient B cells to IL-2, IL-4, IL-13, and IL-15. Blood. 1995;85:38–42. [PubMed] [Google Scholar]
- 32.Izuhara K, Heike T, Otsuka T, Yamaoka K, Mayumi M, Imamura T, Niho Y, Harada N. Signal transduction pathway of interleukin-4 and interleukin-13 in human B cells derived from X-linked severe combined immunodeficiency patients. J Biol Chem. 1996;271:619–622. doi: 10.1074/jbc.271.2.619. [DOI] [PubMed] [Google Scholar]
- 33.Taylor N, Candotti F, Smith S, Oakes SA, Jahn T, Isakov J, Puck JM, O'Shea JJ, Weinberg K, Johnston JA. Interleukin-4 signaling in B lymphocytes from patients with X-linked severe combined immunodeficiency. J Biol Chem. 1996;272:7314–7319. doi: 10.1074/jbc.272.11.7314. [DOI] [PubMed] [Google Scholar]
- 34.Levy F, Kristofic C, Heusser C, Brinkmann V. Role of IL-13 in CD4 T cell–dependent IgE production in atopy. Int Arch Allergy Immunol. 1997;112:49–58. doi: 10.1159/000237431. [DOI] [PubMed] [Google Scholar]
- 35.Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci USA. 1996;93:6231–6235. doi: 10.1073/pnas.93.13.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karras JG, Wang Z, Hou L, Frank DA, Rothstein TL. Induction of STAT protein signaling through the CD40 receptor in B lymphocytes: distinct STAT activation following surface Ig and CD40 receptor engagement. J Immunol. 1997;159:4350–4355. [PubMed] [Google Scholar]
- 37.Brewer JM, Conacher M, Satoskar A, Bluethmann H, Alexander J. In interleukin-4–deficient mice, alum not only generates T helper 1 responses equivalent to Freund's complete adjuvant, but continues to induce T helper 2 cytokine production. Eur J Immunol. 1996;26:2062–2066. doi: 10.1002/eji.1830260915. [DOI] [PubMed] [Google Scholar]